
The Molecular Mechanisms of Cognitive Dysfunction in Long COVID: A Narrative Review
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
3. Neurological and Cognitive Manifestations of Long COVID
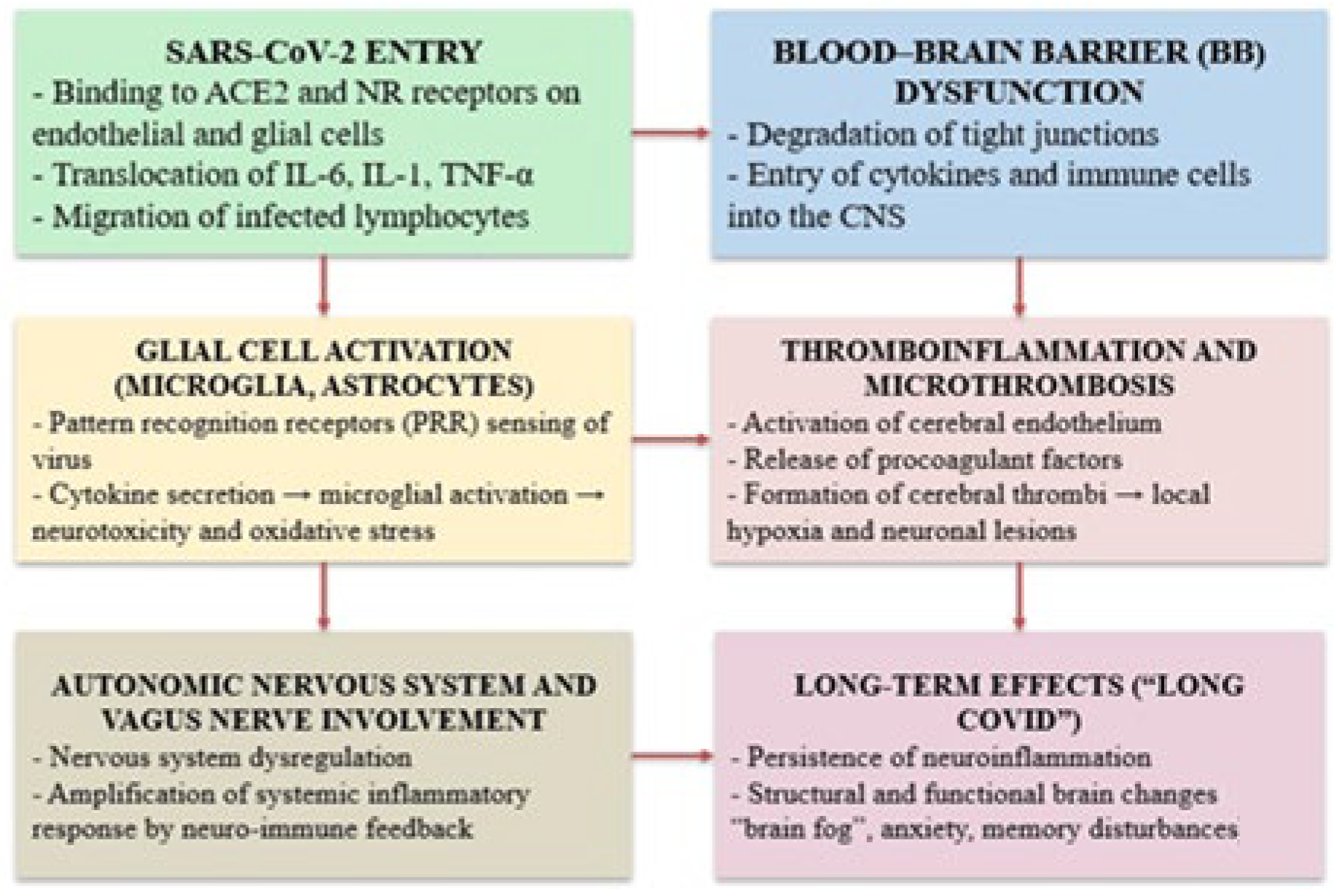
4. Pathogenic Mechanisms Underlying Neurological Dysfunction
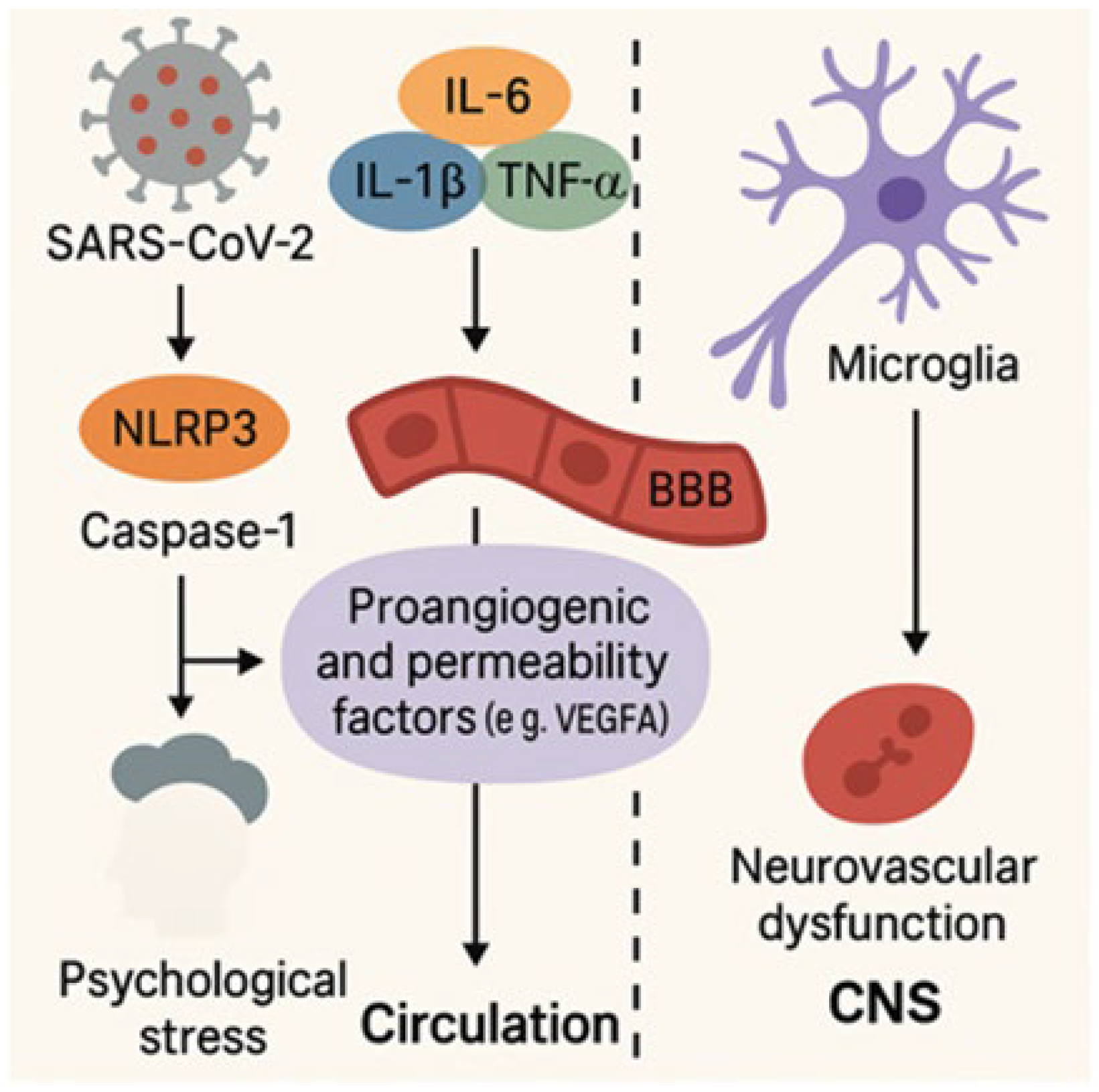
4.1. Neuroinflammation and Its Impact on Brain Function
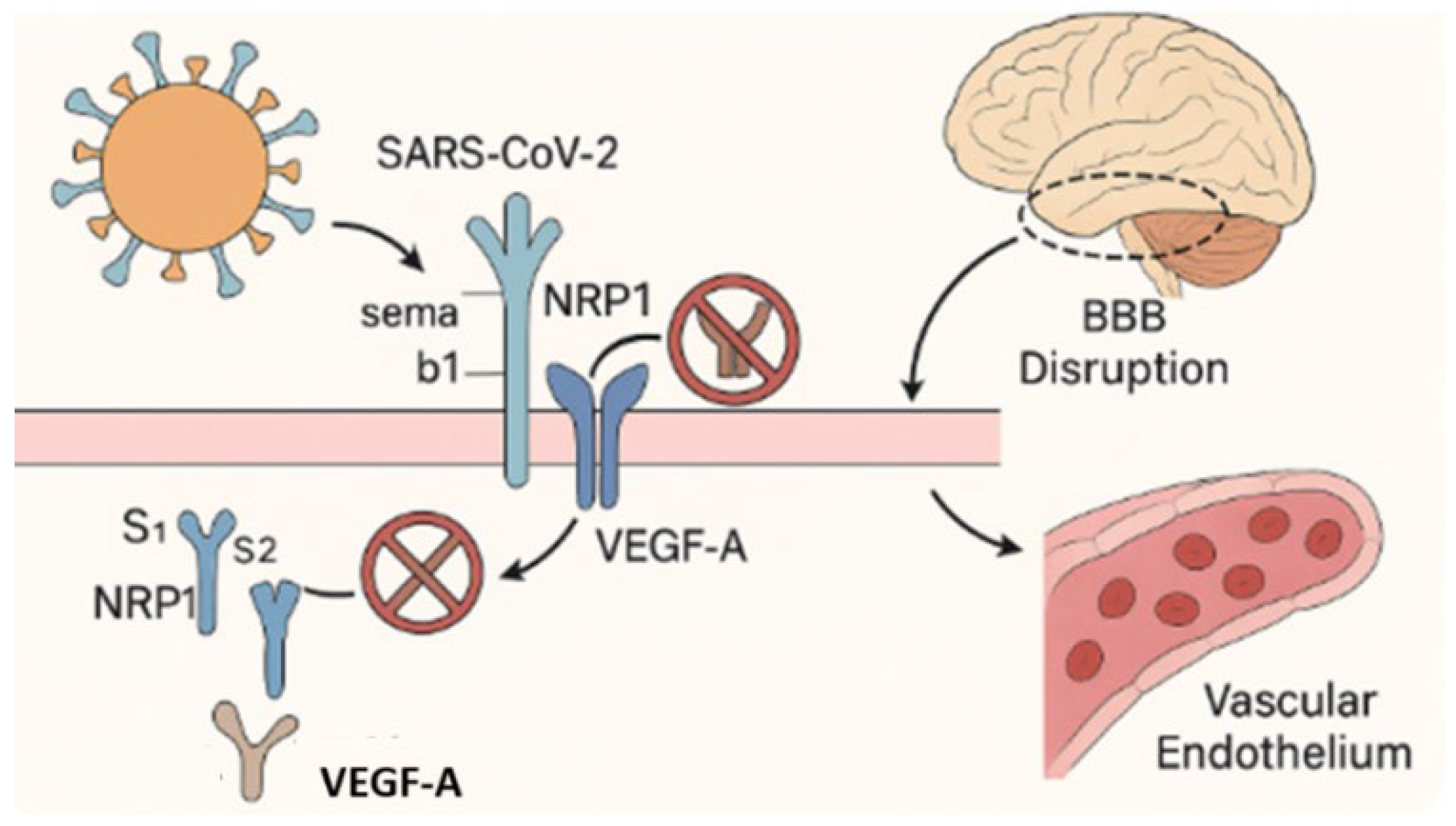
4.2. Blood–Brain Barrier Disruption and Neurovascular Injury in Long COVID
4.3. Endothelial Dysfunction and Cerebral Microvascular Thrombosis in Long COVID
4.4. Neuroendocrine Dysregulation and Autonomic Nervous System Dysfunction in Long COVID
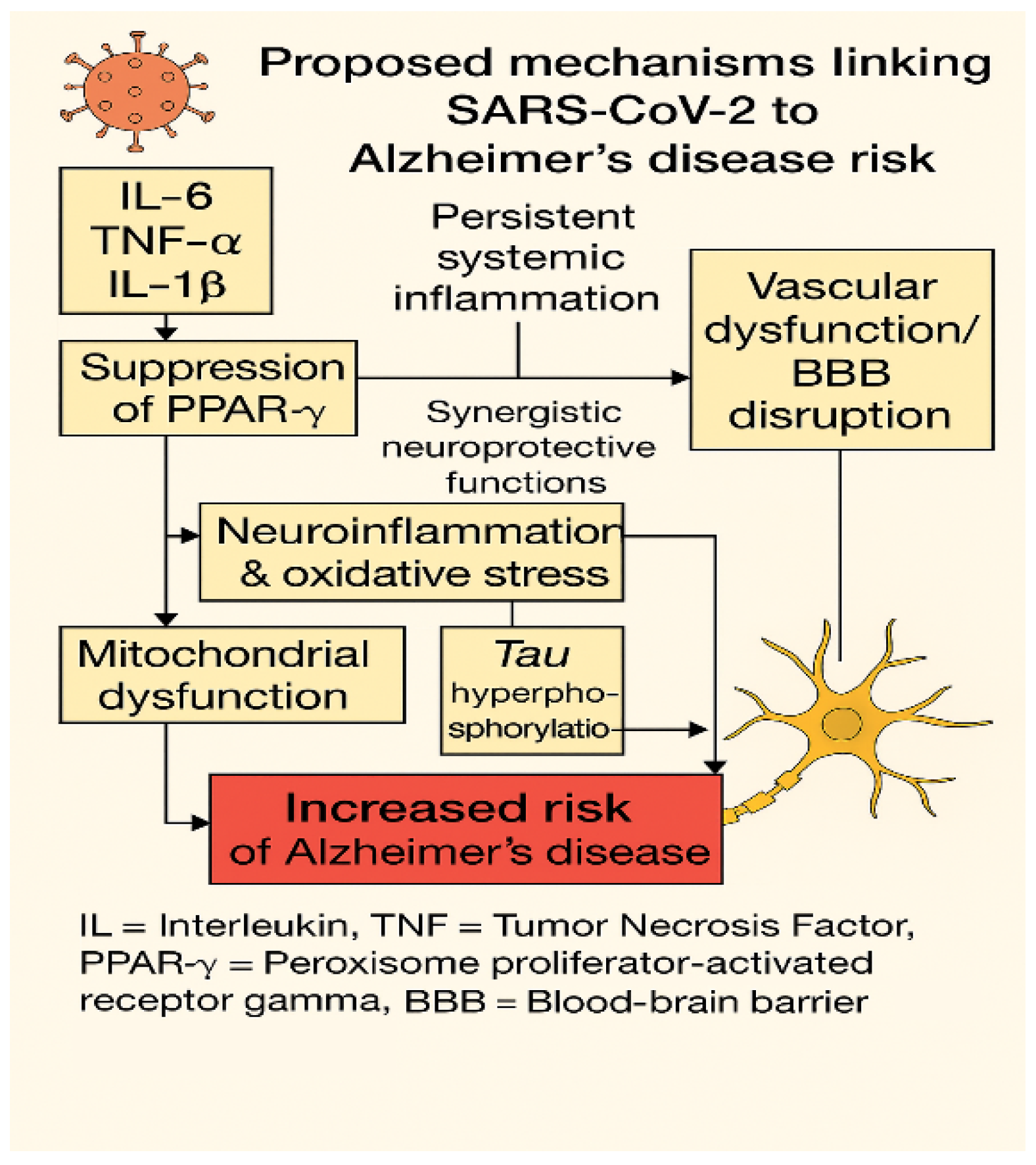
5. Converging Pathogenic Mechanisms Between Long COVID and Alzheimer’s Disease
6. Molecular Markers and Therapeutic Targets
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-converting enzyme 2 |
| ADEVs | Astrocyte-derived extracellular vesicles |
| ANS | Autonomic nervous system |
| APOE-ε4 | Apolipoprotein E epsilon 4 |
| ATP | Adenosine triphosphate |
| BBB | Blood–brain barrier |
| B-CSF | Blood–cerebrospinal fluid barrier |
| CAP | Cholinergic anti-inflammatory pathway |
| CD | Cluster of differentiation |
| CDC | Centers for Disease Control and Prevention |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CXCL | C-X-C motif chemokine ligand |
| ECs | Endothelial cells |
| EV | Extracellular vehicles |
| GFAP | Glial fibrillary acidic protein |
| GM-CSF | Granulocyte–macrophage colony-stimulating factor |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HRV | Heart rate variability |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IFN | Interferon |
| IL | Interleukin |
| KKS | Kallikrein–kinin system |
| LGALS3 | Galectin-3 |
| miRNAs | MicroRNA |
| ME/CFS | Myalgic encephalomyelitis/chronic fatigue syndrome |
| MMSE | Mini-mental state examination |
| MoCA | Montreal cognitive assessment |
| MMP-9 | Matrix metalloproteinase 9 |
| NFL | Neurofilament light chain |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NOD-, LRR-, and pyrin domain-containing protein 3 |
| NRP1 | Neuropilin-1 |
| NDEVs | Neuron-derived extracellular vesicles |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PASC | Post-acute sequelae of SARS-CoV-2 infection |
| PBMCs | Peripheral blood mononuclear cells |
| POTS | Postural orthostatic tachycardia syndrome |
| PRRs | Pattern recognition receptors |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| ROS | Reactive oxygen species |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SDMT Symbol | Digit modalities test |
| S1/S2 | Subunit 1/Subunit 2 (spike protein cleavage sites) |
| S100B | S100 calcium-binding protein β |
| TGF-β | Transforming growth factor beta |
| TIAs | Transient ischemic attacks |
| TMPRSS2 | Transmembrane protease serine 2 |
| TNF-α | Tumor necrosis factor alpha |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF-A | Vascular endothelial growth factor A |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| WHO | World Health Organization |
References
- Servier, C.; Porcher, R.; Pane, I.; Ravaud, P.; Tran, V.T. Trajectories of the evolution of post-COVID-19 condition, up to two years after symptoms onset. Int. J. Infect. Dis. 2023, 133, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major Findings, Mechanisms and Recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Al-Aly, Z.; Davis, H.; McCorkell, L.; Wulf-Hanson, S.; Iwasaki, A.; Topol, E.J. Long COVID science, research and policy. Nat. Med. 2024, 30, 2148–2164. [Google Scholar] [CrossRef]
- Reese, J.T.; Blau, H.; Casiraghi, E.; Bergquist, T.; Loomba, J.J.; Callahan, T.J.; Laraway, B.; Antonescu, C.; Coleman, B.; Gargano, M.; et al. Generalisable long COVID subtypes: Findings from the NIH N3C and RECOVER programmes. EBioMedicine 2023, 87, 104413. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Post-COVID-19 Condition. Available online: https://www.who.int/europe/news-room/fact-sheets/item/post-COVID-19-condition (accessed on 15 February 2025).
- Brown, D.A.; O’Brien, K.K. Conceptualising Long COVID as an episodic health condition. BMJ Glob. Health 2021, 6, e007004. [Google Scholar] [CrossRef]
- Arbula, S.; Pisanu, E.; Bellavita, G.; Menichelli, A.; Lunardelli, A.; Furlanis, G.; Manganotti, P.; Cappa, S.; Rumiati, R. Insights into attention and memory difficulties in post-COVID syndrome using standardized neuropsychological tests and experimental cognitive tasks. Sci. Rep. 2023, 13, 54613. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, S.G.; Garbarino, S.; Bellucci, M.; Schenone, C.; Candiani, V.; Di Lillo, S.; Campi, C.; Barisione, E.; Aloè, T.; Tagliabue, E.; et al. Neurological Long COVID in the Outpatient Clinic: Is It so Long? Eur. J. Neurol. 2025, 32, e16510. [Google Scholar] [CrossRef] [PubMed]
- Taruffi, L.; Muccioli, L.; Mitolo, M.; Ferri, L.; Descovich, C.; Mazzoni, S.; Michelucci, R.; Lodi, R.; Liguori, R.; Cortelli, P.; et al. Neurological Manifestations of Long COVID: A Single-Center One-Year Experience. Neuropsychiatr. Dis. Treat. 2023, 19, 311–319. [Google Scholar] [CrossRef]
- Holdsworth, D.A.; Chamley, R.; Barker-Davies, R.; O’Sullivan, O.; Ladlow, P.; Mitchell, J.L.; Dewson, D.; Mills, D.; May, S.L.J.; Cranley, M.; et al. Comprehensive clinical assessment identifies specific neurocognitive deficits in working-age patients with long-COVID. PLoS ONE 2022, 17, e0267392. [Google Scholar] [CrossRef]
- Aljadah, M.; Khan, N.; Beyer, A.M.; Chen, Y.; Blanker, A.; Widlansky, M.E. Clinical Implications of COVID-19-Related Endothelial Dysfunction. JACC Adv. 2024, 3, 101070. [Google Scholar] [CrossRef]
- Becker, J.H.; Lin, J.J.; Doernberg, M.; Stone, K.; Navis, A.; Festa, J.R.; Wisnivesky, J.P. Assessment of Cognitive Function in Patients After COVID-19 Infection. JAMA Netw. Open 2021, 4, e2130645. [Google Scholar] [CrossRef] [PubMed]
- Crivelli, L.; Palmer, K.; Calandri, I.; Guekht, A.; Beghi, E.; Carroll, W.; Frontera, J.; García-Azorín, D.; Westenberg, E.; Winkler, A.S.; et al. Changes in cognitive functioning after COVID-19: A systematic review and meta-analysis. Alzheimers Dement. 2022, 18, 1047–1066. [Google Scholar] [CrossRef] [PubMed]
- Panagea, E.; Messinis, L.; Petri, M.C.; Liampas, I.; Anyfantis, E.; Nasios, G.; Patrikelis, P.; Kosmidis, M. Neurocognitive Impairment in Long COVID: A Systematic Review. Arch. Clin. Neuropsychol. 2025, 40, 125–149. [Google Scholar] [CrossRef]
- Chen, A.K.; Wang, X.; McCluskey, L.P.; Morgan, J.C.; Switzer, J.A.; Mehta, R.; Tingen, M.; Su, S.; Harris, R.A.; Hess, D.C.; et al. Neuropsychiatric sequelae of long COVID-19: Pilot results from the COVID-19 neurological and molecular prospective cohort study in Georgia, USA. Brain Behav. Immun. Health 2022, 24, 100491. [Google Scholar] [CrossRef]
- Junco, B.; Samano Martin Del Campo, D.; Karakeshishyan, V.; Bass, D.; Sobczak, E.; Swafford, E.; Bolanos, A.; Rooks, J.; Baumel, B.S.; Ramos, A.R.; et al. Long-Term Brain Fog and Cognitive Impairment in Previously Hospitalized COVID-19 Patients. PLoS ONE 2024, 19, e0309102. [Google Scholar] [CrossRef] [PubMed]
- de Erausquin, G.A.; Snyder, H.; Carrillo, M.; Hosseini, A.A.; Brugha, T.S.; Seshadri, S. The chronic neuropsychiatric sequelae of COVID-19: The need for a prospective study of viral impact on brain functioning. Alzheimers Dement. 2021, 17, 1056–1065. [Google Scholar] [CrossRef]
- Ferrucci, R.; Dini, M.; Groppo, E.; Rosci, C.; Reitano, M.R.; Bai, F.; Poletti, B.; Brugnera, A.; Silani, V.; Monforte, A.D.; et al. Long-Lasting Cognitive Abnormalities after COVID-19. Brain Sci. 2021, 11, 235. [Google Scholar] [CrossRef]
- Rodriguez-Morales, A.J.; Cardona-Ospina, J.A.; Gutiérrez-Ocampo, E.; Villamizar-Peña, R.; Holguin-Rivera, Y.; Escalera-Antezana, J.P.; Alvarado-Arnez, L.E.; Bonilla-Aldana, D.K.; Franco-Paredes, C.; Henao-Martinez, A.F.; et al. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Med. Infect. Dis. 2020, 34, 101623. [Google Scholar] [CrossRef]
- Davis, H.E.; Assaf, G.S.; McCorkell, L.; Wei, H.; Low, R.J.; Re’em, Y.; Redfield, S.; Austin, J.P.; Akrami, A. Characterizing Long COVID in an International Cohort: 7 Months of Symptoms and Their Impact. eClinicalMedicine 2021, 38, 101019. [Google Scholar] [CrossRef]
- Damiano, R.F.; Caruso, M.J.G.; Cincoto, A.V.; Rocca, C.C.d.A.; Serafim, A.d.P.; Bacchi, P.; Guedes, B.F.; Brunoni, A.R.; Pan, P.M.; Nitrini, R.; et al. Post-COVID-19 psychiatric and cognitive morbidity: Preliminary findings from a Brazilian cohort study. Gen. Hosp. Psychiatry 2022, 75, 38–45. [Google Scholar] [CrossRef]
- Del Brutto, O.H.; Wu, S.; Mera, R.M.; Costa, A.F.; Recalde, B.Y.; Issa, N.P. Cognitive decline among individuals with history of mild symptomatic SARS-CoV-2 infection: A longitudinal prospective study nested to a population cohort. Eur. J. Neurol. 2021, 28, 3245–3253. [Google Scholar] [CrossRef] [PubMed]
- Cysique, L.A.; Jakabek, D.; Bracken, S.G.; Allen-Davidian, Y.; Heng, B.; Chow, S.; Dehhaghi, M.; Pires, A.S.; Darley, D.R.; Byrne, A.; et al. The Kynurenine Pathway Relates to Post-Acute COVID-19 Objective Cognitive Impairment and PASC. Ann. Clin. Transl. Neurol. 2023, 10, 1338–1352. [Google Scholar] [CrossRef]
- Taquet, M.; Sillett, R.; Zhu, L.; Mendel, J.; Camplisson, I.; Dercon, Q.; Harrison, P.J. Neurological and Psychiatric Risk Trajectories after SARS-CoV-2 Infection: An Analysis of 2-Year Retrospective Cohort Studies Including 1,284,437 Patients. Lancet Psychiatry 2022, 9, 815–827. [Google Scholar] [CrossRef]
- Boesl, F.; Audebert, H.; Endres, M.; Prüss, H.; Franke, C. A Neurological Outpatient Clinic for Patients with Post-COVID-19 Syndrome—A Report on the Clinical Presentations of the First 100 Patients. Front Neurol. 2021, 12, 738405. [Google Scholar] [CrossRef]
- Brown, L.A.; Ballentine, E.; Zhu, Y.; McGinley, E.L.; Pezzin, L.; Abramoff, B. The Unique Contribution of Depression to Cognitive Impairment in Post-Acute Sequelae of SARS-CoV-2 Infection. Brain Behav. Immun. Health 2022, 22, 100460. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Pöppelle, I.; Ottiger, M.; Gehrke, M.; Thiel, J.; Peter, N.; Martus, P.; Zickler, S. Long-Term Course and Factors Influencing Work Ability and Return to Work in Post-COVID Patients 12 Months after Inpatient Rehabilitation. J. Occup. Med. Toxicol. 2024, 19, 43. [Google Scholar] [CrossRef] [PubMed]
- O’Mahoney, L.L.; Routen, A.; Gillies, C.; Jenkins, S.A.; Almaqhawi, A.; Ayoubkhani, D.; Banerjee, A.; Brightling, C.; Calvert, M.; Cassambai, S.; et al. The Risk of Long COVID Symptoms: A Systematic Review and Meta-Analysis of Controlled Studies. Nat. Commun. 2025, 16, 4249. [Google Scholar] [CrossRef]
- Hampshire, A.; Azor, A.; Atchison, C.; Trender, W.; Hellyer, P.J.; Giunchiglia, V.; Husain, M.; Cooke, G.S.; Cooper, E.; Lound, A.; et al. Cognition and Memory after COVID-19 in a Large Community Sample. N. Engl. J. Med. 2024, 390, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Martin, E.M.; Reuken, P.A.; Scholcz, A.; Ganse-Dumrath, A.; Srowig, A.; Utech, I.; Kozik, V.; Radscheidt, M.; Brodoehl, S.; et al. Long COVID Is Associated with Severe Cognitive Slowing: A Multicentre Cross-Sectional Study. EClinicalMedicine 2024, 68, 102434. [Google Scholar] [CrossRef]
- Quan, M.; Wang, X.; Gong, M.; Wang, Q.; Li, Y.; Jia, J. Post-COVID cognitive dysfunction: Current status and research recommendations for high risk population. Lancet Reg. Health West Pac. 2023, 38, 100836. [Google Scholar] [CrossRef]
- Etter, M.M.; Martins, T.A.; Kulsvehagen, L.; Pitsch, J.; Sahanic, S.; Zraunig, J.; Trummer, C.; Rass, V.; Fischer, M.; Aichner, M.; et al. Severe Neuro-COVID Is Associated with Peripheral Immune Signatures, Autoimmunity and Neurodegeneration: A Prospective Cross-Sectional Study. Nat. Commun. 2022, 13, 6777. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.L.; Imam, M.H.; Zehra, W.; Jamil, S. Neuro-Inflammatory Pathways in COVID-19-Induced Central Nervous System Injury: Implications for Prevention and Treatment Strategies. Exp. Neurol. 2024, 382, 114984. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef] [PubMed]
- DeOre, B.J.; Tran, K.A.; Andrews, A.M.; Ramirez, S.H.; Galie, P.A. SARS-CoV-2 Spike Protein Disrupts Blood–Brain Barrier Integrity via RhoA Activation. J. Neuroimmune Pharmacol. 2021, 16, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Popa, E.; Tetia, T.; Poroch, M.; Ungureanu, M.; Cosmescu, A.; Barbacariu, L.; Slanina, A.M.; Bacusca, A.; Petroae, A.; Novac, O.; et al. The Effects of the COVID-19 Pandemic on Mental Health: A Web-Based Study Among Romanian Adults. Cureus 2022, 14, e31331. [Google Scholar] [CrossRef]
- Guo, Y.; Hu, K.; Li, Y.; Lu, C.; Ling, K.; Cai, C.; Wang, W.; Ye, D. Targeting TNF-α for COVID-19: Recent Advances and Controversies. Front. Public Health 2022, 10, 833967. [Google Scholar] [CrossRef]
- Dehhaghi, M.; Heydari, M.; Panahi, H.K.S.; Lewin, S.R.; Heng, B.; Brew, B.J.; Guillemin, G.J. The Roles of the Kynurenine Pathway in COVID-19 Neuropathogenesis. Infection 2024, 52, 2043–2059. [Google Scholar] [CrossRef]
- Mazza, M.G.; Palladini, M.; De Lorenzo, R.; Magnaghi, C.; Poletti, S.; Furlan, R.; Ciceri, F.; Rovere-Querini, P.; Benedetti, F. Persistent Psychopathology and Neurocognitive Impairment in COVID-19 Survivors: Effect of Inflammatory Biomarkers at Three-Month Follow-Up. Brain Behav. Immun. 2021, 94, 138–147. [Google Scholar] [CrossRef]
- Fotuhi, M.; Mian, A.; Meysami, S.; Raji, C.A. Neurobiology of COVID-19. J. Alzheimers Dis. 2020, 76, 3–19. [Google Scholar] [CrossRef]
- Leng, A.; Shah, M.; Ahmad, S.A.; Premraj, L.; Wildi, K.; Li Bassi, G.; Pardo, C.A.; Choi, A.; Cho, S.M. Pathogenesis Underlying Neurological Manifestations of Long COVID Syndrome and Potential Therapeutics. Cells 2023, 12, 816. [Google Scholar] [CrossRef]
- Mavroudis, I.; Petridis, F.; Petroaie, A.D.; Ciobica, A.; Kamal, F.Z.; Honceriu, C.; Iordache, A.; Ionescu, C.; Novac, B.; Novac, O. Exploring Symptom Overlaps: Post-COVID-19 Neurological Syndrome and Post-Concussion Syndrome in Athletes. Biomedicines 2024, 12, 1587. [Google Scholar] [CrossRef]
- Choudhury, N.A.; Mukherjee, S.; Singer, T.; Venkatesh, A.; Perez Giraldo, G.S.; Jimenez, M.; Miller, J.; Lopez, M.; Hanson, B.A.; Bawa, A.P.; et al. Neurologic Manifestations of Long COVID Disproportionately Affect Young and Middle-Age Adults. Ann. Neurol. 2025, 97, 369–383. [Google Scholar] [CrossRef] [PubMed]
- O’Mahoney, L.L.; Routen, A.; Gillies, C.; Hutchinson, J.; Hawes, H.; Davies, M.J.; Zaccardi, F.; Khunti, K. The Prevalence and Long-Term Health Effects of Long COVID among Hospitalised and Non-Hospitalised Populations: A Systematic Review and Meta-Analysis. eClinicalMedicine 2022, 55, 101762. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.L.R.; Griffanti, L.; Duff, E.; et al. SARS-CoV-2 Is Associated with Changes in Brain Structure in UK Biobank. Nature 2022, 604, 697–707. [Google Scholar] [CrossRef]
- Pacheco-Jaime, L.; Garcia-Vicente, C.; Ariza, M.; Cano, N.; Garolera, M.; Carreras-Vidal, L.; Roura, I.; Capdevila-Lacasa, C.; Oltra, J.; Pardo, J.; et al. Structural Brain Changes in Post-COVID Condition and Its Relationship with Cognitive Impairment. Brain Commun. 2025, 7, fcaf070. [Google Scholar] [CrossRef]
- Sudo, F.K.; Pinto, T.P.; Oliveira, M.F.; Yamamoto, F.I.; Lopes, M.A.; Medeiros, M.M.; Tavares, J.G.P.; Nitrini, R.; Smid, J.; Grinberg, L.T.; et al. Cognitive, Behavioral, Neuroimaging and Inflammatory Biomarkers after Hospitalization for COVID-19 in Brazil. Brain Behav. Immun. 2024, 115, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu-Bercu, A.; Lobiuc, A.; Căliman-Sturdza, O.A.; Oiţă, R.C.; Iavorschi, M.; Pavăl, N.-E.; Șoldănescu, I.; Dimian, M.; Covasa, M. Long COVID: Molecular Mechanisms and Detection Techniques. Int. J. Mol. Sci. 2024, 25, 408. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host–Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef]
- Fekete, M.; Lehoczki, A.; Szappanos, Á.; Toth, A.; Mahdi, M.; Sótonyi, P.; Benyó, Z.; Yabluchanskiy, A.; Tarantini, S.; Ungvari, Z. Cerebromicrovascular Mechanisms Contributing to Long COVID: Implications for Neurocognitive Health. GeroScience 2025, 47, 745–779. [Google Scholar] [CrossRef]
- Jafari Khaljiri, H.; Jamalkhah, M.; Amini Harandi, A.; Pakdaman, H.; Moradi, M.; Mowla, A. Comprehensive Review on Neuro-COVID-19 Pathophysiology and Clinical Consequences. Neurotox. Res. 2021, 39, 1613–1629. [Google Scholar] [CrossRef]
- Proulx, S.T.; Engelhardt, B. Central Nervous System Zoning: How Brain Barriers Establish Subdivisions for CNS Immune Privilege and Immune Surveillance. J. Intern. Med. 2022, 291, 779–800. [Google Scholar] [CrossRef] [PubMed]
- Bouayed, J.; Bohn, T. The Link between Microglia and the Severity of COVID-19: The “Two-Hit” Hypothesis. J. Inflamm. Res. 2021, 14, 4217–4228. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Gudowska-Sawczuk, M.; Mroczko, B. The Role of Neuropilin-1 (NRP-1) in SARS-CoV-2 Infection: Review. J. Clin. Med. 2021, 10, 2772. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.R.; Akter, R.; Neelotpol, S.; Mayesha, I.I.; Afrose, A. The Neuropathological Impacts of COVID-19: Challenges and Alternative Treatment Options for Alzheimer’s-Like Brain Changes in Severely SARS-CoV-2 Infected Patients. Am. J. Alzheimers Dis. Other Demen. 2023, 38, 15333175231214974. [Google Scholar] [CrossRef]
- Wei, Z.D.; Liang, K.; Shetty, A.K. Role of Microglia, Decreased Neurogenesis and Oligodendrocyte Depletion in Long COVID-Mediated Brain Impairments. Aging Dis. 2023, 14, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Kanberg, N.; Ashton, N.J.; Andersson, L.M.; Yilmaz, A.; Lindh, M.; Nilsson, S.; Price, R.W.; Blennow, K.; Zetterberg, H.; Gisslén, M. Neurochemical Evidence of Astrocytic and Neuronal Injury Commonly Found in COVID-19. Neurology 2020, 95, e1754–e1759. [Google Scholar] [CrossRef]
- Peluso, M.J.; Deeks, S.G.; Mustapic, M.; Kapogiannis, D.; Henrich, T.J.; Lu, S.; Goldberg, S.A.; Hoh, R.; Chen, J.Y.; Martinez, E.O.; et al. SARS-CoV-2 and Mitochondrial Proteins in Neural-Derived Exosomes of COVID-19. Ann. Neurol. 2022, 91, 772–781. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Brain Mechanisms Involved in Post COVID Syndrome: A Narrative Review. Neurochem. J. 2024, 18, 397–405. [Google Scholar] [CrossRef]
- Slama Schwok, A.; Henri, J. Long Neuro-COVID-19: Current Mechanistic Views and Therapeutic Perspectives. Biomedicines 2024, 14, 1081. [Google Scholar] [CrossRef]
- Dey, R.; Bishayi, B. Microglial Inflammatory Responses to SARS-CoV-2 Infection: A Comprehensive Review. Cell Mol. Neurobiol. 2024, 44, 2. [Google Scholar] [CrossRef] [PubMed]
- Garber, C.; Soung, A.; Vollmer, L.L.; Kanmogne, M.; Last, A.; Brown, J.; Klein, R.S. T Cells Promote Microglia-Mediated Synaptic Elimination and Cognitive Dysfunction during Recovery from Neuropathogenic Flaviviruses. Nat. Neurosci. 2019, 22, 1276–1288. [Google Scholar] [CrossRef]
- Luo, E.Y.; Chang, R.C.-C.; Gilbert-Jaramillo, J. SARS-CoV-2 Infection in Microglia and Its Sequelae: What Do We Know So Far? Brain Behav. Immun. Health 2024, 12, 100888. [Google Scholar] [CrossRef] [PubMed]
- Chagas, L.d.S.; Serfaty, C.A. The Influence of Microglia on Neuroplasticity and Long-Term Cognitive Sequelae in Long COVID: Impacts on Brain Development and Beyond. Int. J. Mol. Sci. 2024, 25, 3819. [Google Scholar] [CrossRef] [PubMed]
- Syage, A.; Pachow, C.; Cheng, Y.; Mangale, V.; Green, K.N.; Lane, T.E. Microglia Influence Immune Responses and Restrict Neurologic Disease in Response to Central Nervous System Infection by a Neurotropic Murine Coronavirus. Front. Cell. Neurosci. 2023, 17, 1291255. [Google Scholar] [CrossRef]
- Thakur, K.T.; Miller, E.H.; Glendinning, M.D.; Al-Dalahmah, O.; Banu, M.A.; Boehme, A.K.; Przedborski, S. COVID-19 Neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain 2021, 144, 2696–2708. [Google Scholar] [CrossRef]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.; Maat, C.A.; Schmartz, G.P.; Wyss-Coray, T. Dysregulation of Brain and Choroid Plexus Cell Types in Severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef]
- Anderson, F.L.; Biggs, K.E.; Rankin, B.E.; Havrda, M.C. NLRP3 Inflammasome in Neurodegenerative Disease. Transl. Res. 2023, 252, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Potere, N.; Del Buono, M.G.; Caricchio, R.; Cremer, P.C.; Vecchié, A.; Porreca, E.; Dalla Gasperina, D.; Dentali, F.; Abbate, A.; Bonaventura, A. Interleukin-1 and the NLRP3 Inflammasome in COVID-19: Pathogenetic and Therapeutic Implications. EBioMedicine 2022, 85, 104299. [Google Scholar] [CrossRef]
- Nazarinia, D.; Behzadifard, M.; Gholampour, J.; Karimi, R.; Gholampour, M. Eotaxin-1 (CCL11) in Neuroinflammatory Disorders and Possible Role in COVID-19 Neurologic Complications. Acta Neurol. Belg. 2022, 122, 865–869. [Google Scholar] [CrossRef]
- Bernard, I.; Limonta, D.; Mahal, L.K.; Hobman, T.C. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses 2020, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Wellford, S.A.; Moseman, E.A. Olfactory immune response to SARS-CoV-2. Cell Mol. Immunol. 2024, 21, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Moutal, A.; Martin, L.F.; Boinon, L.; Gomez, K.; Ran, D.; Zhou, Y.; Stratton, H.J.; Cai, S.; Luo, S.; Gonzalez, K.B.; et al. SARS-CoV-2 Spike Protein Co-Opts VEGF-A/Neuropilin-1 Receptor Signaling to Induce Analgesia. Pain 2021, 162, 243–252. [Google Scholar] [CrossRef]
- Saleki, K.; Alijanizadeh, P.; Azadmehr, A. Is neuropilin-1 the neuroimmune initiator of multi-system hyperinflammation in COVID-19. Biomed. Pharmacother. 2023, 167, 115558. [Google Scholar] [CrossRef]
- Mone, P.; de Donato, A.; Varzideh, F.; Jankauskas, S.; Kansakar, U.; Santulli, G. Functional Role of miR-34a in Diabetes and Frailty. Front. Aging 2022, 3, 949924. [Google Scholar] [CrossRef] [PubMed]
- Evers, P.; Uguccioni, S.M.; Ahmed, N.; Francis, M.E.; Kelvin, A.A.; Pezacki, J.P. miR-24-3p Is Antiviral Against SARS-CoV-2 by Downregulating Critical Host Entry Factors. Viruses 2024, 16, 1844. [Google Scholar] [CrossRef]
- Madsen, H.B.; Durhuus, J.A.; Andersen, O.; Straten, P.T.; Rahbech, A.; Desler, C. Mitochondrial Dysfunction in Acute and Post-Acute Phases of COVID-19 and Risk of Non-Communicable Diseases. NPJ Metab. Health Dis. 2024, 2, 36. [Google Scholar] [CrossRef]
- Bannazadeh Baghi, H.; Bayat, M.; Mehrasa, P.; Rezaee, S.A. Regulatory Role of MicroRNAs in Virus-Mediated Inflammation. J. Inflamm. 2024, 21, 43. [Google Scholar] [CrossRef]
- Agnello, L.; Gambino, C.M.; Ciaccio, A.M.; Masucci, A.; Vassallo, R.; Tamburello, M.; Scazzone, C.; Lo Sasso, B.; Ciaccio, M. Molecular Biomarkers of Neurodegenerative Disorders: A Practical Guide to Their Appropriate Use and Interpretation in Clinical Practice. Int. J. Mol. Sci. 2024, 25, 4323. [Google Scholar] [CrossRef]
- Bland, A.R.; Barraclough, M.; Trender, W.R.; Varesi, A.; Carson, A.; Kumar, V.; Koychev, I.; Husain, M.; Harrison, N.A. Profiles of Objective and Subjective Cognitive Function in Post-COVID Syndrome, COVID-19 Recovered, and COVID-19 Naïve Individuals. Sci. Rep. 2024, 14, 13368. [Google Scholar] [CrossRef]
- Duggan, M.R.; Parikh, V. Microglia and Modifiable Life Factors: Potential Contributions to Cognitive Resilience in Aging. Behav. Brain Res. 2021, 405, 113207. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.; Yang, J.; Simanauskaite, J.; Choi, M.; Castellanos, D.M.; Chang, R.; Sun, J.; Jagadeesan, N.; Parfitt, K.D.; Cribbs, D.H.; et al. Biologic TNF-α Inhibitors Reduce Microgliosis, Neuronal Loss, and Tau Phosphorylation in a Transgenic Mouse Model of Tauopathy. J. Neuroinflamm. 2021, 18, 312. [Google Scholar] [CrossRef] [PubMed]
- Guasp, M.; Muñoz-Sánchez, G.; Martínez-Hernández, E.; Santana, D.; Carbayo, Á.; Naranjo, L.; Bolós, U.; Framil, M.; Saiz, A.; Balasa, M.; et al. CSF biomarkers in COVID-19 associated encephalopathy and encephalitis predict long-term outcome. Front. Immunol. 2022, 13, 866153. [Google Scholar] [CrossRef]
- Bohmwald, K.; Diethelm-Varela, B.; Rodríguez-Guilarte, L.; Ríos, M.; Kalergis, A.M. Pathophysiological, immunological, and inflammatory features of long COVID. Front. Immunol. 2024, 15, 1341600. [Google Scholar] [CrossRef] [PubMed]
- Angriman, F.; Ferreyro, B.L.; Burry, L.; Fan, E.; Ferguson, N.D.; Husain, S.; Keshavjee, S.H.; Lupia, E.; Munshi, L.; Renzi, S.; et al. Interleukin-6 Receptor Blockade in Patients with COVID-19: Placing Clinical Trials into Context. Lancet Respir. Med. 2021, 9, 655–664. [Google Scholar] [CrossRef]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi, M.; Akarid, K.; El Khatib, K. The Pro-Inflammatory Cytokines in COVID-19 Pathogenesis: What Goes Wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef]
- Shankar-Hari, M.; Vale, C.L.; Godolphin, P.J.; Fisher, D.; Higgins, J.P.T.; Spiga, F.; Savović, J.; Tierney, J.; Baron, G.; Benbenishty, J.; et al. Association Between Administration of IL-6 Antagonists and Mortality Among Patients Hospitalized for COVID-19: A Meta-analysis. JAMA 2021, 326, 499–518. [Google Scholar] [CrossRef]
- Lokau, J.; Garbers, C.; Vicente, R.; Dittrich, A.; Meltendorf, S.; Lingel, H.; Münster-Kühnel, A.; Brunner-Weinzierl, M.C.; Garbers, C. Long-term Increase in Soluble Interleukin-6 Receptor Levels in COVID-19 Convalescents. Front. Immunol. 2025, 16, 123456. [Google Scholar] [CrossRef]
- Mohd Zawawi, Z.; Kalyanasundram, J.; Mohd Zain, R.; Thayan, R.; Basri, D.F.; Yap, W.B. Prospective Roles of Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19: Prognosis, Therapeutic and Management. Int. J. Mol. Sci. 2023, 24, 6142. [Google Scholar] [CrossRef]
- Kruger, A.; Joffe, D.; Lloyd-Jones, G.; Khan, M.A.; Šalamon, Š.; Laubscher, G.J.; Putrino, D.; Kell, D.B.; Pretorius, E. Vascular Pathogenesis in Acute and Long COVID: Current Insights and Therapeutic Outlook. Semin. Thromb. Hemost. 2025, 51, 256–271. [Google Scholar] [CrossRef]
- Greene, C.; Connolly, R.; Brennan, D.; Farrell, R.; O’Sullivan, M.; Kelly, Á.; Doyle, S.L.; O’Halloran, K.D.; Mills, K.H.G.; Campbell, M.; et al. Blood-Brain Barrier Disruption and Sustained Systemic Inflammation in Individuals with Long COVID-Associated Cognitive Impairment. Nat. Neurosci. 2024, 27, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The Spatial and Cell-Type Distribution of SARS-CoV-2 Receptor ACE2 in the Human and Mouse Brains. Front. Neurol. 2021, 11, 573095. [Google Scholar] [CrossRef] [PubMed]
- Ceasovschih, A.; Sorodoc, V.; Shor, A.; Haliga, R.E.; Roth, L.; Lionte, C.; Onofrei Aursulesei, V.; Sirbu, O.; Culis, N.; Shapieva, A.; et al. Distinct Features of Vascular Diseases in COVID-19. J. Inflamm. Res. 2023, 16, 2783–2800. [Google Scholar] [CrossRef]
- Takeda, M. Proteolytic Activation of SARS-CoV-2 Spike Protein and Its Implication in Viral Entry. Microbiol. Immunol. 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, R.S.; Sanchez Sevilla Uruchurtu, A.; Siggins, M.K.; Liew, F.; Russell, C.D.; Moore, S.C.; Fairfield, C.; Carter, E.; Abrams, S.; Short, C.-E.; et al. Inflammatory Profiles Across the Spectrum of Disease Reveal a Distinct Role for GM-CSF in Severe COVID-19. Sci. Immunol. 2021, 6, eabg9873. [Google Scholar] [CrossRef]
- Hernández-Parra, H.; Reyes-Hernández, O.D.; Figueroa-González, G.; González-Del Carmen, M.; González-Torres, M.; Peña-Corona, S.I.; Florán, B.; Cortés, H.; Leyva-Gómez, G. Alteration of the Blood–Brain Barrier by COVID-19 and Its Implication in the Permeation of Drugs into the Brain. Front. Cell Neurosci. 2023, 17, 1125109. [Google Scholar] [CrossRef]
- Wang, J.; Liu, F.; Chen, S. Astrocytes and the Psychiatric Sequelae of COVID-19: What We Learned from the Pandemic. Neurochem. Res. 2022, 48, 1015–1025. [Google Scholar] [CrossRef]
- Agafonova, A.; Cosentino, A.; Musso, N.; Prinzi, C.; Russo, C.; Pellitteri, R.; Anfuso, C.D.; Lupo, G. Hypoxia-Induced Inflammation in In Vitro Model of Human Blood–Brain Barrier: Modulatory Effects of the Olfactory Ensheathing Cell-Conditioned Medium. Int. J. Mol. Sci. 2024, 25, 1264. [Google Scholar] [CrossRef]
- Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the Blood–Brain Barrier. Int. J. Mol. Sci. 2021, 22, 2681. [Google Scholar] [CrossRef]
- Salomão, R.; Assis, V.; de Sousa Neto, I.V.; Petriz, B.; Babault, N.; Durigan, J.L.Q.; de Cássia Marqueti, R. Involvement of Matrix Metalloproteinases in COVID-19: Molecular Targets, Mechanisms, and Insights for Therapeutic Interventions. Biology 2023, 12, 843. [Google Scholar] [CrossRef]
- Jarius, S.; Pache, F.; Körtvelyessy, P.; Jelčić, I.; Stettner, M.; Franciotta, D.; Keller, E.; Neumann, B.; Ringelstein, M.; Senel, M.; et al. Cerebrospinal Fluid Findings in COVID-19: A Multicenter Study of 150 Lumbar Punctures in 127 Patients. J. Neuroinflamm. 2022, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Schwabenland, M.; Salié, H.; Tanevski, J.; Killmer, S.; Franke, C.; Glatzel, M.; Korte, L.; Kallfass, T.; Kranich, J.; Madlung, M.; et al. Deep Spatial Profiling Reveals Subcellular Neuroinvasion of SARS-CoV-2 in Human Brain Tissue. Immunity 2021, 54, 5559–5573.e18. [Google Scholar] [CrossRef]
- Yang, R.C.; Huang, K.; Zhang, H.P.; Li, L.; Zhang, Y.-F.; Tan, C.; Chen, H.-C.; Jin, M.-L.; Wang, X.-R. SARS-CoV-2 Productively Infects Human Brain Microvascular Endothelial Cells. J. Neuroinflamm. 2022, 19, 149. [Google Scholar] [CrossRef]
- Choi, J.Y.; Park, J.H.; Jo, C.; Kim, K.C.; Koh, Y.H. SARS-CoV-2 Spike S1 Subunit Protein-Mediated Increase of Beta-Secretase 1 (BACE1) Impairs Human Brain Vessel Cells. Biochem. Biophys. Res. Commun. 2022, 626, 66–71. [Google Scholar] [CrossRef]
- Hosp, J.A.; Reisert, M.; Dressing, A.; Rieger, J.; Pape, S.; Kellner, E.; Klöppel, S.; Frisch, S.; Ertl-Wagner, B.; Bäzner, H.; et al. Cerebral Microstructural Alterations in Post-COVID Condition Are Related to Cognitive Impairment, Olfactory Dysfunction and Fatigue. Nat. Commun. 2024, 15, 4256. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Xiao, S.; Chen, G.; Chen, P.; Yang, Z.; Tang, X.; Huang, L.; Wang, Y. COVID-19 Is Associated with Changes in Brain Function and Structure: A Multimodal Meta-Analysis of Neuroimaging Studies. Neurosci. Biobehav. Rev. 2024, 157, 105792. [Google Scholar] [CrossRef]
- Wirth, K.J.; Scheibenbogen, C.; Paul, F. An Attempt to Explain the Neurological Symptoms of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Transl. Med. 2021, 19, 471. [Google Scholar] [CrossRef]
- Butowt, R.; von Bartheld, C.S. Anosmia in COVID-19: Underlying Mechanisms and Assessment of an Olfactory Route to Brain Infection. Neuroscientist 2021, 27, 582–603. [Google Scholar] [CrossRef]
- Birnhuber, A.; Fließer, E.; Gorkiewicz, G.; Zacharias, M.; Seeliger, B.; David, S.; Welte, T.; Schmidt, M.; Olschewski, H.; Kwapiszewska, G.; et al. Between Inflammation and Thrombosis: Endothelial Cells in COVID-19. Eur. Respir. J. 2021, 58, 2100377. [Google Scholar] [CrossRef]
- Tarnawski, A.S.; Ahluwalia, A. Endothelial Cells and Blood–Brain Barrier Injury in COVID-19. World J. Gastroenterol. 2022, 28, 275–289. [Google Scholar] [CrossRef]
- Urata, R.; Ikeda, K.; Yamazaki, E.; Kobayashi, Y.; Morimoto, H.; Seki, S.; Matsumoto, Y.; Nishida, K.; Komuro, I.; Hirata, Y.; et al. Senescent Endothelial Cells Are Predisposed to SARS-CoV-2 Infection and Subsequent Endothelial Dysfunction. Sci. Rep. 2022, 12, 11855. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Spudich, S.; Nath, A. Nervous System Consequences of COVID-19. Science 2022, 375, 267–269. [Google Scholar] [CrossRef]
- Turner, S.; Khan, M.A.; Putrino, D.; Woodcock, A.; Kell, D.B.; Pretorius, E. Long COVID: Pathophysiological Factors and Abnormalities of Coagulation. Trends Endocrinol. Metab. 2023, 34, 321–344. [Google Scholar] [CrossRef]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent Clotting Protein Pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) Is Accompanied by Increased Levels of Antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef]
- Patel, A.; Shah, A.; Shah, R.; Soin, D.; Rawal, J. Coagulation Biomarkers in Long COVID: A Review. Thromb. Res. 2022, 213, 152–158. [Google Scholar] [CrossRef]
- Graham, E.L.; Clark, J.R.; Orban, Z.S.; Lim, P.H.; Szymanski, A.L.; Taylor, C.; DiBiase, R.M.; Jia, D.T.; Balabanov, R.; Ho, S.U.; et al. Persistent Neurologic Symptoms and Cognitive Dysfunction in Non-Hospitalized COVID-19 “Long Haulers”. Ann. Clin. Transl. Neurol. 2021, 8, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-J.; Liu, S.-H.; Manachevakul, S.; Lee, T.-A.; Kuo, C.-T.; Bello, D. Biomarkers in Long COVID-19: A Systematic Review. Front. Med. 2023, 10, 1085988. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Levi, M. Endothelial Dysfunction and Immunothrombosis as Key Pathogenic Mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef]
- Popa, E.; Boanca, M.; Bacusca, A.I.; Ungureanu, M.; Bologa, C.; Ceasovschih, A.; Coman, A.E. The Peroxisome Proliferator-Activated Receptor-Alpha (PPARA) and Expression of CD36 Receptor Scavenger on Circulating Cells as Biomarkers of Atherosclerosis. Atherosclerosis 2023, 379 (Suppl. S1), S15. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Q.; Han, B.; Chen, Y.; Qiao, X.; Wang, L. CD36 Promotes NLRP3 Inflammasome Activation via the mtROS Pathway in Renal Tubular Epithelial Cells of Diabetic Kidneys. Cell Death Dis. 2021, 12, 523. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Xu, Y.; Tan, Y.; Shi, H.; Jin, P.; Li, Y.; Teng, J.; Liu, H.; Pan, H.; Hu, Q.; et al. CD36 Mediates SARS-CoV-2-Envelope-Protein-Induced Platelet Activation and Thrombosis. Nat. Commun. 2023, 14, 5077. [Google Scholar] [CrossRef]
- Ambrosino, P.; Calcaterra, I.L.; Mosella, M.; Formisano, R.; D’Anna, S.E.; Bachetti, T.; Marcuccio, G.; Galloway, B.; Mancini, F.P.; Papa, A.; et al. Endothelial Dysfunction in COVID-19: A Unifying Mechanism and a Potential Therapeutic Target. Biomedicines 2022, 10, 812. [Google Scholar] [CrossRef]
- Haffke, M.; Freitag, H.; Rudolf, G.; Endres, M.; Scherbakov, N.; Dreher, M.; Dahlem, D.; Bobbert, T.; Bauer, J.M.; Doehner, W. Mild-to-Moderate COVID-19 Convalescents May Present Prolonged Endothelial Dysfunction. J. Clin. Med. 2022, 11, 6461. [Google Scholar] [CrossRef]
- Dani, M.; Dirksen, A.; Taraborrelli, P.; Torocastro, M.; Panagopoulos, D.; Sutton, R.; Lim, P.B. Autonomic Dysfunction in ‘Long COVID’: Rationale, Physiology and Management Strategies. Clin. Med. 2021, 21, e63–e67. [Google Scholar] [CrossRef] [PubMed]
- Tanking, C.; Lakkananurak, C.; Srisakvarakul, C.; Jitpreeda, A.; Threechod, K.; Sukitpunyaroj, D. Postural Orthostatic Tachycardia Syndrome and Other Autonomic Dysfunctions Following COVID-19: Incidence, Characteristics, and Associated Factors. J. Arrhythmia 2024, 40, e13001. [Google Scholar] [CrossRef]
- Mastrorosa, F.; Giannini, M.; Pignataro, G.; Montalbano, G.; Zanza, C.; Piccioni, A.; De Luca, L.; Savioli, G. Connecting Dots of Long COVID-19 Pathogenesis: A Vagus Nerve–Hypothalamic-Pituitary-Adrenal–Mitochondrial Axis Dysfunction. Front. Cell. Infect. Microbiol. 2024, 14, 1501949. [Google Scholar] [CrossRef]
- Vitale-Cross, L.; Szalayova, I.; Scoggins, A.; Palkovits, M.; Mezey, E. SARS-CoV-2 entry sites are present in all structural elements of the human glossopharyngeal and vagal nerves: Clinical implications. eBioMedicine 2022, 78, 103981. [Google Scholar] [CrossRef]
- Woo, M.S.; Shafiq, M.; Fitzek, A.; Dottermusch, M.; Altmeppen, H.; Mohammadi, B.; Mayer, C.; Bal, L.C.; Raich, L.; Matschke, J.; et al. Vagus nerve inflammation contributes to dysautonomia in COVID-19. Acta Neuropathol. 2023, 146, 387–394. [Google Scholar] [CrossRef]
- Giunta, S.; Xia, S.; Pelliccioni, G.; Olivieri, F. Autonomic nervous system imbalance during aging contributes to impair endogenous anti-inflammaging strategies. GeroScience 2024, 46, 113–127. [Google Scholar] [CrossRef]
- Marques, K.C.; Quaresma, J.A.S.; Falcão, L.F.M. Cardiovascular Autonomic Dysfunction in “Long COVID”: Pathophysiology, Heart Rate Variability, and Inflammatory Markers. Front. Cardiovasc. Med. 2023, 10, 1256512. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.R.T.; Medeiros, C.A.M.; Ferreira, L.D.B.; de Souza, C.F.; de Oliveira, L.C.; Leite, J.R. Impact of COVID-19 on heart rate variability in post-COVID individuals compared to a control group. Sci. Rep. 2024, 14, 9067. [Google Scholar] [CrossRef]
- Shah, B.; Kunal, S.; Bansal, A.; Jain, J.; Poundrik, S.; Shetty, M.K.; Batra, V.; Chaturvedi, V.; Yusuf, J.; Mukhopadhyay, S.; et al. Heart rate variability as a marker of cardiovascular dysautonomia in post-COVID-19 syndrome using artificial intelligence. Indian Pacing Electrophysiol. J. 2022, 22, 4–11. [Google Scholar] [CrossRef]
- Almulla, A.F.; Thipakorn, Y.; Zhou, B.; Vojdani, A.; Paunova, R.; Maes, M. The Tryptophan Catabolite or Kynurenine Pathway in Long COVID Disease: A Systematic Review and Meta-Analysis. Neuroscience 2024, 563, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Cappelletti, G.; Carsana, E.V.; Lunghi, G.; Breviario, S.; Vanetti, C.; Di Fonzo, A.B.; Frattini, E.; Magni, M.; Zecchini, S.; Clerici, M.; et al. SARS-CoV-2 hampers dopamine production in iPSC-derived dopaminergic neurons. Exp. Mol. Pathol. 2023, 134, 104874. [Google Scholar] [CrossRef]
- Wong, A.C.; Devason, A.S.; Umana, I.C.; Cox, T.O.; Dohnalová, L.; Litichevskiy, L.; Perla, J.; Lundgren, P.; Etwebi, Z.; Izzo, L.T.; et al. Serotonin reduction in post-acute sequelae of viral infection. Cell 2023, 186, 4851–4867.e20. [Google Scholar] [CrossRef]
- Eroğlu, İ.; Eroğlu, B.Ç.; Güven, G.S. Altered tryptophan absorption and metabolism could underlie long-term symptoms in survivors of coronavirus disease 2019 (COVID-19). Nutrition 2021, 90, 111308. [Google Scholar] [CrossRef]
- Badawy, A.A. The kynurenine pathway of tryptophan metabolism: A neglected therapeutic target of COVID-19 pathophysiology and immunotherapy. Biosci. Rep. 2023, 43, BSR20230595. [Google Scholar] [CrossRef] [PubMed]
- Fanciulli, A.; Leys, F.; Krbot Skorić, M.; Reis Carneiro, D.; Calandra-Buonaura, G.; Camaradou, J.; Chiaro, G.; Cortelli, P.; Falup-Pecurariu, C.; Granata, R.; et al. Impact of the COVID-19 pandemic on clinical autonomic practice in Europe: A survey of the European Academy of Neurology and the European Federation of Autonomic Societies. Eur. J. Neurol. 2023, 30, 1712–1726. [Google Scholar] [CrossRef]
- Phu, T.V.; Tran, T.T.; Reinis, A.; Nguyen, T.N.; Ta, N.T.D.; Nguyen, T.V. Postural Orthostatic Tachycardia Syndrome-like Symptoms Following COVID-19 Vaccination: A Review. Health Technol. Assess. Brain 2023, 31, 9–17. [Google Scholar] [CrossRef]
- Yavropoulou, M.P.; Tsokos, G.C.; Chrousos, G.P.; Sfikakis, P.P. Protracted stress-induced hypocortisolemia may account for the clinical and immune manifestations of Long COVID. Clin. Immunol. 2022, 245, 109133. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, D.; Chen, D.G.; Ng, R.H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J.; et al. Multiple Early Factors Anticipate Post-Acute COVID-19 Sequelae. Cell 2022, 185, 881–895.e20. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.A. Long COVID: Hormone Imbalances and/or Rather Complex Immune Dysregulations? J. Endocr. Soc. 2024, 8, bvae043. [Google Scholar] [CrossRef] [PubMed]
- Sypniewski, D.; Matyjek, M.; Dobrowolska, A.; Sypniewska, G. The Importance of Hypocortisolemia in Long COVID–New Lessons from ME/CFS. Endocr. Metab. Immune Disord. Drug Targets 2024, 92, 13–22. [Google Scholar]
- Ruiz-Pablos, M.; Paiva, B.; Zabaleta, A. Hypocortisolemic ASIA: A Vaccine- and Chronic Infection-Induced Syndrome Behind the Origin of Long COVID and Myalgic Encephalomyelitis. Front. Immunol. 2024, 15, 1422940. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Gubbi, S.; Koch, C.A. COVID-19 and chronic fatigue syndrome: An endocrine perspective. J. Clin. Transl. Endocrinol. 2022, 27, 100284. [Google Scholar] [CrossRef]
- Sunada, N.; Honda, H.; Nakano, Y.; Yamamoto, K.; Tokumasu, K.; Sakurada, Y.; Matsuda, Y.; Hasegawa, T.; Otsuka, Y.; Obika, M.; et al. Hormonal trends in patients suffering from long COVID symptoms. Endocr. J. 2022, 69, 1173–1181. [Google Scholar] [CrossRef]
- Coman, A.E.; Ceasovschih, A.; Petroaie, A.D.; Popa, E.; Lionte, C.; Bologa, C.; Haliga, R.E.; Cosmescu, A.; Slănină, A.M.; Bacușcă, A.I.; et al. The Significance of Low Magnesium Levels in COVID-19 Patients. Medicina 2023, 59, 279. [Google Scholar] [CrossRef]
- Saleh, H.A.; Yousef, M.H.; Abdelnaser, A. The Anti-Inflammatory Properties of Phytochemicals and Their Effects on Epigenetic Mechanisms Involved in TLR4/NF-κB-Mediated Inflammation. Front. Immunol. 2021, 12, 606069. [Google Scholar] [CrossRef]
- Sharan, P.; Vellapandian, C. Hypothalamic–Pituitary–Adrenal (HPA) Axis: Unveiling the Potential Mechanisms Involved in Stress-Induced Alzheimer’s Disease and Depression. Cureus 2024, 16, e67595. [Google Scholar] [CrossRef]
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. PPARγ/PGC1α Signaling as a Potential Therapeutic Target for Mitochondrial Biogenesis in Neurodegenerative Disorders. Pharmacol. Ther. 2021, 219, 107705. [Google Scholar] [CrossRef] [PubMed]
- Pszczołowska, M.; Walczak, K.; Misków, W.; Antosz, K.; Batko, J.; Karska, J.; Leszek, J. Molecular Cross-Talk between Long COVID-19 and Alzheimer’s Disease. Geroscience 2024, 46, 2885–2899. [Google Scholar] [CrossRef]
- Rahmani, B.; Ghashghayi, E.; Zendehdel, M.; Baghbanzadeh, A.; Khodadadi, M. Molecular Mechanisms Highlighting the Potential Role of COVID-19 in the Development of Neurodegenerative Diseases. Physiol. Int. 2022, 109, 135–162. [Google Scholar] [CrossRef]
- Fernández-Castañeda, A.; Lu, P.; Geraghty, A.C.; Song, E.; Lee, M.H.; Wood, J.; Yalçın, B.; Taylor, K.; Wrenn, J.; Fitzpatrick, K.; et al. Mild respiratory SARS-CoV-2 infection can cause multi-lineage cellular dysregulation and myelin loss in the brain. Cell 2022, 185, 2452–2468.e16. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in Human and Mouse Brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, Y. Tau and Neuroinflammation in Alzheimer’s Disease: Interplay Mechanisms and Clinical Translation. J. Neuroinflamm. 2023, 20, 165. [Google Scholar] [CrossRef]
- Mayer, C.; Riera-Ponsati, L.; Kauppinen, S.; Klitgaard, H.; Erler, J.T.; Hansen, S.N. Targeting the NRF2 Pathway for Disease Modification in Neurodegenerative Diseases: Mechanisms and Therapeutic Implications. Front. Pharmacol. 2024, 15, 1437939. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. Ther. Adv. Neurol. Disord. 2020, 13, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, R.; Yadav, S.; Hurt, R.T.; Mueller, M.R.; Aakre, C.A.; Gilman, E.A.; Grach, S.L.; Overgaard, J.; Snyder, M.R.; Collins, N.M.; et al. Pro Inflammatory Cytokines Profiles of Patients with Long COVID Differ Between Variant Epochs. J. Prim. Care Community Health 2024, 15, 21501319241254751. [Google Scholar] [CrossRef]
- Green, R.; Mayilsamy, K.; McGill, A.R.; Martinez, T.E.; Chandran, B.; Blair, L.J.; Bickford, P.C.; Mohapatra, S.S. SARS-CoV-2 Infection Increases the Gene Expression Profile for Alzheimer’s Disease Risk. Mol. Ther. Methods Clin. Dev. 2022, 27, 217–229. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, L.; Lin, Z.; Long, H.; Zhang, X.; He, X.; Ma, S.; Wang, Y.; Deng, H.; Chen, J.; et al. APOE Interacts with ACE2 Inhibiting SARS-CoV-2 Cellular Entry and Inflammation in COVID-19 Patients. Signal Transduct. Target. Ther. 2022, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, C.-A.; Tomoaia-Cotisel, M.; Sevastre-Berghian, A.; Tomoaia, G.; Mocanu, A.; Pal-Racz, C.; Toma, V.-A.; Roman, I.; Ujica, M.-A.; Pop, L.-C. A Review on Current Aspects of Curcumin-Based Effects in Relation to Neurodegenerative, Neuroinflammatory and Cerebrovascular Diseases. Molecules 2025, 30, 43. [Google Scholar] [CrossRef]
- Li, Z.Q.; Lin, H.S.; Huang, X.P.; Zhang, S.Q.; Shu, X.; Wu, X.N. Research Progress on the Role of PGC1α in Mitochondrial Dysfunction Associated with Alzheimer’s Disease. Ageing Neur. Dis. 2023, 3, 14. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Profumo, E.; Tucci, P.; Saso, L. A Perspective on Nrf2 Signaling Pathway for Neuroinflammation: A Potential Therapeutic Target in Alzheimer’s and Parkinson’s Diseases. Front. Cell. Neurosci. 2021, 15, 787258. [Google Scholar] [CrossRef]
- Lionte, C.; Sorodoc, V.; Haliga, R.E.; Bologa, C.; Ceasovschih, A.; Petris, O.R.; Coman, A.E.; Stoica, A.; Sirbu, O.; Puha, G.; et al. Inflammatory and Cardiac Biomarkers in Relation with Post-Acute COVID-19 and Mortality: What We Know after Successive Pandemic Waves. Diagnostics 2022, 12, 1373. [Google Scholar] [CrossRef] [PubMed]
- Duindam, H.B.; Mengel, D.; Kox, M.; Göpfert, J.C.; Kessels, R.P.C.; Synofzik, M.; Pickkers, P.; Abdo, W.F. Systemic Inflammation Relates to Neuroaxonal Damage Associated with Long-Term Cognitive Dysfunction in COVID-19 Patients. Brain Behav. Immun. 2024, 117, 510–520. [Google Scholar] [CrossRef]
- Lee, C. Collaborative Power of Nrf2 and PPARγ Activators against Metabolic and Drug-Induced Oxidative Injury. Oxid. Med. Cell. Longev. 2017, 2017, 1378175. [Google Scholar] [CrossRef]
- Mavroudis, I.; Chatzikonstantinou, S.; Petridis, F.; Kazis, D.; Kamal, F.Z.; Diaconu, S.; Visternicu, M.; Ciobica, A.; Iordache, A.; Stadoleanu, C.; et al. Unveiling the Functional Component in Post-Concussive Cognitive Impairment: A Novel Modelling Approach. Brain Broad Res. Artif. Intell. Neurosci. 2025, 16, 83–94. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Biomarker | Pathophysiological Role | References |
|---|---|---|---|
| Fibrotic/ inflammatory | TGF-β | Linked to BBB disruption and neurodegeneration | [48,92,106,108] |
| Inflammatory | IL-6 | Proinflammatory cytokine; contributes to glial activation and cognitive symptoms | [48,86,88,89,166] |
| IL-1β | Mediator of cytokine storm; inflammasome activation | [48,70,71] | |
| TNF-α | Promotes microglial synaptic loss and neuroinflammation | [37,69,70] | |
| MicroRNA | miR-155 | Contributes to neuroinflammation through NF-κB signaling | [48,76,77]. |
| miR-146a | Regulates glial activation and oxidative stress | [48,76,77]. | |
| miR-24 | Suppresses NRP1; modulates the BBB and inflammation | [48,76,77]. | |
| Neuroendocrine | Dopamine | Affects motivation and executive function; deficient in Long COVID | [136] |
| Serotonin | Modulates mood, memory, vagal tone; depleted via the kynurenine pathway | [137] | |
| Cortisol | Reflects HPA axis function; hypocortisolism is linked to fatigue and cognitive impairment | [142,145] | |
| Neuroinflammatory | GFAP | Astrocytic injury marker; elevated in both Long COVID and AD | [33,48,58,74,82] |
| FKBP5 | Associated with tau phosphorylation and neuroinflammation | [48,51]. | |
| LGALS3 | Marker of astrocyte-mediated neurotoxicity; implicated in AD and Long COVID | [119] | |
| NFL | Axonal injury marker; correlates with cognitive symptoms | [33,39,47,48], | |
| Stress-related | KLF4 | Regulates oxidative stress response and apoptosis | [48,51]. |
| Vascular | ICAM-1 | Adhesion molecule involved in endothelial activation and leukocyte infiltration | [92,104,105,110,111,112,113] |
| VEGF-A | Increases vascular permeability; linked to BBB disruption | [48,72,74] | |
| S100B | Astrocyte-derived; reflects BBB breakdown and CNS injury | [92] | |
| VCAM-1 | Promotes immune cell adhesion and microvascular injury | [92,104,105,110,111,112,113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popa, E.; Popa, A.E.; Poroch, M.; Poroch, V.; Ungureanu, M.I.; Slanina, A.M.; Bacusca, A.; Coman, E.A. The Molecular Mechanisms of Cognitive Dysfunction in Long COVID: A Narrative Review. Int. J. Mol. Sci. 2025, 26, 5102. https://doi.org/10.3390/ijms26115102
Popa E, Popa AE, Poroch M, Poroch V, Ungureanu MI, Slanina AM, Bacusca A, Coman EA. The Molecular Mechanisms of Cognitive Dysfunction in Long COVID: A Narrative Review. International Journal of Molecular Sciences. 2025; 26(11):5102. https://doi.org/10.3390/ijms26115102
Chicago/Turabian StylePopa, Elena, Andrei Emilian Popa, Mihaela Poroch, Vladimir Poroch, Monica Iuliana Ungureanu, Ana Maria Slanina, Agnes Bacusca, and Elena Adorata Coman. 2025. "The Molecular Mechanisms of Cognitive Dysfunction in Long COVID: A Narrative Review" International Journal of Molecular Sciences 26, no. 11: 5102. https://doi.org/10.3390/ijms26115102
APA StylePopa, E., Popa, A. E., Poroch, M., Poroch, V., Ungureanu, M. I., Slanina, A. M., Bacusca, A., & Coman, E. A. (2025). The Molecular Mechanisms of Cognitive Dysfunction in Long COVID: A Narrative Review. International Journal of Molecular Sciences, 26(11), 5102. https://doi.org/10.3390/ijms26115102