
HIF-1α-Mediated Disruption of Cellular Junctions: The Impact of Hypoxia on the Tumor Microenvironment and Invasion

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Molecular Basis of Hypoxic Signaling
2.1. HIF-1α: Master Regulator of Hypoxic Response in Cancer
2.2. Cross-Talk with PI3K-Akt, MAPK, and ROS
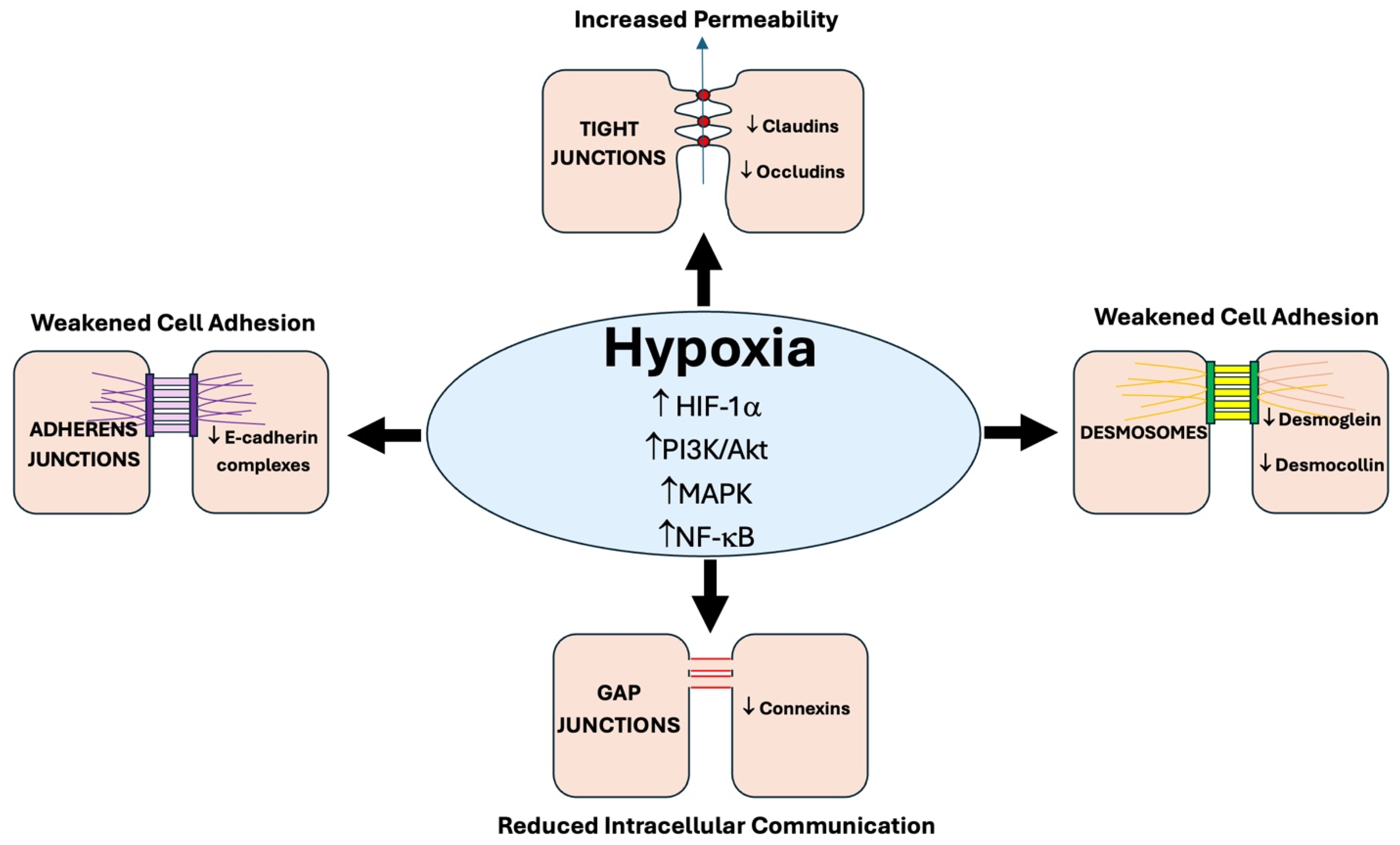
3. Hypoxia-Induced Disruption of Cell–Cell Junctions
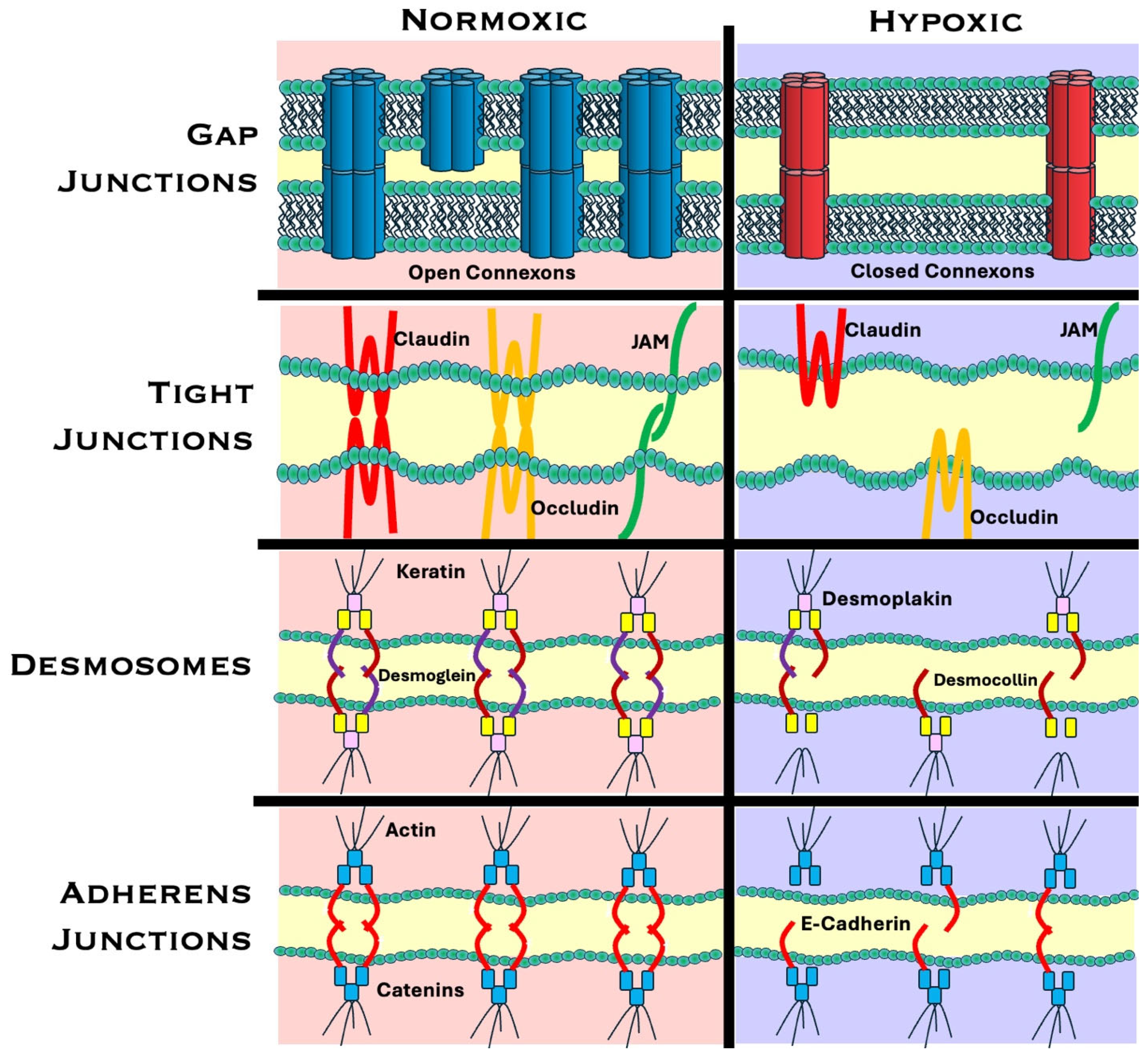
3.1. Gap Junctions: Structure and Function
3.1.1. Hypoxia and Gap Junctions
3.1.2. Mechanisms of Hypoxia-Induced Gap Junction Alterations
Transcriptional Regulation of Connexin Genes
Post-Translational Modifications
Gap Junction Channel Dysfunction
3.2. Adherens Junctions
3.2.1. Hypoxia-Induced Adherens Junction Alterations
3.2.2. Adherens Junctions and EMT
3.3. Tight Junctions
Hypoxia-Induced Tight Junction Alterations
3.4. Desmosomes
Hypoxia-Induced Desmosome Alterations
3.5. Alterations in Junctional Proteins and Cell Polarity
4. Pathophysiological Consequences of Junctional Breakdown
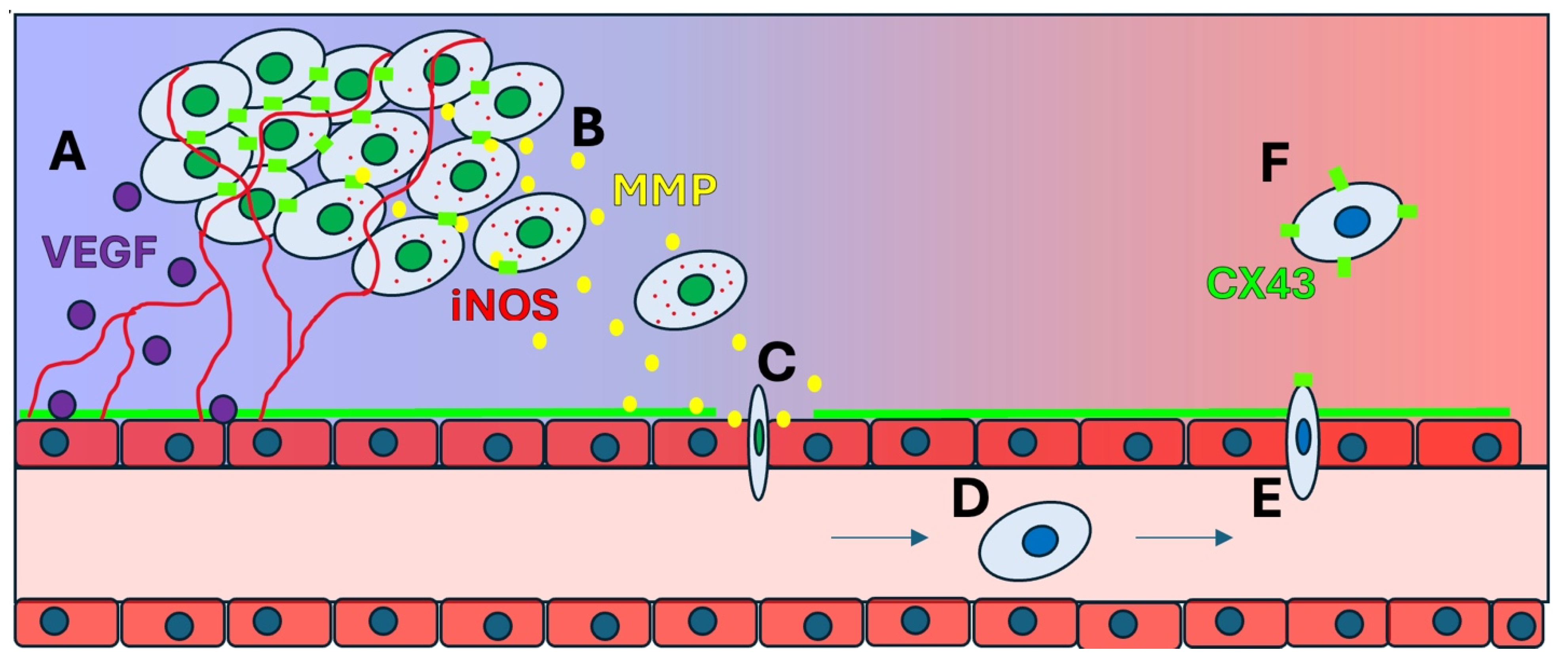
4.1. MMPs and ECM Remodeling
4.2. Invasion and Migration
4.3. The Role of iNOS and NO in Tumor Progression
4.4. COX-2 as a Driver of Angiogenesis and Inflammation
4.5. Immunological Impact of Hypoxia in the Tumor Microenvironment
5. Therapeutic Implications and Approaches
5.1. Resistance to Chemotherapy
5.2. Resistance to Radiation Therapy
5.3. HIF-1α as a Therapeutic Target
5.4. Therapy Aimed at Gap Junction Restoration
5.5. Therapy Directed at Reversing the EMT
5.6. Current Therapeutic Strategies
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAM15 | Disintegrin and metalloprotease |
| Akt | Ak strain transforming |
| ATF4 | Activating transcription factor 4 |
| circWSB1 | RNA molecule called circular RNA, derived from the WSB1 gene |
| CLDN1 | Claudin-1 |
| CLDN4 | Claudin 4 |
| COX-2 | Cyclooxygenase-2 |
| CTL | Cytotoxic T Lymphocytes |
| Cx | Connexin |
| Cx26 | Connexin 26 |
| Cx43 | Connexin 43 |
| DSG2 | Desmoglein-2 |
| DSGs | Desmogleins |
| DSP | Desmoplakin |
| E-cadherin | Epithelial cadherin |
| EC2 | Second extracellular domain in DSG2 |
| ECM | Extracellular membrane |
| EMT | Epithelial-to-mesenchymal transition |
| ERK | Extracellular Signal-Regulated Kinase |
| EZH2 | Enhancer of Zeste homolog 2 |
| GJ | Gap junction |
| GJA1 | Gap junction alpha-1 protein |
| GJIC | Gap junction intercellular communication |
| HIF | Hypoxia-inducible factor |
| HIF-1 | Hypoxia-inducible factor 1 |
| HIF-2a | Hypoxia-inducible factor 2-alpha |
| HRE | Hypoxia-response elements |
| iNOS | Nitric Oxide Synthase |
| JAM3 | Junctional adhesion molecule 3 |
| KC21 | Peptide that inhibits angiogenesis and retinal neovascularization |
| m6A | N6-methyladenosine |
| MAPK | Mitogen-activated protein kinases |
| MARVELD3 | MARVEL domain containing 3, gene that encodes a protein involved in tight junction assembly |
| MCF7 | Michigan Cancer Foundation-7 |
| MET | Mesenchymal-epithelial transition |
| MiR-23a | MicroRNA 23a |
| miRNA | MicroRNA |
| MMPs | Matrix metalloproteinases |
| mRNA | Messenger RNA |
| mTOR | Mammalian target of rapamycin |
| N-cadherin | Neural cadherin |
| NDR2 | Nodal-related 2, gene encodes a protein kinase important for neuronal development |
| NK | Natural Killer Cells |
| NO | Nitric Oxide |
| NSCLC | Non-small cell lung cancer |
| Par3 | Partitioning-defective 3, controls cell polarity, cell migration, proliferation, and development |
| PD-L1 | Programmed Cell Death Ligand |
| PHDs | Prolyl hydroxylase domain proteins |
| PI3K | Phosphoinositide 3-kinase |
| PRC2 | Polycomb Repressive Complex 2 |
| RONS | Reactive oxygen and nitrogen species |
| ROS | Reactive oxygen species |
| SNAI1 | Snail |
| Snail | EMT associated transcription factor, Snail family transcriptional repressor 1 |
| Src | Proto-oncogene tyrosine-protein kinase |
| SUZ12 | SUZ12 Polycomb repressive complex 2 subunit |
| TIMPs | Tissue inhibitors of metalloproteinases |
| TME | Tumor microenvironment |
| TWIST1 | EMT associated transcription factor, twist family transcription factor 1 |
| Twist | EMT associated transcription factor, twist family transcription factor 1 |
| USP10-p53 | Gene that encodes an ubiquitin-specific peptidase, p53 is a tumor suppressor protein |
| V-src | Transforming oncogene of the avian Rous Sarcoma virus |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFA | Vascular Endothelial Growth Factor A |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| ZEB | Zinc finger E-box binding homeobox gene |
| ZEB1 | Transcription factor regulating gene expression and cell behavior in relation to EMT |
| ZO-1 | Zonula occludens-1, plays an important role in tight junction structure and function |
References
- Brahimi-Horn, M.C.; Chiche, J.; Pouyssegur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Du, J.X.; Zhu, D.; Ren, S.Z.; Chen, K.; Zhu, H.L. Recent Research on Methods to Improve Tumor Hypoxia Environment. Oxidative Med. Cell. Longev. 2020, 2020, 5721258. [Google Scholar] [CrossRef]
- Sorensen, B.S.; Horsman, M.R. Tumor Hypoxia: Impact on Radiation Therapy and Molecular Pathways. Front. Oncol. 2020, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Chan, D.A.; Giaccia, A.J. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 2007, 26, 333–339. [Google Scholar] [CrossRef]
- Lal, A.; Peters, H.; St Croix, B.; Haroon, Z.A.; Dewhirst, M.W.; Strausberg, R.L.; Kaanders, J.H.; van der Kogel, A.J.; Riggins, G.J. Transcriptional response to hypoxia in human tumors. J. Natl. Cancer Inst. 2001, 93, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef]
- Brahimi-Horn, C.; Berra, E.; Pouyssegur, J. Hypoxia: The tumor’s gateway to progression along the angiogenic pathway. Trends Cell Biol. 2001, 11, S32–S36. [Google Scholar] [CrossRef]
- Luo, Z.; Tian, M.; Yang, G.; Tan, Q.; Chen, Y.; Li, G.; Zhang, Q.; Li, Y.; Wan, P.; Wu, J. Hypoxia signaling in human health and diseases: Implications and prospects for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 218. [Google Scholar] [CrossRef]
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef]
- Zeng, W.; Liu, P.; Pan, W.; Singh, S.R.; Wei, Y. Hypoxia and hypoxia inducible factors in tumor metabolism. Cancer Lett. 2015, 356 Pt A, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Melincovici, C.S.; Bosca, A.B.; Susman, S.; Marginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)—Key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar] [PubMed]
- Elebiyo, T.C.; Rotimi, D.; Evbuomwan, I.O.; Maimako, R.F.; Iyobhebhe, M.; Ojo, O.A.; Oluba, O.M.; Adeyemi, O.S. Reassessing vascular endothelial growth factor (VEGF) in anti-angiogenic cancer therapy. Cancer Treat. Res. Commun. 2022, 32, 100620. [Google Scholar] [CrossRef]
- Islam, S.M.T.; Won, J.; Khan, M.; Mannie, M.D.; Singh, I. Hypoxia-inducible factor-1 drives divergent immunomodulatory functions in the pathogenesis of autoimmune diseases. Immunology 2021, 164, 31–42. [Google Scholar] [CrossRef]
- Malkov, M.I.; Lee, C.T.; Taylor, C.T. Regulation of the Hypoxia-Inducible Factor (HIF) by Pro-Inflammatory Cytokines. Cells 2021, 10, 2340. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Xie, L. Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology 2014, 146, 630–642. [Google Scholar] [CrossRef]
- Cimmino, F.; Avitabile, M.; Lasorsa, V.A.; Montella, A.; Pezone, L.; Cantalupo, S.; Visconte, F.; Corrias, M.V.; Iolascon, A.; Capasso, M. HIF-1 transcription activity: HIF1A driven response in normoxia and in hypoxia. BMC Med. Genet. 2019, 20, 37. [Google Scholar] [CrossRef] [PubMed]
- Span, P.N.; Bussink, J. Biology of hypoxia. Semin. Nucl. Med. 2015, 45, 101–109. [Google Scholar] [CrossRef]
- Tan, T.; Shi, P.; Abbas, M.N.; Wang, Y.; Xu, J.; Chen, Y.; Cui, H. Epigenetic modification regulates tumor progression and metastasis through EMT (Review). Int. J. Oncol. 2022, 60, 70. [Google Scholar] [CrossRef]
- Liu, H.; Li, X.Z.; Peng, M.; Ji, W.; Zhao, L.; Li, L.; Zhang, L.; Si, J.Q.; Ma, K.T. Role of gap junctions in the contractile response to agonists in the mesenteric resistance artery of rats with acute hypoxia. Mol. Med. Rep. 2017, 15, 1823–1831. [Google Scholar] [CrossRef]
- Goodenough, D.A.; Paul, D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576. [Google Scholar] [CrossRef]
- Kotini, M.; Mayor, R. Connexins in migration during development and cancer. Dev. Biol. 2015, 401, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Kutova, O.M.; Pospelov, A.D.; Balalaeva, I.V. The Multifaceted Role of Connexins in Tumor Microenvironment Initiation and Maintenance. Biology 2023, 12, 204. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2017, 17, 74. [Google Scholar] [CrossRef]
- Zhou, M.; Zheng, M.; Zhou, X.; Tian, S.; Yang, X.; Ning, Y.; Li, Y.; Zhang, S. The roles of connexins and gap junctions in the progression of cancer. Cell Commun. Signal 2023, 21, 8. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta Biomembr. 2018, 1860, 83–90. [Google Scholar] [CrossRef]
- Sosinsky, G.E.; Nicholson, B.J. Structural organization of gap junction channels. Biochim. Biophys. Acta 2005, 1711, 99–125. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef]
- Zucker, S.N.; Nicholson, B.J. Mutagenic approaches to modifying gap junction phenotype. Curr. Drug Targets 2002, 3, 441–453. [Google Scholar] [CrossRef]
- Paunikar, S.; Tamagnone, L. Connexin-43 in Cancer: Above and Beyond Gap Junctions! Cancers 2024, 16, 4191. [Google Scholar] [CrossRef]
- Bagati, A.; Hutcherson, T.C.; Koch, Z.; Pechette, J.; Dianat, H.; Higley, C.; Chiu, L.; Song, Y.; Shah, J.; Chazen, E.; et al. Novel combination therapy for melanoma induces apoptosis via a gap junction positive feedback mechanism. Oncotarget 2020, 11, 3443–3458. [Google Scholar] [CrossRef]
- Oliveira, M.C.; Cordeiro, R.M.; Bogaerts, A. Effect of lipid oxidation on the channel properties of Cx26 hemichannels: A molecular dynamics study. Arch. Biochem. Biophys. 2023, 746, 109741. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H. The HIF pathway in cancer. Semin. Cell Dev. Biol. 2005, 16, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Spray, D.C.; Hanstein, R.; Lopez-Quintero, S.V.; Stout, R.F., Jr.; Suadicani, S.O.; Thi, M.M. Gap junctions and Bystander Effects: Good Samaritans and executioners. Wiley Interdiscip. Rev. Membr. Transp. Signal 2013, 2, 1–15. [Google Scholar] [CrossRef]
- Hompland, T.; Fjeldbo, C.S.; Lyng, H. Tumor Hypoxia as a Barrier in Cancer Therapy: Why Levels Matter. Cancers 2021, 13, 499. [Google Scholar] [CrossRef] [PubMed]
- Zefferino, R.; Piccoli, C.; Gioia, S.D.; Capitanio, N.; Conese, M. Gap Junction Intercellular Communication in the Carcinogenesis Hallmarks: Is This a Phenomenon or Epiphenomenon? Cells 2019, 8, 896. [Google Scholar] [CrossRef]
- Yang, Z.J.; Bi, Q.C.; Gan, L.J.; Zhang, L.L.; Wei, M.J.; Hong, T.; Liu, R.; Qiu, C.L.; Han, X.J.; Jiang, L.P. Exosomes Derived from Glioma Cells under Hypoxia Promote Angiogenesis through Up-regulated Exosomal Connexin 43. Int. J. Med. Sci. 2022, 19, 1205–1215. [Google Scholar] [CrossRef]
- Han, X.J.; Zhang, W.F.; Wang, Q.; Li, M.; Zhang, C.B.; Yang, Z.J.; Tan, R.J.; Gan, L.J.; Zhang, L.L.; Lan, X.M.; et al. HIF-1alpha promotes the proliferation and migration of pulmonary arterial smooth muscle cells via activation of Cx43. J. Cell Mol. Med. 2021, 25, 10663–10673. [Google Scholar] [CrossRef]
- Gillies, R.J.; Schornack, P.A.; Secomb, T.W.; Raghunand, N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia 1999, 1, 197–207. [Google Scholar] [CrossRef]
- Singh, A.K.; Cancelas, J.A. Gap Junctions in the Bone Marrow Lympho-Hematopoietic Stem Cell Niche, Leukemia Progression, and Chemoresistance. Int. J. Mol. Sci. 2020, 21, 796. [Google Scholar] [CrossRef]
- McNair, A.J.; Wilson, K.S.; Martin, P.E.; Welsh, D.J.; Dempsie, Y. Connexin 43 plays a role in proliferation and migration of pulmonary arterial fibroblasts in response to hypoxia. Pulm. Circ. 2020, 10, 2045894020937134. [Google Scholar] [CrossRef]
- Chandrasekhar, A.; Bera, A.K. Hemichannels: Permeants and their effect on development, physiology and death. Cell Biochem. Funct. 2012, 30, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.K.; Chen, M.C.; Leong, H.F.; Kuo, Y.L.; Kuo, C.Y.; Lee, C.H. Connexin 43 suppresses tumor angiogenesis by down-regulation of vascular endothelial growth factor via hypoxic-induced factor-1alpha. Int. J. Mol. Sci. 2014, 16, 439–451. [Google Scholar] [CrossRef]
- Peracchia, C. Calcium Role in Gap Junction Channel Gating: Direct Electrostatic or Calmodulin-Mediated? Int. J. Mol. Sci. 2024, 25, 9789. [Google Scholar] [CrossRef]
- Rodriguez-Candela Mateos, M.; Carpintero-Fernandez, P.; Freijanes, P.S.; Mosquera, J.; Nebril, B.A.; Mayan, M.D. Insights into the role of connexins and specialized intercellular communication pathways in breast cancer: Mechanisms and applications. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189173. [Google Scholar] [CrossRef] [PubMed]
- Troyanovsky, S.M. Adherens junction: The ensemble of specialized cadherin clusters. Trends Cell Biol. 2023, 33, 374–387. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Pouyssegur, J. Hypoxia in cancer cell metabolism and pH regulation. Essays Biochem. 2007, 43, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Bhattacharya, S.; Kalyan, G.; Hazra, S. Cadherin profiling for therapeutic interventions in Epithelial Mesenchymal Transition (EMT) and tumorigenesis. Exp. Cell Res. 2018, 368, 137–146. [Google Scholar] [CrossRef]
- Imai, T.; Horiuchi, A.; Wang, C.; Oka, K.; Ohira, S.; Nikaido, T.; Konishi, I. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am. J. Pathol. 2003, 163, 1437–1447. [Google Scholar] [CrossRef]
- Indra, I.; Hong, S.; Troyanovsky, R.; Kormos, B.; Troyanovsky, S. The adherens junction: A mosaic of cadherin and nectin clusters bundled by actin filaments. J. Investig. Dermatol. 2013, 133, 2546–2554. [Google Scholar] [CrossRef]
- Amack, J.D. Cellular dynamics of EMT: Lessons from live in vivo imaging of embryonic development. Cell Commun. Signal 2021, 19, 79. [Google Scholar] [CrossRef]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2015, 54, 554–565. [Google Scholar] [CrossRef]
- Raykhel, I.; Ronkainen, V.P.; Myllyharju, J.; Manninen, A. HIF2alpha-dependent Dock4/Rac1-signaling regulates formation of adherens junctions and cell polarity in normoxia. Sci. Rep. 2024, 14, 12153. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Fanlo, L.; Siles, L.; Montes-Moreno, S.; Moros, A.; Chiva-Blanch, G.; Estruch, R.; Martinez, A.; Colomer, D.; Gyorffy, B.; et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. 2014, 21, 247–257. [Google Scholar] [CrossRef]
- Monga, S.P. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 2015, 148, 1294–1310. [Google Scholar] [CrossRef] [PubMed]
- Hapke, R.Y.; Haake, S.M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 2020, 487, 10–20. [Google Scholar] [CrossRef]
- Kyuno, D.; Takasawa, A.; Kikuchi, S.; Takemasa, I.; Osanai, M.; Kojima, T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef]
- Corallino, S.; Malabarba, M.G.; Zobel, M.; Di Fiore, P.P.; Scita, G. Epithelial-to-Mesenchymal Plasticity Harnesses Endocytic Circuitries. Front. Oncol. 2015, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell Res. 2017, 358, 39–44. [Google Scholar] [CrossRef]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.; Moon, E.H.; Kim, G.J.; Choi, J. Hypoxia disrupt tight junctions and promote metastasis of oral squamous cell carcinoma via loss of par3. Cancer Cell Int. 2023, 23, 79. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Chen, Y.; Yin, A. JAM3 promotes cervical cancer metastasis by activating the HIF-1alpha/VEGFA pathway. BMC Womens Health 2024, 24, 293. [Google Scholar] [CrossRef] [PubMed]
- Tabaries, S.; Siegel, P.M. The role of claudins in cancer metastasis. Oncogene 2017, 36, 1176–1190. [Google Scholar] [CrossRef]
- Osanai, M.; Takasawa, A.; Murata, M.; Sawada, N. Claudins in cancer: Bench to bedside. Pflug. Arch. 2017, 469, 55–67. [Google Scholar] [CrossRef]
- Wang, D.W.; Zhang, W.H.; Danil, G.; Yang, K.; Hu, J.K. The role and mechanism of claudins in cancer. Front. Oncol. 2022, 12, 1051497. [Google Scholar] [CrossRef]
- Mattern, J.; Roghi, C.S.; Hurtz, M.; Knauper, V.; Edwards, D.R.; Poghosyan, Z. ADAM15 mediates upregulation of Claudin-1 expression in breast cancer cells. Sci. Rep. 2019, 9, 12540. [Google Scholar] [CrossRef]
- Chang, P.H.; Chen, M.C.; Tsai, Y.P.; Tan, G.Y.T.; Hsu, P.H.; Jeng, Y.M.; Tsai, Y.F.; Yang, M.H.; Hwang-Verslues, W.W. Interplay between desmoglein2 and hypoxia controls metastasis in breast cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2014408118. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Lin, Y.S.; Pan, Y.C.; Tsai, P.H.; Wu, C.Y.; Kuo, P.L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Wen, Y.; Chen, S.; Zhou, C.; Wu, F. Single-cell transcriptomics reveal metastatic CLDN4+ cancer cells underlying the recurrence of malignant pleural effusion in patients with advanced non-small-cell lung cancer. Clin. Transl. Med. 2024, 14, e1649. [Google Scholar] [CrossRef]
- Kojima, T.; Takasawa, A.; Kyuno, D.; Ito, T.; Yamaguchi, H.; Hirata, K.; Tsujiwaki, M.; Murata, M.; Tanaka, S.; Sawada, N. Downregulation of tight junction-associated MARVEL protein marvelD3 during epithelial-mesenchymal transition in human pancreatic cancer cells. Exp. Cell Res. 2011, 317, 2288–2298. [Google Scholar] [CrossRef]
- Wei, D.-F.; Tang, M.-K.; Liu, Y.; Zhang, C.-Y.; Qin, L.-J. Effect of Hypoxia Inducible Factor-1 Alpha on Brain Metastasis from Lung Cancer and Its Mechanism. J. Sichuan Univ. 2019, 50, 188–192. [Google Scholar]
- Perl, A.L.; Pokorny, J.L.; Green, K.J. Desmosomes at a glance. J. Cell Sci. 2024, 137, jcs261899. [Google Scholar] [CrossRef]
- Najor, N.A. Desmosomes in Human Disease. Annu. Rev. Pathol. 2018, 13, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Dusek, R.L.; Godsel, L.M.; Green, K.J. Discriminating roles of desmosomal cadherins: Beyond desmosomal adhesion. J. Dermatol. Sci. 2007, 45, 7–21. [Google Scholar] [CrossRef]
- Augustin, R.C.; Delgoffe, G.M.; Najjar, Y.G. Characteristics of the Tumor Microenvironment That Influence Immune Cell Functions: Hypoxia, Oxidative Stress, Metabolic Alterations. Cancers 2020, 12, 3802. [Google Scholar] [CrossRef]
- Huang, Z.; Tang, Y.; Zhang, J.; Huang, J.; Cheng, R.; Guo, Y.; Kleer, C.G.; Wang, Y.; Xue, L. Hypoxia makes EZH2 inhibitor not easy-advances of crosstalk between HIF and EZH2. Life Metab. 2024, 3, loae017. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.S.; Lee, Y.N.; Wang, S.W.; Wu, Y.J.; Su, C.H.; Hsieh, C.L.; Tien, T.Y.; Wang, B.J.; Chen, M.C.; Chen, C.W.; et al. KC21 Peptide Inhibits Angiogenesis and Attenuates Hypoxia-Induced Retinopathy. J. Cardiovasc. Transl. Res. 2019, 12, 366–377. [Google Scholar] [CrossRef]
- Wang, J.; Yang, C.; Xu, H.; Fan, X.; Jia, L.; Du, Y.; Liu, S.; Wang, W.; Zhang, J.; Zhang, Y.; et al. The Interplay Between HIF-1alpha and EZH2 in Lung Cancer and Dual-Targeted Drug Therapy. Adv. Sci. 2024, 11, e2303904. [Google Scholar] [CrossRef]
- Bai, X.; Huang, J.; Jin, Y.; Chen, J.; Zhou, S.; Dong, L.; Han, X.; He, X. M6A RNA methylation in biliary tract cancer: The function roles and potential therapeutic implications. Cell Death Discov. 2024, 10, 83. [Google Scholar] [CrossRef]
- Lesko, A.C.; Goss, K.H.; Yang, F.F.; Schwertner, A.; Hulur, I.; Onel, K.; Prosperi, J.R. The APC tumor suppressor is required for epithelial cell polarization and three-dimensional morphogenesis. Biochim. Biophys. Acta 2015, 1853, 711–723. [Google Scholar] [CrossRef]
- Bruzzone, S.; Guida, L.; Zocchi, E.; Franco, L.; De Flora, A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001, 15, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.; Xu, X.; Park, H.; Wei, C.J.; Chang, S.; Chatterjee, B.; Lo, C. Connexin43 modulates cell polarity and directional cell migration by regulating microtubule dynamics. PLoS ONE 2011, 6, e26379. [Google Scholar] [CrossRef]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef]
- Borradori, L.; Sonnenberg, A. Structure and function of hemidesmosomes: More than simple adhesion complexes. J. Investig. Dermatol. 1999, 112, 411–418. [Google Scholar] [CrossRef]
- Zeng, S.G.; Lin, X.; Liu, J.C.; Zhou, J. Hypoxia-induced internalization of connexin 26 and connexin 43 in pulmonary epithelial cells is involved in the occurrence of non-small cell lung cancer via the P53/MDM2 signaling pathway. Int. J. Oncol. 2019, 55, 845–859. [Google Scholar] [CrossRef]
- Amar, S.; Smith, L.; Fields, G.B. Matrix metalloproteinase collagenolysis in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864 Pt A, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31 (Suppl. S1), 177–183. [Google Scholar] [CrossRef]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Merchant, N.; Nagaraju, G.P.; Rajitha, B.; Lammata, S.; Jella, K.K.; Buchwald, Z.S.; Lakka, S.S.; Ali, A.N. Matrix metalloproteinases: Their functional role in lung cancer. Carcinogenesis 2017, 38, 766–780. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Kannikal, J.; Nath, N. Macrophage Reprogramming and Cancer Therapeutics: Role of iNOS-Derived NO. Cells 2021, 10, 3194. [Google Scholar] [CrossRef]
- Belgorosky, D.; Girouard, J.; Langle, Y.V.; Hamelin-Morrissete, J.; Marino, L.; Aguero, E.I.; Malagrino, H.; Reyes-Moreno, C.; Eijan, A.M. Relevance of iNOS expression in tumor growth and maintenance of cancer stem cells in a bladder cancer model. J. Mol. Med. 2020, 98, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, V.; Basudhar, D.; Bharadwaj, G.; No, J.H.; Ridnour, L.A.; Cheng, R.Y.S.; Fujita, M.; Thomas, D.D.; Anderson, S.K.; McVicar, D.W.; et al. Molecular Mechanisms of Nitric Oxide in Cancer Progression, Signal Transduction, and Metabolism. Antioxid. Redox Signal. 2019, 30, 1124–1143. [Google Scholar] [CrossRef]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Ben-Batalla, I.; Cubas-Cordova, M.; Udonta, F.; Wroblewski, M.; Waizenegger, J.S.; Janning, M.; Sawall, S.; Gensch, V.; Zhao, L.; Martinez-Zubiaurre, I.; et al. Cyclooxygenase-2 blockade can improve efficacy of VEGF-targeting drugs. Oncotarget 2015, 6, 6341–6358. [Google Scholar] [CrossRef]
- De Paz Linares, G.A.; Opperman, R.M.; Majumder, M.; Lala, P.K. Prostaglandin E2 Receptor 4 (EP4) as a Therapeutic Target to Impede Breast Cancer-Associated Angiogenesis and Lymphangiogenesis. Cancers 2021, 13, 942. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112. [Google Scholar] [CrossRef]
- Fu, Z.; Mowday, A.M.; Smaill, J.B.; Hermans, I.F.; Patterson, A.V. Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy. Cells 2021, 10, 1006. [Google Scholar] [CrossRef]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia Enhances Immunosuppression by Inhibiting CD4+ Effector T Cell Function and Promoting Treg Activity. Cell Physiol. Biochem. 2017, 41, 1271–1284. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef]
- Vuillefroy de Silly, R.; Dietrich, P.Y.; Walker, P.R. Hypoxia and antitumor CD8(+) T cells: An incompatible alliance? Oncoimmunology 2016, 5, e1232236. [Google Scholar] [CrossRef]
- Bouleftour, W.; Rowinski, E.; Louati, S.; Sotton, S.; Wozny, A.S.; Moreno-Acosta, P.; Mery, B.; Rodriguez-Lafrasse, C.; Magne, N. A Review of the Role of Hypoxia in Radioresistance in Cancer Therapy. Med. Sci. Monit. 2021, 27, e934116. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.E.; Yakimova, A.O. Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing. Cancers 2021, 13, 1102. [Google Scholar] [CrossRef] [PubMed]
- Menegakis, A.; Klompmaker, R.; Vennin, C.; Arbusa, A.; Damen, M.; van den Broek, B.; Zips, D.; van Rheenen, J.; Krenning, L.; Medema, R.H. Resistance of Hypoxic Cells to Ionizing Radiation Is Mediated in Part via Hypoxia-Induced Quiescence. Cells 2021, 10, 610. [Google Scholar] [CrossRef]
- Zdrowowicz, M.; Spisz, P.; Hac, A.; Herman-Antosiewicz, A.; Rak, J. Influence of Hypoxia on Radiosensitization of Cancer Cells by 5-Bromo-2′-deoxyuridine. Int. J. Mol. Sci. 2022, 23, 1429. [Google Scholar] [CrossRef]
- Mancuso, M.; Pasquali, E.; Leonardi, S.; Rebessi, S.; Tanori, M.; Giardullo, P.; Borra, F.; Pazzaglia, S.; Naus, C.C.; Di Majo, V.; et al. Role of connexin43 and ATP in long-range bystander radiation damage and oncogenesis in vivo. Oncogene 2011, 30, 4601–4608. [Google Scholar] [CrossRef]
- Bui, B.P.; Nguyen, P.L.; Lee, K.; Cho, J. Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers 2022, 14, 6054. [Google Scholar] [CrossRef]
- Giaccia, A.; Siim, B.G.; Johnson, R.S. HIF-1 as a target for drug development. Nat. Rev. Drug Discov. 2003, 2, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Bery, F.; Figiel, S.; Kouba, S.; Fontaine, D.; Gueguinou, M.; Potier-Cartereau, M.; Vandier, C.; Guibon, R.; Bruyere, F.; Fromont, G.; et al. Hypoxia Promotes Prostate Cancer Aggressiveness by Upregulating EMT-Activator Zeb1 and SK3 Channel Expression. Int. J. Mol. Sci. 2020, 21, 4786. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Zhan, Z.; Gan, L.; Bai, O. Mechanisms of HDACs in cancer development. Front. Immunol. 2025, 16, 1529239. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Springer, M.; Burakgazi, Z.A.; Domukhovska, A.; Nafchi, B.; Beary, M.C.; Acquisto, A.; Acquisto, J.; Komarov, V.; Jensen, M.; Gulledge, B.; et al. HIF-1α-Mediated Disruption of Cellular Junctions: The Impact of Hypoxia on the Tumor Microenvironment and Invasion. Int. J. Mol. Sci. 2025, 26, 5101. https://doi.org/10.3390/ijms26115101
Springer M, Burakgazi ZA, Domukhovska A, Nafchi B, Beary MC, Acquisto A, Acquisto J, Komarov V, Jensen M, Gulledge B, et al. HIF-1α-Mediated Disruption of Cellular Junctions: The Impact of Hypoxia on the Tumor Microenvironment and Invasion. International Journal of Molecular Sciences. 2025; 26(11):5101. https://doi.org/10.3390/ijms26115101
Chicago/Turabian StyleSpringer, Michael, Zeynep Aydin Burakgazi, Anastasiia Domukhovska, Ben Nafchi, Michael C. Beary, Arielle Acquisto, Juliette Acquisto, Vladyslav Komarov, Madison Jensen, Brandon Gulledge, and et al. 2025. "HIF-1α-Mediated Disruption of Cellular Junctions: The Impact of Hypoxia on the Tumor Microenvironment and Invasion" International Journal of Molecular Sciences 26, no. 11: 5101. https://doi.org/10.3390/ijms26115101
APA StyleSpringer, M., Burakgazi, Z. A., Domukhovska, A., Nafchi, B., Beary, M. C., Acquisto, A., Acquisto, J., Komarov, V., Jensen, M., Gulledge, B., Poplavskyi, M., Uddin, M. G., Rayan, G., & Zucker, S. N. (2025). HIF-1α-Mediated Disruption of Cellular Junctions: The Impact of Hypoxia on the Tumor Microenvironment and Invasion. International Journal of Molecular Sciences, 26(11), 5101. https://doi.org/10.3390/ijms26115101