
Streamlined Postprocessing of NMR Structures with the Molecular Restrainer: A Universal Tool for High-Quality Protein–Ligand Models and Non-Standard Amino Acid Residues

 , ,
, , 
Abstract
1. Introduction
2. Results
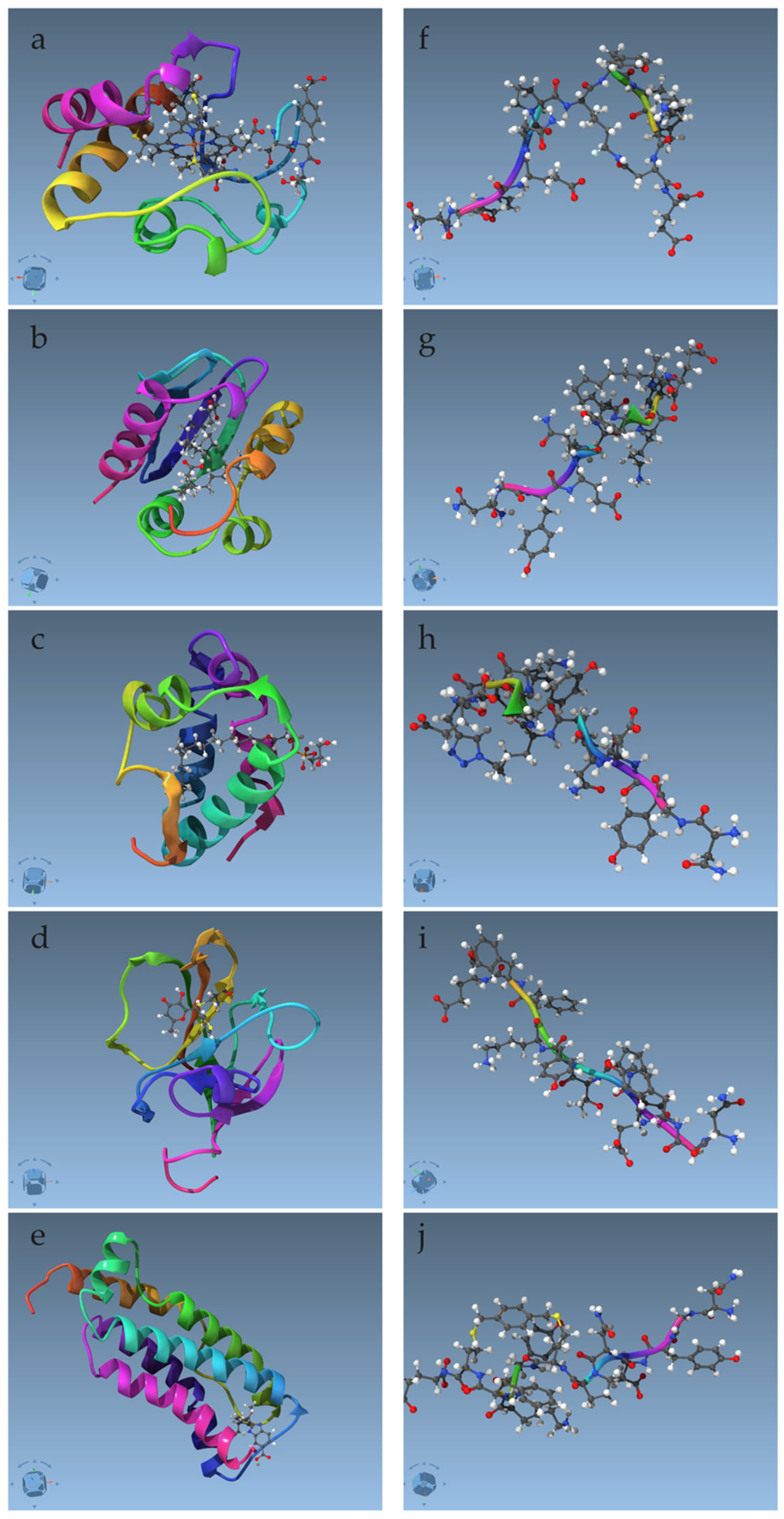
2.1. Characterization of the Data
2.2. Performance of the Refinement
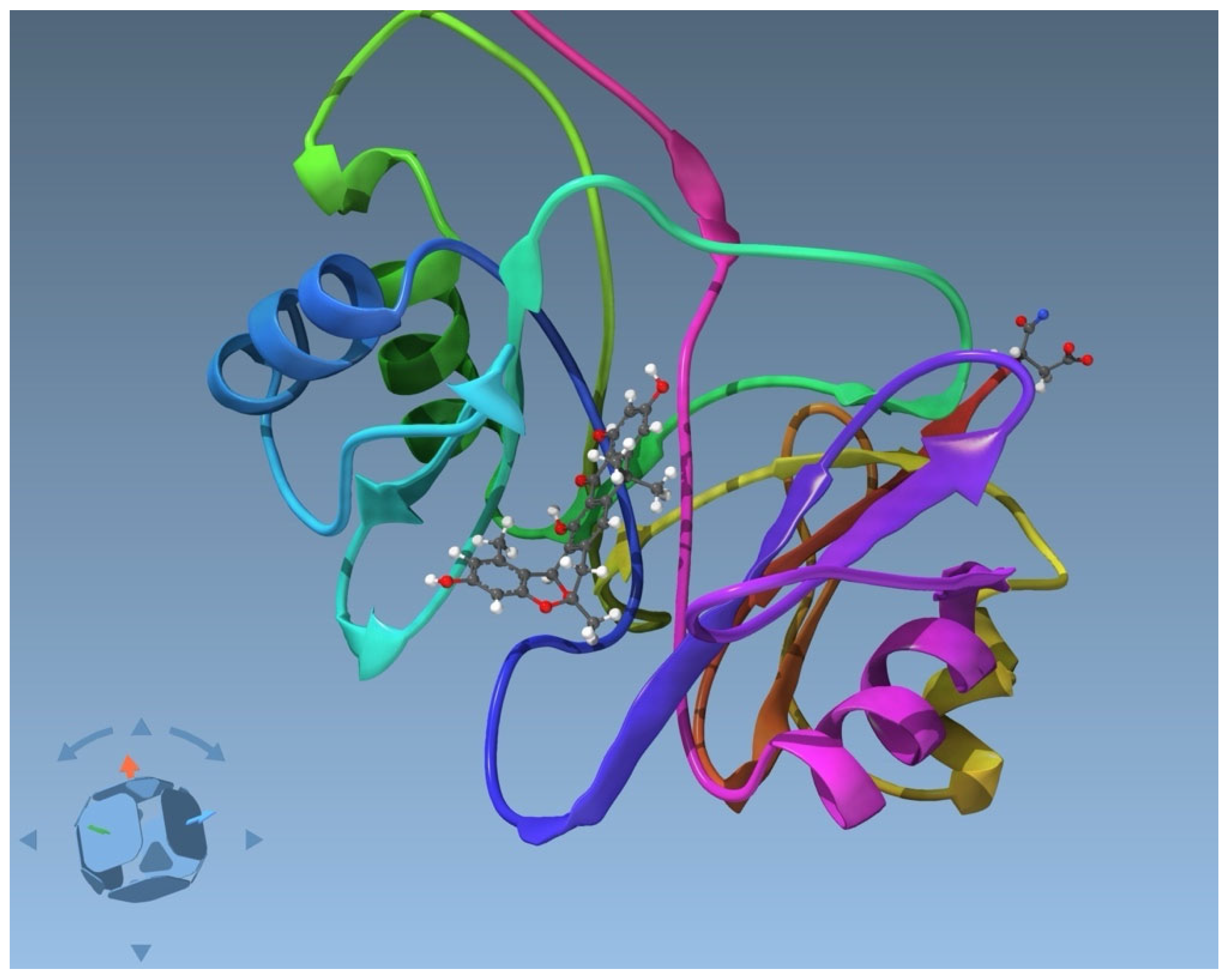
2.2.1. Proteins and Complexes
2.2.2. Peptides with Non-Standard Residues
3. Discussion
3.1. Justification of the Use of UFF
3.1.1. General Versus Specific Parameters
3.1.2. More Accurate Parameters for Structure Refinement?
3.2. Strengths and Temporary Weaknesses of the Molecular Restrainer
4. Materials and Methods
4.1. Treatment of the Structure File, NMR-Specific Data
4.2. Distance Restraints, Force Field, and Structure Minimization
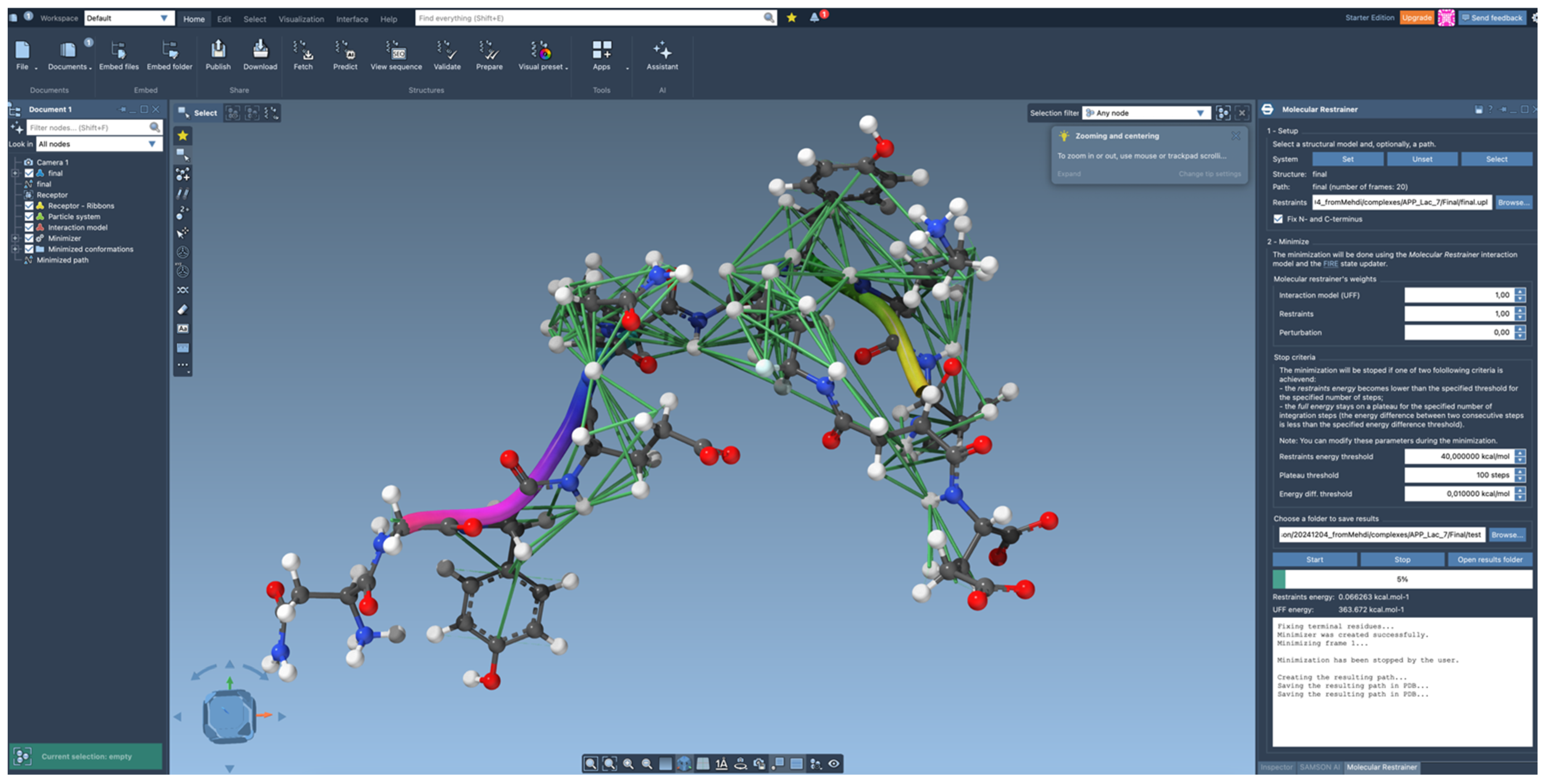
4.3. Algorithm for Structure Refinement
4.4. Further Information
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Williamson, M.P.; Havel, T.F.; Wüthrich, K. Solution conformation of proteinase inhibitor IIA from bull seminal plasma by 1H nuclear magnetic resonance and distance geometry. J. Mol. Biol. 1985, 182, 295–315. [Google Scholar] [CrossRef]
- Fesik, S.W.; O’Donnell, T.J.; Gampe, R.T.; Olejniczak, E.T. Determining the structure of a glycopeptide Ac2-Lys-D-Ala-D-Ala complex using NMR parameters and molecular modeling. J. Am. Chem. Soc. 1986, 108, 3165–3170. [Google Scholar] [CrossRef]
- Hodsdon, M.E.; Ponder, J.W.; Cistola, D.P. The NMR Solution Structure of Intestinal Fatty Acid-binding Protein Complexed with Palmitate: Application of a Novel Distance Geometry Algorithm. J. Mol. Biol. 1996, 264, 585–602. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Kuszewski, J.J.; Tjandra, N.; Marius Clore, G. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003, 160, 65–73. [Google Scholar] [CrossRef]
- Güntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 2015, 62, 453–471. [Google Scholar] [CrossRef]
- Rieping, W.; Habeck, M.; Bardiaux, B.; Bernard, A.; Malliavin, T.E.; Nilges, M. ARIA2: Automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 2007, 23, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Allain, F.; Mareuil, F.; Ménager, H.; Nilges, M.; Bardiaux, B. ARIAweb: A server for automated NMR structure calculation. Nucleic Acids Res. 2020, 48, W41–W47. [Google Scholar] [CrossRef] [PubMed]
- Guerry, P.; Duong, V.D.; Herrmann, T. CASD-NMR 2: Robust and accurate unsupervised analysis of raw NOESY spectra and protein structure determination with UNIO. J. Biomol. NMR 2015, 62, 473–480. [Google Scholar] [CrossRef]
- Huang, Y.J.; Tejero, R.; Powers, R.; Montelione, G.T. A topology-constrained distance network algorithm for protein structure determination from NOESY data. Proteins Struct. Funct. Bioinform. 2006, 62, 587–603. [Google Scholar] [CrossRef]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS Biomolecular Simulation Program Package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; vanderSpoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, E.M.; Güntert, P. NMR structure calculation for all small molecule ligands and non-standard residues from the PDB Chemical Component Dictionary. J. Biomol. NMR 2015, 63, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Kagami, L.; Wilter, A.; Diaz, A.; Vranken, W. The ACPYPE web server for small-molecule MD topology generation. Bioinformatics 2023, 39, btad350. [Google Scholar] [CrossRef]
- Bugnon, M.; Goullieux, M.; Röhrig, U.F.; Perez, M.A.S.; Daina, A.; Michielin, O.; Zoete, V. SwissParam 2023: A Modern Web-Based Tool for Efficient Small Molecule Parametrization. J. Chem. Inf. Model. 2023, 63, 6469–6475. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Koziara, K.B.; Stroet, M.; Malde, A.K.; Mark, A.E. Testing and validation of the Automated Topology Builder (ATB) version 2.0: Prediction of hydration free enthalpies. J. Comput. Aided Mol. Des. 2014, 28, 221–233. [Google Scholar] [CrossRef]
- SAMSON: Integrative Molecular Design. Available online: https://www.samson-connect.net (accessed on 24 April 2025).
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Moreno-Beltrán, B.; Guerra-Castellano, A.; Díaz-Quintana, A.; Del Conte, R.; García-Mauriño, S.M.; Díaz-Moreno, S.; González-Arzola, K.; Santos-Ocaña, C.; Velázquez-Campoy, A.; De la Rosa, M.A.; et al. Structural basis of mitochondrial dysfunction in response to cytochrome c phosphorylation at tyrosine 48. Proc. Natl. Acad. Sci. USA 2017, 114, E3041–E3050. [Google Scholar] [CrossRef]
- Singarapu, K.K.; Ahuja, A.; Potula, P.R.; Ummanni, R. Solution Nuclear Magnetic Resonance Studies of Sterol Carrier Protein 2 Like 2 (SCP2L2) Reveal the Insecticide Specific Structural Characteristics of SCP2 Proteins in Aedes aegypti Mosquitoes. Biochemistry 2016, 55, 4919–4927. [Google Scholar] [CrossRef]
- Shenkarev, Z.O.; Melnikova, D.N.; Finkina, E.I.; Sukhanov, S.V.; Boldyrev, I.A.; Gizatullina, A.K.; Mineev, K.S.; Arseniev, A.S.; Ovchinnikova, T.V. Ligand Binding Properties of the Lentil Lipid Transfer Protein: Molecular Insight into the Possible Mechanism of Lipid Uptake. Biochemistry 2017, 56, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Jaudzems, K.; Kurbatska, V.; Jēkabsons, A.; Bobrovs, R.; Rudevica, Z.; Leonchiks, A. Targeting Bacterial Sortase A with Covalent Inhibitors: 27 New Starting Points for Structure-Based Hit-to-Lead Optimization. ACS Infect. Dis. 2020, 6, 186–194. [Google Scholar] [CrossRef]
- Rübbelke, M.; Hamilton, J.; Binder, F.; Bauer, M.; King, J.; Nar, H.; Zeeb, M. Discovery and Structure-Based Optimization of Fragments Binding the Mixed Lineage Kinase Domain-like Protein Executioner Domain. J. Med. Chem. 2021, 64, 15629–15638. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham Iii, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Autenrieth, F.; Tajkhorshid, E.; Baudry, J.; Luthey-Schulten, Z. Classical force field parameters for the heme prosthetic group of cytochrome c. J. Comput. Chem. 2004, 25, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Bartling, C.R.O.; Alexopoulou, F.; Kuschert, S.; Chin, Y.K.Y.; Jia, X.; Sereikaite, V.; Özcelik, D.; Jensen, T.M.; Jain, P.; Nygaard, M.M.; et al. Comprehensive Peptide Cyclization Examination Yields Optimized APP Scaffolds with Improved Affinity toward Mint2. J. Med. Chem. 2023, 66, 3045–3057. [Google Scholar] [CrossRef]
- Ho, A.; Liu, X.; Südhof, T.C. Deletion of Mint Proteins Decreases Amyloid Production in Transgenic Mouse Models of Alzheimer’s Disease. J. Neurosci. 2008, 28, 14392–14400. [Google Scholar] [CrossRef]
- Yan, X.; Jia, X.; Luo, Z.; Orts, J.; Kobe, B.; Zhao, Y.; Mobli, M.; Qu, X. An enzymatic dual oxa-Diels-Alder reaction constructs the oxygen-bridged tricyclic acetal unit of (-)-anthrabenzoxocinones. Nat. Chem. 2025. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. D 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Rappe, A.K.; Goddard, W.A., III. Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 1991, 95, 3358–3363. [Google Scholar] [CrossRef]
- Artemova, S.; Jaillet, L.; Redon, S. Automatic molecular structure perception for the universal force field. J. Comput. Chem. 2016, 37, 1191–1205. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Seo, B.; Savoie, B.M. Evidence That Less Can Be More for Transferable Force Fields. J. Chem. Inf. Model. 2023, 63, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef] [PubMed]
- Force Field (Chemistry). Wikipedia. 2025. Available online: https://en.wikipedia.org/wiki/Force_field_(chemistry) (accessed on 21 May 2025).
- Monticelli, L.; Tieleman, D.P. Force Fields for Classical Molecular Dynamics. In Biomolecular Simulations; Humana Press: Totowa, NJ, USA, 2013; pp. 197–213. [Google Scholar]
- Mareš, J.; Delgado, P.M. Getting the intermolecular forces correct: Introducing the ASTA strategy for a water model. RSC Adv. 2024, 14, 25712–25727. [Google Scholar] [CrossRef]
- Jing, Z.; Liu, C.; Cheng, S.Y.; Qi, R.; Walker, B.D.; Piquemal, J.-P.; Ren, P. Polarizable Force Fields for Biomolecular Simulations: Recent Advances and Applications. Annu. Rev. Biophys. 2019, 48, 371–394. [Google Scholar] [CrossRef]
- Walker, B.; Liu, C.; Wait, E.; Ren, P. Automation of AMOEBA polarizable force field for small molecules: Poltype 2. J. Comput. Chem. 2022, 43, 1530–1542. [Google Scholar] [CrossRef]
- Antila, H.S.; Dixit, S.; Kav, B.; Madsen, J.J.; Miettinen, M.S.; Ollila, O.H.S. Evaluating Polarizable Biomembrane Simulations against Experiments. J. Chem. Theory Comput. 2024, 20, 4325–4337. [Google Scholar] [CrossRef]
- Bitzek, E.; Koskinen, P.; Gähler, F.; Moseler, M.; Gumbsch, P. Structural Relaxation Made Simple. Phys. Rev. Lett. 2006, 97, 170201. [Google Scholar] [CrossRef]
- Sinelnikova, A.; Spoel, D.v.d. NMR refinement and peptide folding using the GROMACS software. J. Biomol. NMR 2021, 75, 143–149. [Google Scholar] [CrossRef] [PubMed]
- SAMSON Connect: UFF. Available online: https://www.samson-connect.net/extensions/8cbdc8b1-59e1-6459-d68f-b840275dd5e9 (accessed on 24 April 2025).
- SAMSON Connect: FIRE. Available online: https://www.samson-connect.net/extensions/8ec255b0-7326-0bed-366e-40be71c1d160 (accessed on 24 April 2025).

{kind=link}
{kind=link}
{kind=link}
| PDB Code of the Complex | PDB Code of the Ligand | Systematic Name * |
|---|---|---|
| 2N3Y | 1PA | 4-(carboxymethyl)-L-phenylalanine |
| MH0 | mesoheme | |
| 2NBN | PLM | palmitic acid (COOH form!) |
| 5LQV | PGM | 1-myristoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] |
| 6R1V | JPT | 6-(hydroxymethyl)-3-oxidanyl-2-(thiophen-3-ylmethyl)pyran-4-one |
| 7NM2 | UJ5 | 2-[(~[S])-methoxy-(4-propan-2-ylphenyl)methyl]-3~[H]-benzimidazole-5-carboxylic acid (COOH form!) |
| PDB name of the peptide | Non-standard chem. moiety | Further/alternative specification |
| APP-Lac-7 | Lactam | Cyclic amide |
| APP-RCM-21 | Aliphatic double bond | Forming a long aliphatic section of a cycle |
| APP-WT-4 | N/A | N/A |
| APP-TRIAZ-31 | Triazole ring | Five-membered ring as a part of a larger cycle |
| APP-X-lin-57 | Organic sulfide | Thioether-containing cycle |
| Clashscore * | Ramachandran | Side Chain | CYANA TF | |||||
|---|---|---|---|---|---|---|---|---|
| 2N3Y | 0 | 1 | 1.3 | 2.2 | 7.0 | 5.3 | 4.18 | 0.93 |
| 2NBN | 19 | 2 | 0.1 | 1.9 | 30.3 | 4.9 | 4.93 | 2.00 |
| 5LQV | 14 | 4 | 0.2 | 2.0 | 10.2 | 3.5 | 160.57 | 22.23 |
| 6R1V | 7 | 1 | 1.4 | 3.0 | 9.3 | 7.7 | 16.34 | 2.42 |
| 7NM2 | 10 | 2 | 1.2 | 0.4 | 18.3 | 7.9 | 3.74 | 1.82 |
| APP-Lac-7 | 0 | 12 ** | 0.0 | 0.0 | 2.1 | 1.4 | 0.05 | 0.07 |
| APP-RCM-21 | 0 | 0 | 0.0 | 0.0 | 3.6 | 0.7 | 0.19 | 0.46 |
| APP-TRIAZ-31 | 0 | 0 | 0.0 | 0.0 | 20.0 | 20.0 | 0.15 | 0.77 |
| APP-WT-4 | 0 | 1 | 0.0 | 0.0 | 11.4 | 3.3 | 0.03 | 0.04 |
| APP-X-lin-57 | 0 | 0 | 0.0 | 0.0 | 15.0 | 14.3 | 0.22 | 0.30 |
| abx-ABX | 18 | 5 | 0.8 | 1.5 | 17.3 | 9.8 | 7.09 | 9.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mareš, J.; Tarang, G.S.; Marin, D.; Mobli, M.; Redon, S.; Orts, J. Streamlined Postprocessing of NMR Structures with the Molecular Restrainer: A Universal Tool for High-Quality Protein–Ligand Models and Non-Standard Amino Acid Residues. Int. J. Mol. Sci. 2025, 26, 5091. https://doi.org/10.3390/ijms26115091
Mareš J, Tarang GS, Marin D, Mobli M, Redon S, Orts J. Streamlined Postprocessing of NMR Structures with the Molecular Restrainer: A Universal Tool for High-Quality Protein–Ligand Models and Non-Standard Amino Acid Residues. International Journal of Molecular Sciences. 2025; 26(11):5091. https://doi.org/10.3390/ijms26115091
Chicago/Turabian StyleMareš, Jiří, Guneet Singh Tarang, Dmitriy Marin, Mehdi Mobli, Stephane Redon, and Julien Orts. 2025. "Streamlined Postprocessing of NMR Structures with the Molecular Restrainer: A Universal Tool for High-Quality Protein–Ligand Models and Non-Standard Amino Acid Residues" International Journal of Molecular Sciences 26, no. 11: 5091. https://doi.org/10.3390/ijms26115091
APA StyleMareš, J., Tarang, G. S., Marin, D., Mobli, M., Redon, S., & Orts, J. (2025). Streamlined Postprocessing of NMR Structures with the Molecular Restrainer: A Universal Tool for High-Quality Protein–Ligand Models and Non-Standard Amino Acid Residues. International Journal of Molecular Sciences, 26(11), 5091. https://doi.org/10.3390/ijms26115091