Machine Learning-Based Analysis of Differentially Expressed Genes in the Muscle Transcriptome Between Beef Cattle and Dairy Cattle

 , , , and
, , , and
Abstract
1. Introduction
2. Results
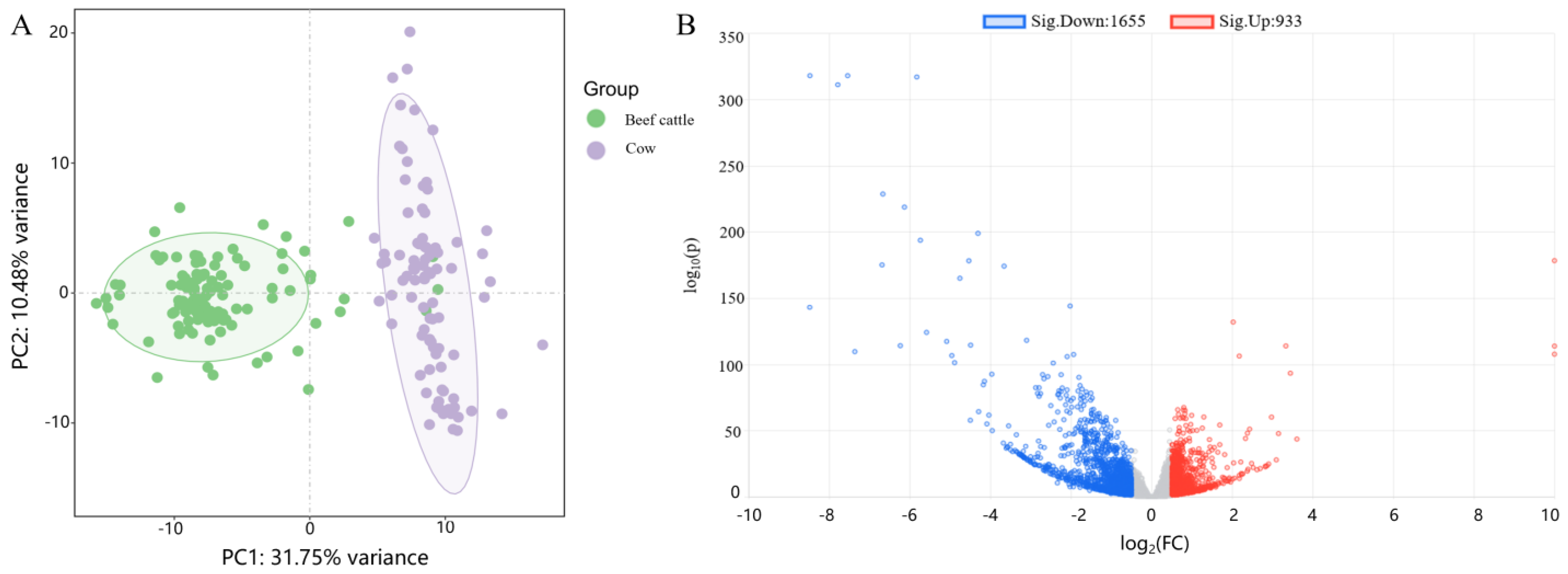
2.1. Data Quality Control and Differential Analysis
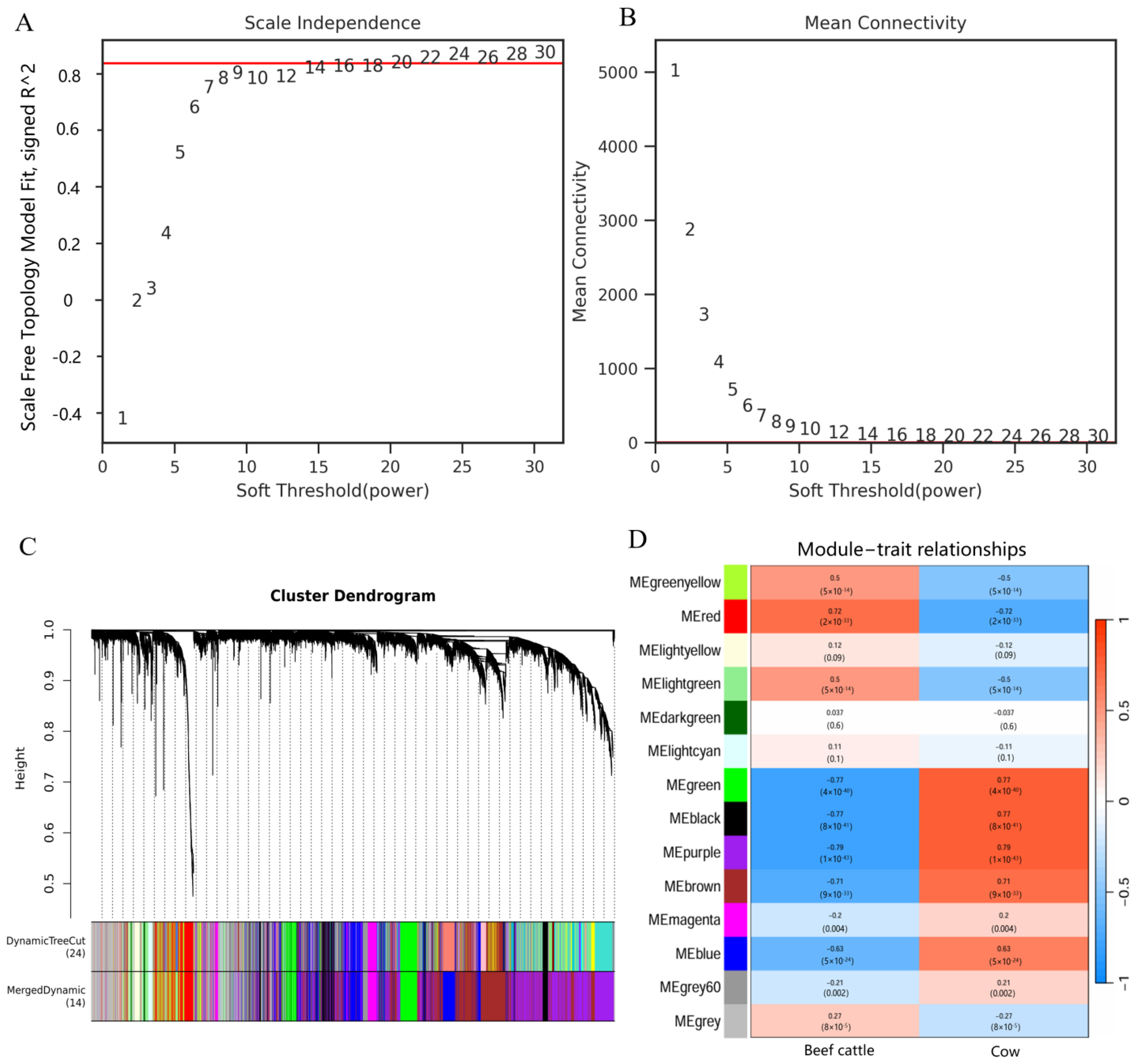
2.2. Weighted Gene Co-Expression Network Analysis (WGCNA) of the Muscle Transcriptome
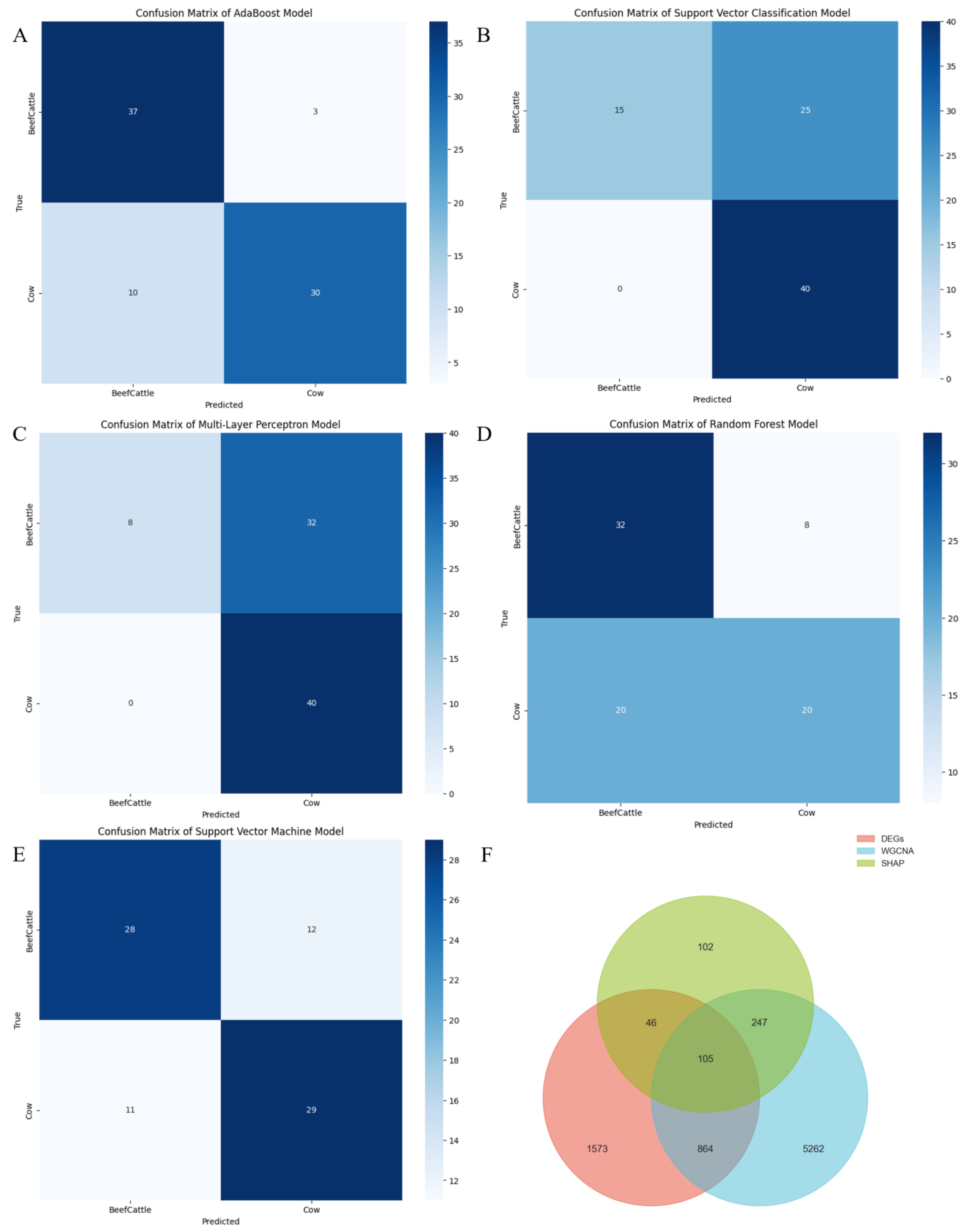
2.3. Evaluate the Classification Performance of Five Machine Learning Models on Beef Cattle and Cow Cattle
2.4. Enrichment Analysis and Network Interaction Relationships
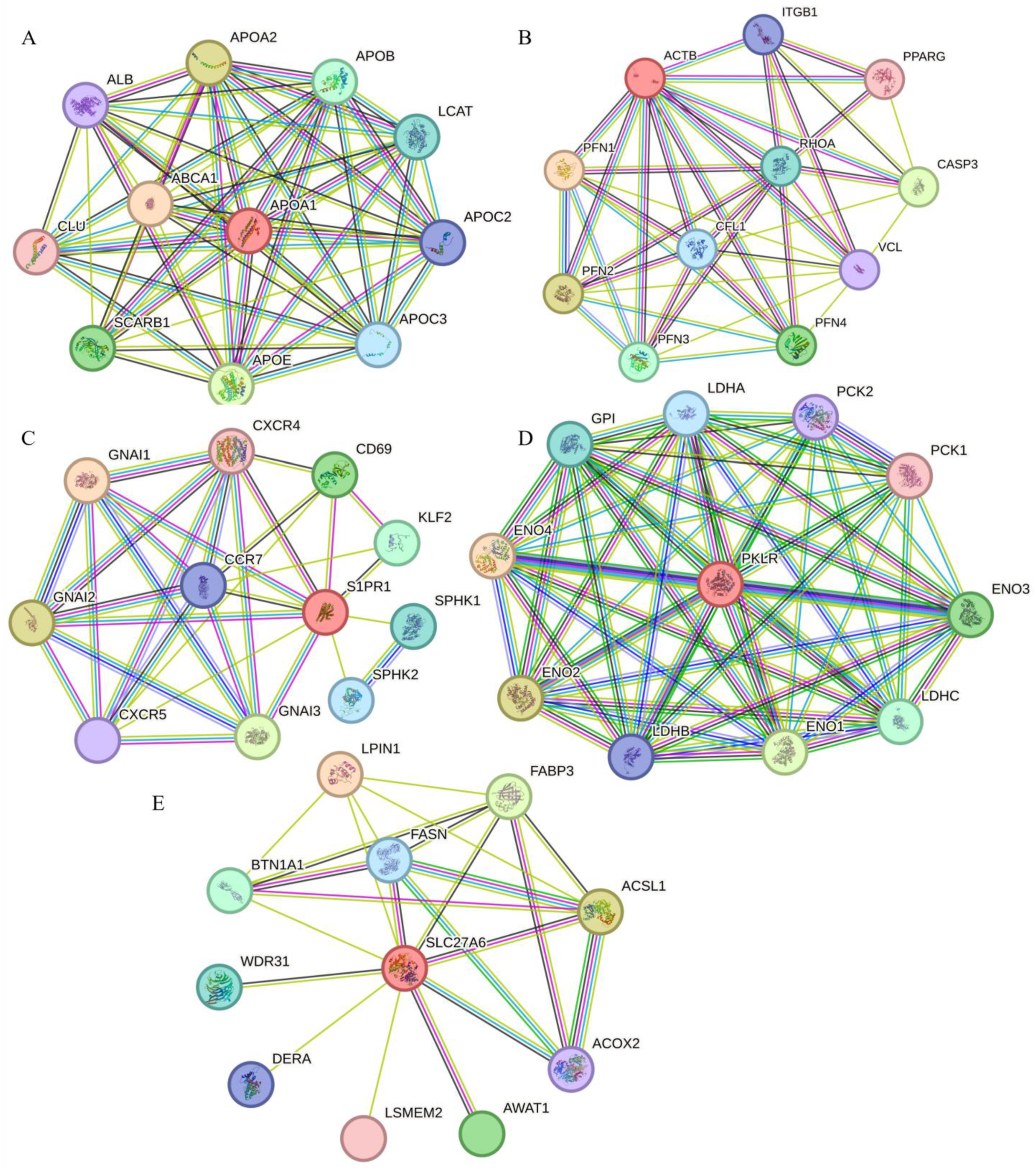
2.5. Analysis of Protein–Protein Interaction Network of Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Source of Transcriptome Data
4.2. Processing and Quality Control of Transcriptome Data
4.3. Differential Analysis of Muscle Transcriptome
4.4. Weighted Gene Co-Expression Network Analysis of Muscle Transcriptome
4.5. Machine Learning and Model Interpretability
4.6. Enrichment Analysis and Network Interaction Analysis
4.7. Protein–Protein Interaction Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenwood, P.L. Review: An overview of beef production from pasture and feedlot globally, as demand for beef and the need for sustainable practices increase. Animal 2021, 15, 100295. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 6, 1344. [Google Scholar] [CrossRef] [PubMed]
- Sahraeian, S.M.E.; Mohiyuddin, M.; Sebra, R.; Tilgner, H.; Afshar, P.T.; Au, K.F.; Asadi, N.B.; Gerstein, M.B.; Wong, W.H.; Snyder, M.P.; et al. Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis. Nat. Commun. 2017, 5, 59. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 1, 15–21. [Google Scholar] [CrossRef]
- Zhang, H.; Liang, Y.; Chen, S.; Xuan, Z.; Jiang, Y.; Li, R.; Cao, Y. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Anim. Genet. 2023, 54, 421–424. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Xia, H.; Li, F.; Peng, L.; Du, Y.; Hua, G.; Yang, L.; Yang Zhou, Y. Over-Expression of Two Different Isoforms of Cattle TUSC5 Showed Opposite Effects on Adipogenesis. Genes 2022, 8, 1444. [Google Scholar] [CrossRef]
- Liu, R.; Liu, X.; Bai, X.; Xiao, C.; Dong, Y. Different expression of lipid metabolism-related genes in Shandong black cattle and Luxi cattle based on transcriptome analysis. Sci. Rep. 2020, 10, 21915. [Google Scholar] [CrossRef]
- Li, Y.; Wang, M.; Li, Q.; Gao, Y.; Li, Q.; Li, J.; Cao, Y. Transcriptome profiling of longissimus lumborum in Holstein bulls and steers with different beef qualities. PLoS ONE 2020, 15, e0235218. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Zhou, X.; Yuan, X.; Zhao, S.; Li, X.; Liu, X. KAML: Improving genomic prediction accuracy of complex traits using machine learning determined parameters. Genome Biol. 2020, 21, 146. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Tan, J.; Greene, C.S. Cross-platform normalization of microarray and RNA-seq data for machine learning applications. PeerJ 2016, 4, e1621. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Liu, T.T.; Pankratz, D.G.; Colby, T.V.; Barth, N.M.; Lynch, D.A.; Walsh, P.S.; Raghu, G.; Kennedy, G.C.; Huang, J. Identification of usual interstitial pneumonia pattern using RNA-Seq and machine learning: Challenges and solutions. BMC Genom. 2018, 19, 101. [Google Scholar] [CrossRef]
- Wang, L.; Xi, Y.; Sung, S.; Qiao, H. RNA-seq assistant: Machine learning based methods to identify more transcriptional regulated genes. BMC Genom. 2018, 19, 546. [Google Scholar] [CrossRef] [PubMed]
- Messad, F.; Louveau, I.; Koffi, B.; Gilbert, H.; Gondret, F. Investigation of muscle transcriptomes using gradient boosting machine learning identifies molecular predictors of feed efficiency in growing pigs. BMC Genom. 2019, 20, 659. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Spurlock, D.M.; Armentano, L.E.; Page, C.D.; VandeHaar, M.J.; Bickhart, D.M.; Weigel, K.A. Random Forests approach for identifying additive and epistatic single nucleotide polymorphisms associated with residual feed intake in dairy cattle. J. Dairy Sci. 2013, 96, 6716–6729. [Google Scholar] [CrossRef]
- Yao, C.; Zhu, X.; Weigel, K.A. Semi-supervised learning for genomic prediction of novel traits with small reference populations: An application to residual feed intake in dairy cattle. Genet. Sel. Evol. 2016, 48, 84. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, S.; Na, R.; Guo, L.; Huo, C.; Zhu, L.; Shi, C.; Na, R.; Gu, M.; Zhang, W. Comparative Transcriptome Analysis of Bovine, Porcine, and Sheep Muscle Using Interpretable Machine Learning Models. Animals 2024, 14, 2947. [Google Scholar] [CrossRef]
- Raja, G.; Ramkumar, B.; Rajendiran, B.; Prathiba, B.; Arumugam, S.B.; Karuppanan, T.; Nkenyereye, K.; Nkenyereye, L.; Dev, K. Synergistic Analysis of Lung Cancer’s Impact on Cardiovascular Disease Using ML-Based Techniques. IEEE J. Biomed. Health Inform. 2024, 13, 1–8. [Google Scholar] [CrossRef]
- Park, S.K.; Sheffler, T.L.; Spurlock, M.E.; Grant, A.L.; Gerrard, D.E. Chronic activation of 5′-AMP-activated protein kinase changes myosin heavy chain expression in growing pigs. J. Anim. Sci. 2009, 10, 3124–3133. [Google Scholar] [CrossRef]
- Zhu, J.Y.; Shi, X.E.; Lu, H.Z.; Xia, B.; Li, Y.F.; Li, X.; Zhang, Q.L.; Yang, G.S. RNA-seq transcriptome analysis of extensor digitorum longus and soleus muscles in large white pigs. Mol. Genet. Genom. 2016, 291, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Li, A.N.; Raza, S.H.A.; Khan, R.; Wang, X.Y.; Wang, S.H.; Wang, G.H.; Zhang, Y.; Zan, L.S. The molecular characteristics of the FAM13A gene and the role of transcription factors ACSL1 and ASCL2 in its core promoter region. Genes 2019, 10, 981. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Zhang, X.; Wang, D.; Jin, G.; Li, B.; Xu, F.; Cheng, J.; Zhang, F.; Wu, S.; et al. The comprehensive liver transcriptome of two cattle breeds with different intramuscular fat content. Biochem. Biophys. Res. Commun. 2017, 3, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, Y.; Wang, J.; Huang, N.; Jiang, Q.; Ju, Z.; Yang, C.; Wei, X.; Xiao, Y.; Zhang, Y.; et al. Selection Signature and CRISPR/Cas9-Mediated Gene Knockout Analyses Reveal ZC3H10 Involved in Cold Adaptation in Chinese Native Cattle. Genes 2022, 13, 1910. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, C.; Cai, H.; Xu, Y.; Lan, X.; Lei, C.; Chen, H. Novel polymorphisms of the APOA2 gene and its promoter region affect body traits in cattle. Genes 2013, 531, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bai, J.; Ju, Z.; Jiang, Q.; Wang, J.; Gao, Y.; Zhang, Y.; Wei, X.; Huang, J. Effect of functional single nucleotide polymorphism g.-572 A > G of apolipoprotein A1 gene on resistance to ketosis in Chinese Holstein cows. Res. Vet. Sci. 2021, 135, 310–316. [Google Scholar] [CrossRef]
- Drazic, A.; Aksnes, H.; Marie, M.; Boczkowska, M.; Varland, S.; Timmerman, E.; Foyn, F.; Glomnes, H.; Rebowski, G.; Impens, F.; et al. NAA80 is actin’s N-terminal acetyltransferase and regulates cytoskeleton assembly and cell motility. Proc. Natl. Acad. Sci. USA 2018, 115, 4399–4404. [Google Scholar] [CrossRef]
- Malheiros, J.M.; Enríquez-Valencia, C.E.; Braga, C.P.; Vieira, J.C.S.; Vieira, D.S.; Pereira, G.L.; Curi, R.A.; Neto, O.M.M.; Oliveira, H.N.; Padilha, P.M.; et al. Application of proteomic to investigate the different degrees of meat tenderness in Nellore breed. J. Proteom. 2021, 30, 104331. [Google Scholar] [CrossRef]
- Rahman, M.M.; Takashima, S.; Kamatari, Y.O.; Badr, Y.; Shimizu, K.; Okada, A.; Inoshima, Y. Putative Internal Control Genes in Bovine Milk Small Extracellular Vesicles Suitable for Normalization in Quantitative Real Time-Polymerase Chain Reaction. Membranes 2021, 11, 933. [Google Scholar] [CrossRef]
- Ma, X.; Wei, D.; Cheng, G.; Li, S.; Wang, L.; Wang, Y.; Wang, X.; Zhang, S.; Wang, H.; Zan, L. Bta-miR-130a/b regulates preadipocyte differentiation by targeting PPARG and CYP2U1 in beef cattle. Mol. Cell. Probes 2018, 42, 10–17. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, A.; Xiao, W.; Mi, S.; Hu, L.; Brito, L.F.; Guo, G.; Yan, Q.; Chen, S.; Wang, Y. Genetic parameters and genome-wide association analyses for lifetime productivity in Chinese Holstein cattle. J. Dairy Sci. 2024, 107, 9638–9655. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Gondro, C.; Park, H.S.; Cho, Y.M.; Chai, H.H.; Seong, H.H.; Yang, B.S.; Hong, S.K.; Chang, W.K.; Lee, S.H. Identification of recently selected mutations driven by artificial selection in hanwoo. Asian-Australas. J. Anim. Sci. 2013, 26, 603. [Google Scholar] [CrossRef] [PubMed]
- Hoda, A.; Bixheku, X.; Çekani, M.L. Computational analysis of non-synonymous single nucleotide polymorphism in the bovine PKLR geneComputational analysis of bovine PKLR gene. J. Biomol. Struct. Dyn. 2024, 42, 4168–4255. [Google Scholar] [CrossRef]
- Du, A.; Zhao, F.; Liu, Y.; Xu, L.; Chen, K.; Sun, D.; Han, B. Genetic polymorphisms of PKLR gene and their associations with milk production traits in Chinese Holstein cows. Front. Genet. 2022, 2, 1002706. [Google Scholar] [CrossRef]
- Brym, P.; Sazanov, A.A.; Kaminski, S. Associations between 60 SNPs identified by APEX microarray and growth rate, meatiness and selection index in boars. Genetika 2011, 47, 651. [Google Scholar] [CrossRef]
- Gimeno, R.E.; Ortegon, A.M.; Patel, S.; Punreddy, S.; Ge, P.; Sun, Y.; Lodish, H.F.; Stahl, A. Characterization of a heart-specific fatty acid transport protein. J. Biol. Chem. 2003, 278, 16039–16044. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, Z.; Yang, Z.; Jiang, H.; Chu, S.; Mao, Y.; Li, M.; Chen, Z.; Aboragah, A.; Loor, J.J.; et al. Abundance of solute carrier family 27 member 6 (SLC27A6) in the bovine mammary gland alters fatty acid metabolism. Food Funct. 2021, 12, 4909–4920. [Google Scholar] [CrossRef]
- Nafikov, R.A.; Schoonmaker, J.P.; Korn, K.T.; Noack, K.; Garrick, D.J.; Koehler, K.J.; Minick-Bormann, J.; Reecy, J.M.; Spurlock, D.E.; Beitz, D.C. Association of polymorphisms in solute carrier family 27, isoform A6 (SLC27A6) and fatty acid-binding protein-3 and fatty acid-binding protein-4 (FABP3 and FABP4) with fatty acid composition of bovine milk. J. Dairy Sci. 2013, 96, 6007–6021. [Google Scholar] [CrossRef] [PubMed]
- Sasazaki, S.; Kawaguchi, F.; Nakajima, A.; Yamamoto, R.; Akiyama, T.; Kohama, N.; Yoshida, E.; Kobayashi, E.; Honda, T.; Oyama, K.; et al. Detection of candidate polymorphisms around the QTL for fat area ratio to rib eye area on BTA7 using whole-genome resequencing in Japanese Black cattle. Anim. Sci. J. 2020, 91, e13335. [Google Scholar] [CrossRef]
- Fuerniss, L.K.; Johnson, B.J. Semi-automated technique for bovine skeletal muscle fiber cross-sectional area and myosin heavy chain determination. J. Anim. Sci. 2023, 101, skad205. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Song, S.M.; Ahn, C.H.; Kim, G.D. Muscle fiber typing in Bovine and porcine skeletal muscles using immunofluorescence with monoclonal antibodies specific to myosin heavy chain isoforms. Food Sci. Anim. Resour. 2020, 40, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yang, B.; Song, G.; Zhang, X.; Wang, Z.; Mo, Z.; Zan, L.; Wang, H. Effect of bovine myosin heavy chain 3 on proliferation and differentiation of myoblast. Anim. Biotechnol. 2023, 34, 4337–4346. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Yang, Y.; Zhou, L.; Baiyin, B.; Liu, Z.; Guo, L.; Ma, F.; Wang, J.; Chai, Y.; Shi, C.; et al. Candidate Genes and Gene Networks Change with Age in Japanese Black Cattle by Blood Transcriptome Analysis. Genes 2023, 14, 504. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- Bunkhumpornpat, C.; Boonchieng, E.; Chouvatut, V.; Lipsky, D. FLEX-SMOTE: Synthetic over-sampling technique that flexibly adjusts to different minority class distributions. Patterns 2024, 5, 101073. [Google Scholar] [CrossRef]
- Huo, C.; Zhang, C.; Lu, J.; Su, X.; Qi, X.; Guo, Y.; Bao, Y.; Jia, H.; Cao, G.; Na, R.; et al. A deep learning tissue classifier based on differential co-expression genes predicts the pregnancy outcomes of cattle. Biol. Reprod. 2025, 112, 550–562. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Precision | Recall | F1-Score | Accuracy |
|---|---|---|---|---|
| AdaBoost | 0.85 | 0.84 | 0.84 | 0.84 |
| SVM | 0.71 | 0.71 | 0.71 | 0.71 |
| MLP | 0.78 | 0.60 | 0.52 | 0.60 |
| SVC | 0.81 | 0.69 | 0.65 | 0.69 |
| RF | 0.66 | 0.65 | 0.64 | 0.65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Guo, Y.; Huo, C.; Zhu, L.; Shi, C.; Na, R.; Gu, M.; Zhang, W. Machine Learning-Based Analysis of Differentially Expressed Genes in the Muscle Transcriptome Between Beef Cattle and Dairy Cattle. Int. J. Mol. Sci. 2025, 26, 5046. https://doi.org/10.3390/ijms26115046
Li S, Guo Y, Huo C, Zhu L, Shi C, Na R, Gu M, Zhang W. Machine Learning-Based Analysis of Differentially Expressed Genes in the Muscle Transcriptome Between Beef Cattle and Dairy Cattle. International Journal of Molecular Sciences. 2025; 26(11):5046. https://doi.org/10.3390/ijms26115046
Chicago/Turabian StyleLi, Shuai, Yaqiang Guo, Chenxi Huo, Lin Zhu, Caixia Shi, Risu Na, Mingjuan Gu, and Wenguang Zhang. 2025. "Machine Learning-Based Analysis of Differentially Expressed Genes in the Muscle Transcriptome Between Beef Cattle and Dairy Cattle" International Journal of Molecular Sciences 26, no. 11: 5046. https://doi.org/10.3390/ijms26115046
APA StyleLi, S., Guo, Y., Huo, C., Zhu, L., Shi, C., Na, R., Gu, M., & Zhang, W. (2025). Machine Learning-Based Analysis of Differentially Expressed Genes in the Muscle Transcriptome Between Beef Cattle and Dairy Cattle. International Journal of Molecular Sciences, 26(11), 5046. https://doi.org/10.3390/ijms26115046