
The Role of Extracellular Vesicles in the Pathogenesis of Metabolic Dysfunction-Associated Steatotic Liver Disease and Other Liver Diseases



Abstract
1. Introduction
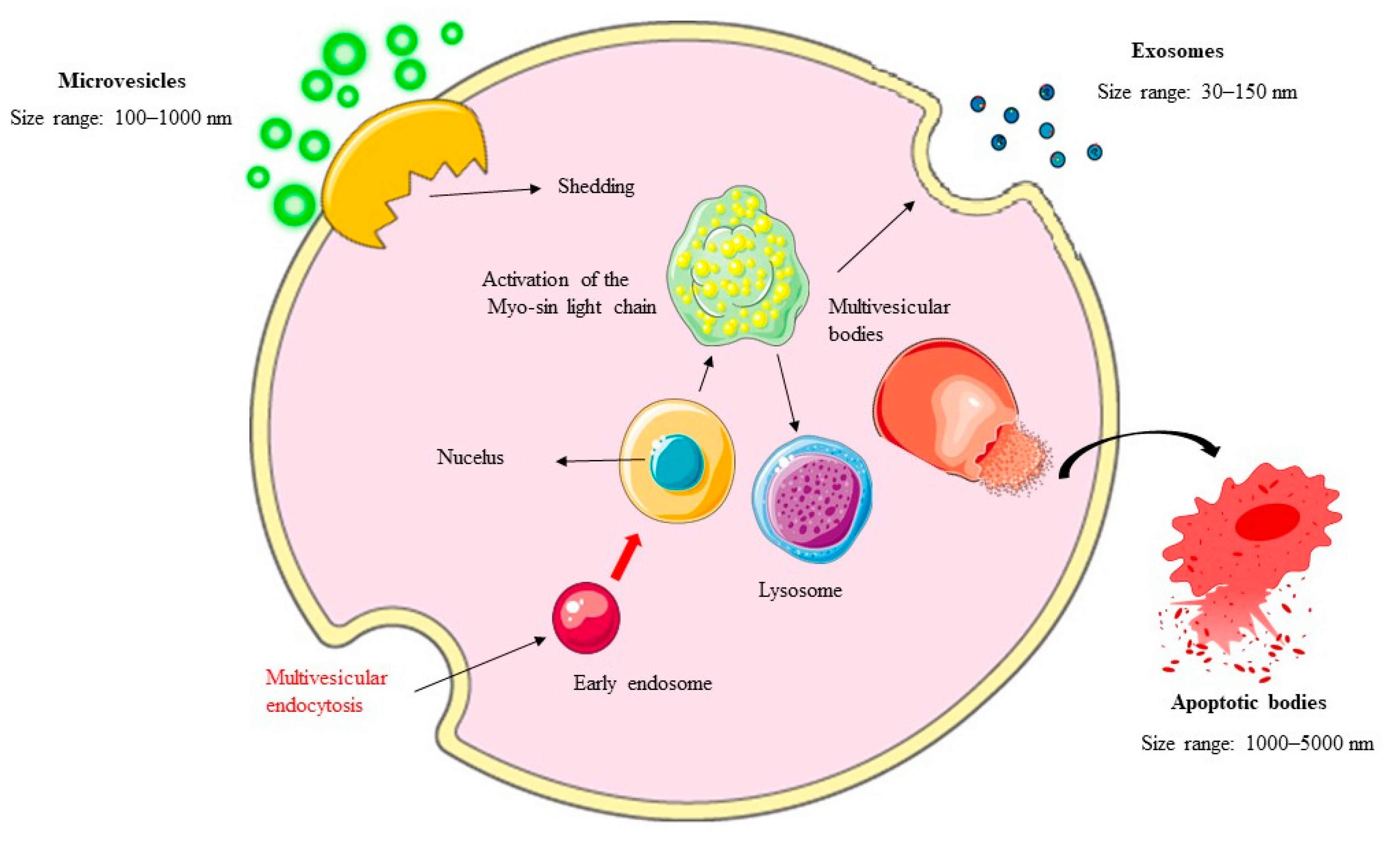
2. EVs: Biogenesis and Classification
2.1. Exosomes: Formation and Functional Diversity
2.2. Microvesicles (MVs): Direct Budding and Pathophysiological Roles
2.3. Apoptotic Bodies: Markers of Programmed Cell Death
2.4. Other Types of EVs: Ectosomes, Exomeres, and Supermeres
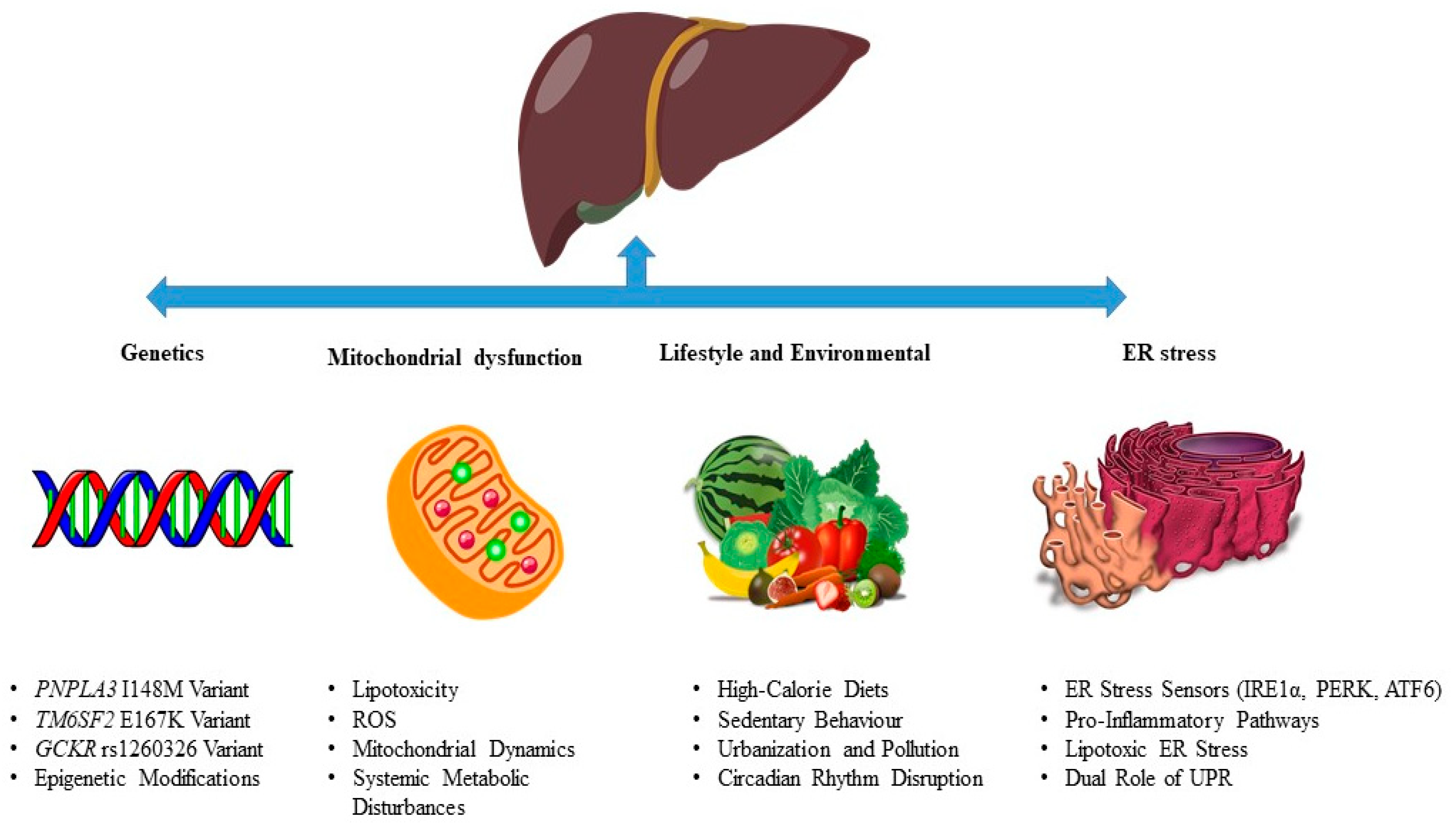
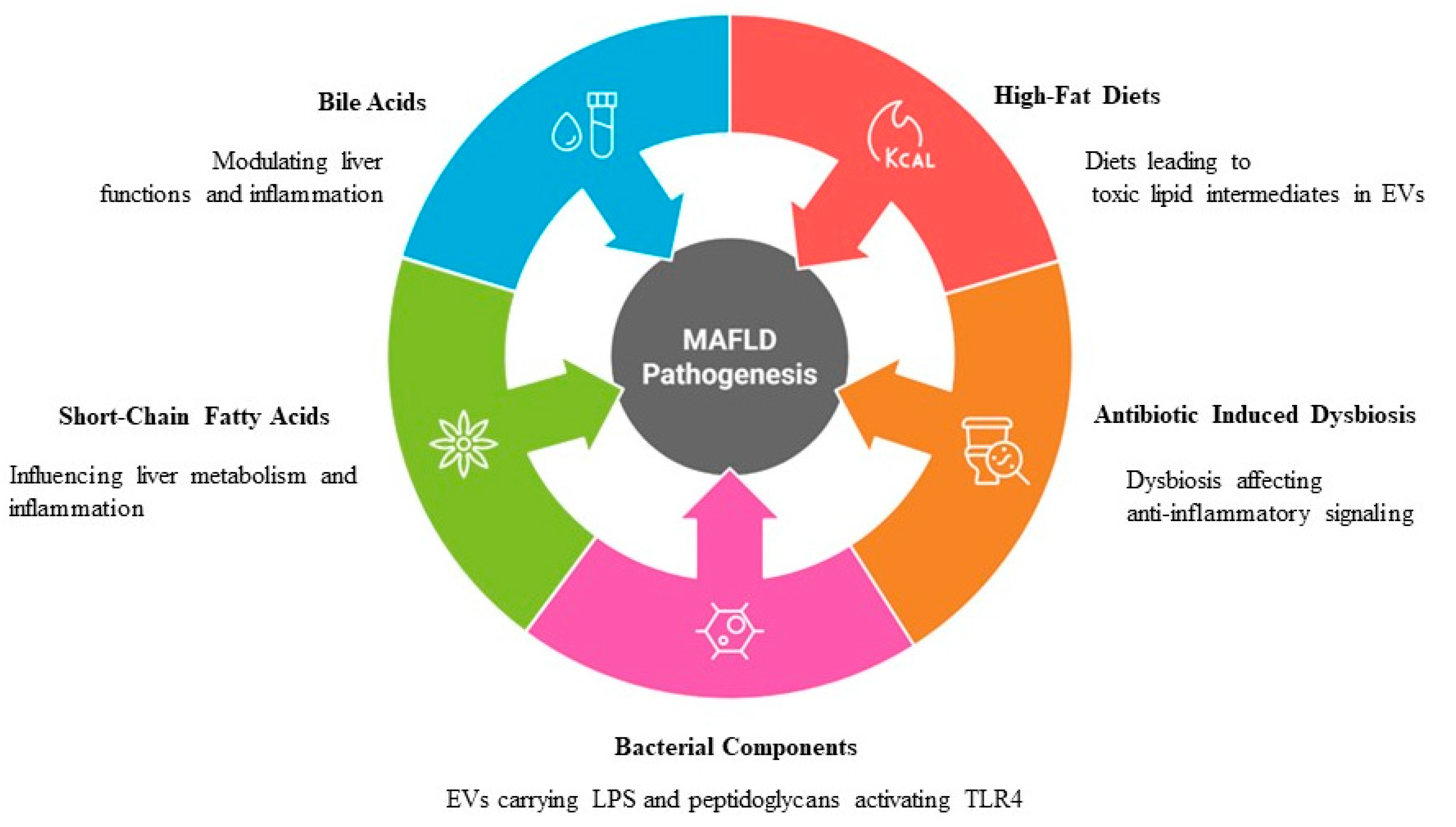
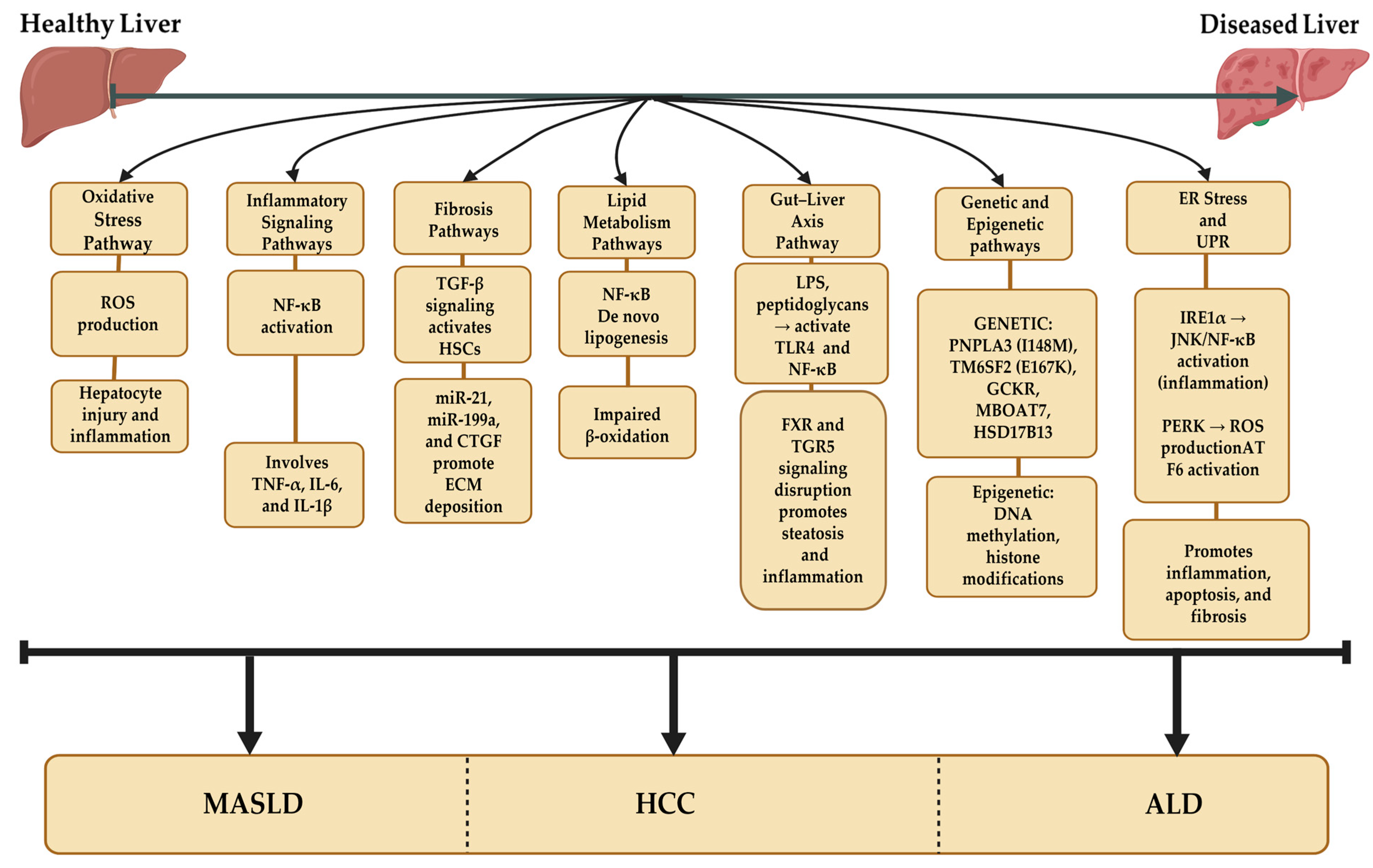
3. MASLD Pathogenesis
3.1. Genetics in MASLD Pathogenesis
3.2. Lifestyle and Environmental Influences in MASLD Pathogenesis
3.3. Mitochondrial Dysfunction in MASLD Pathogenesis
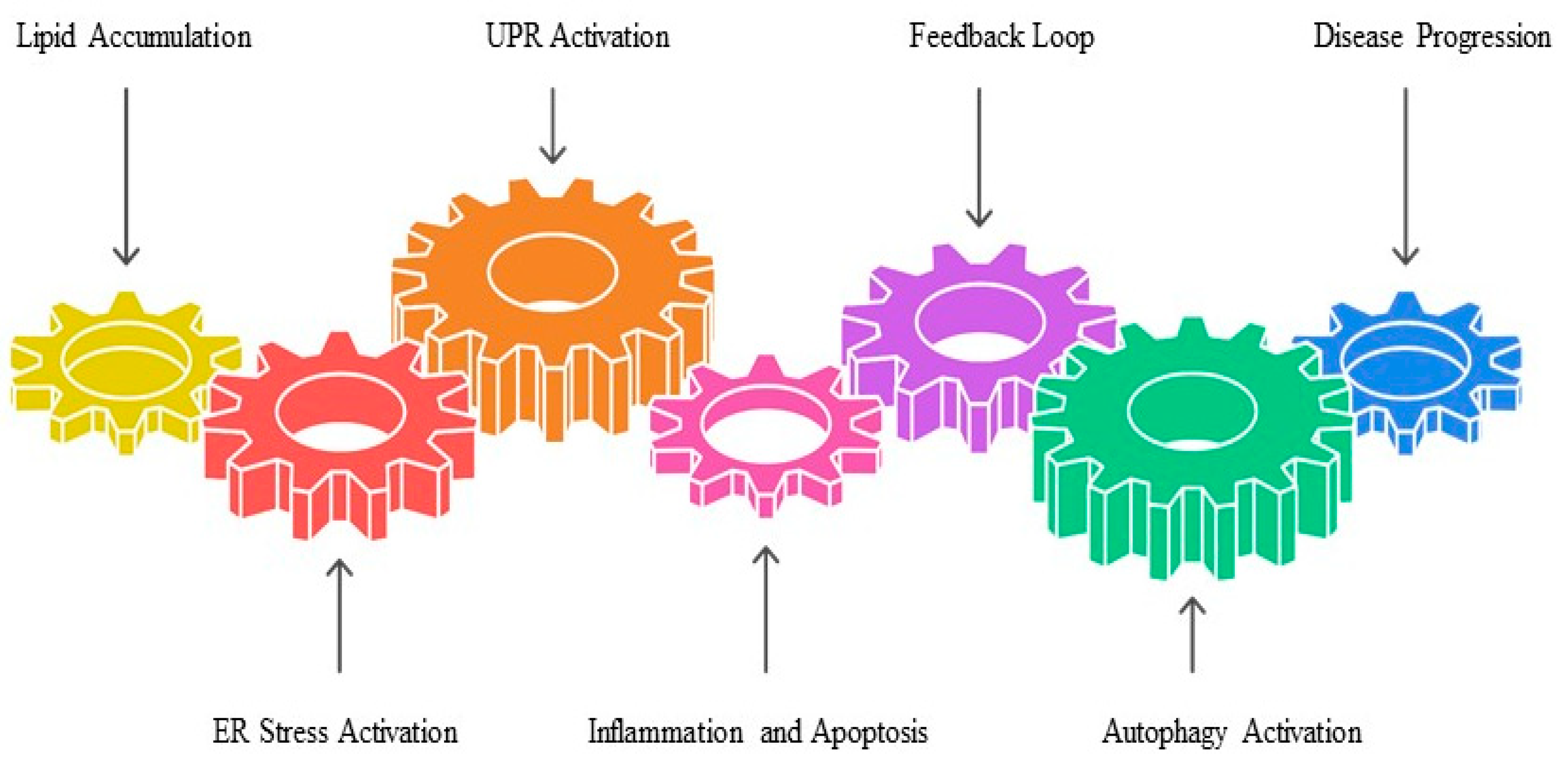
3.4. ER Stress and the Unfolded Protein Response (UPR) in MASLD Pathogenesis
4. The Role of EVs in MASLD Pathogenesis
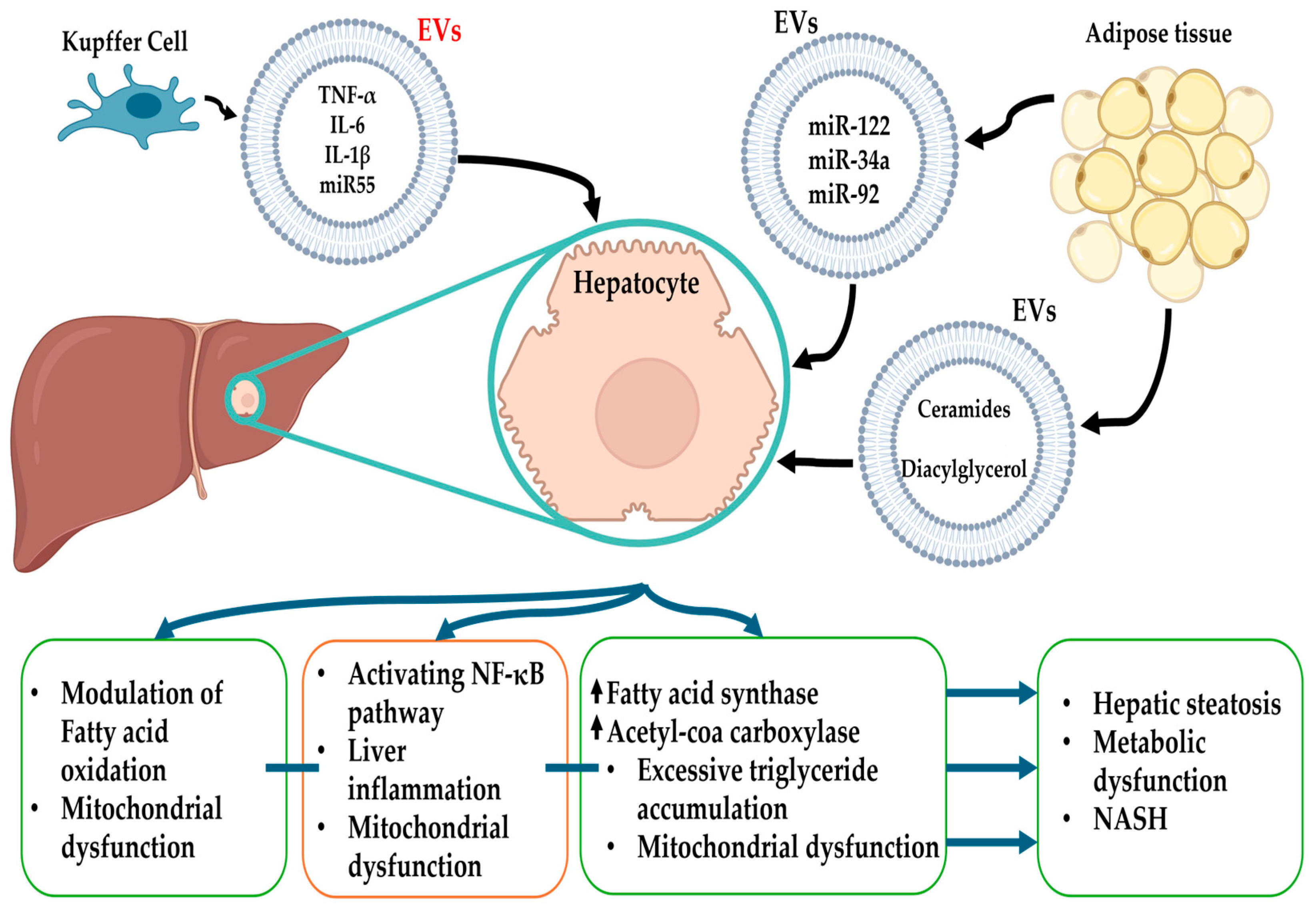
4.1. Lipid Metabolism
4.2. Inflammation
4.3. Fibrosis and Remodeling
4.4. Gut Microbiota and EVs
5. Extracellular Vesicles in Other Liver Diseases
5.1. EVs in Viral Hepatitis
5.2. EVs in ALD
5.3. EVs in DILI
5.4. EVs in HCC
6. Biomarkers and Therapeutic Potential
6.1. EVs as Biomarkers
6.2. Therapeutic Strategies Targeting EV-Mediated Pathways
6.3. Challenges and Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Méndez-Sánchez, N.; Bugianesi, E.; Gish, R.G.; Lammert, F.; Tilg, H.; Nguyen, M.H.; Sarin, S.K.; Fabrellas, N.; Zelber-Sagi, S.; Fan, J.G.; et al. Global multi-stakeholder endorsement of the MAFLD definition. Lancet Gastroenterol. Hepatol. 2022, 7, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.E.; Koh, T.J.L.; Tang, A.S.P.; Quek, J.; Yong, J.N.; Tay, P.; Tan, D.J.H.; Lim, W.H.; Lin, S.Y.; Huang, D.; et al. Global Prevalence and Clinical Characteristics of Metabolic-associated Fatty Liver Disease: A Meta-Analysis and Systematic Review of 10 739 607 Individuals. J. Clin. Endocrinol. Metab. 2022, 107, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ayada, I.; Zhang, X.; Wang, L.; Li, Y.; Wen, T.; Ma, Z.; Bruno, M.J.; de Knegt, R.J.; Cao, W.; et al. Estimating Global Prevalence of Metabolic Dysfunction-Associated Fatty Liver Disease in Overweight or Obese Adults. Clin. Gastroenterol. Hepatol. 2022, 20, e573–e582. [Google Scholar] [CrossRef]
- Méndez-Sánchez, N.; Brouwer, W.P.; Lammert, F.; Yilmaz, Y. Metabolic dysfunction associated fatty liver disease in healthy weight individuals. Hepatol. Int. 2024, 18, 884–896. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Sheron, N.; Zelber-Sagi, S.; Carrieri, P.; Dusheiko, G.; Bugianesi, E.; Pryke, R.; Hutchinson, S.J.; Sangro, B.; Martin, N.K.; et al. The EASL-Lancet Liver Commission: Protecting the next generation of Europeans against liver disease complications and premature mortality. Lancet 2022, 399, 61–116. [Google Scholar] [CrossRef]
- Vaz, K.; Clayton-Chubb, D.; Majeed, A.; Lubel, J.; Simmons, D.; Kemp, W.; Roberts, S.K. Current understanding and future perspectives on the impact of changing NAFLD to MAFLD on global epidemiology and clinical outcomes. Hepatol. Int. 2023, 17, 1082–1097. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Paik, J.M.; Henry, L.; Yang, J.; Fernandes, G.; Stepanova, M.; Nader, F. The Growing Economic and Clinical Burden of Nonalcoholic Steatohepatitis (NASH) in the United States. J. Clin. Exp. Hepatol. 2023, 13, 454–467. [Google Scholar] [CrossRef]
- van Meer, S.; van Erpecum, K.J.; Sprengers, D.; Klümpen, H.J.; Jansen, P.L.; Ijzermans, J.N.; Siersema, P.D.; de Man, R.A.; Verheij, J. Hepatocellular carcinoma in noncirrhotic livers is associated with steatosis rather than steatohepatitis: Potential implications for pathogenesis. Eur. J. Gastroenterol. Hepatol. 2016, 28, 955–962. [Google Scholar] [CrossRef]
- Schwartz, J.M.; Reinus, J.F. Prevalence and natural history of alcoholic liver disease. Clin. Liver Dis. 2012, 16, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Miteva, D.; Peshevska-Sekulovska, M.; Snegarova, V.; Peruhova, M.; Vasilev, G.H.; Vasilev, G.V.; Sekulovski, M.; Lazova, S.; Gulinac, M.; Tomov, L. Microbiome and Genetic Factors in the Pathogenesis of Liver Diseases. Gastroenterol. Insights 2023, 14, 575–597. [Google Scholar] [CrossRef]
- Bashir, A.; Duseja, A.; De, A.; Mehta, M.; Tiwari, P. Non-alcoholic fatty liver disease development: A multifactorial pathogenic phenomena. Liver Res. 2022, 6, 72–83. [Google Scholar] [CrossRef]
- Liu, G.; Yin, X.-M. The role of extracellular vesicles in liver pathogenesis. Am. J. Pathol. 2022, 192, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Escudero-Cernuda, S.; Eiro, N.; Fraile, M.; Vizoso, F.J.; Fernández-Colomer, B.; Fernández-Sánchez, M.L. Limitations and challenges in the characterization of extracellular vesicles from stem cells and serum. Mikrochim. Acta 2025, 192, 311. [Google Scholar] [CrossRef]
- Mo, W.; Peng, Y.; Zheng, Y.; Zhao, S.; Deng, L.; Fan, X. Extracellular vesicle-mediated bidirectional communication between the liver and other organs: Mechanistic exploration and prospects for clinical applications. J. Nanobiotechnol. 2025, 23, 190. [Google Scholar] [CrossRef]
- Hernández, A.; Arab, J.P.; Reyes, D.; Lapitz, A.; Moshage, H.; Bañales, J.M.; Arrese, M. Extracellular Vesicles in NAFLD/ALD: From Pathobiology to Therapy. Cells 2020, 9, 817. [Google Scholar] [CrossRef]
- Zhou, X.; Xie, F.; Wang, L.; Zhang, L.; Zhang, S.; Fang, M.; Zhou, F. The function and clinical application of extracellular vesicles in innate immune regulation. Cell. Mol. Immunol. 2020, 17, 323–334. [Google Scholar] [CrossRef]
- Sun, N.; Lee, Y.T.; Zhang, R.Y.; Kao, R.; Teng, P.C.; Yang, Y.; Yang, P.; Wang, J.J.; Smalley, M.; Chen, P.J.; et al. Purification of HCC-specific extracellular vesicles on nanosubstrates for early HCC detection by digital scoring. Nat. Commun. 2020, 11, 4489. [Google Scholar] [CrossRef]
- Słomka, A.; Mocan, T.; Wang, B.; Nenu, I.; Urban, S.K.; Gonzales-Carmona, M.; Schmidt-Wolf, I.G.H.; Lukacs-Kornek, V.; Strassburg, C.P.; Spârchez, Z.; et al. EVs as Potential New Therapeutic Tool/Target in Gastrointestinal Cancer and HCC. Cancers 2020, 12, 3019. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.S.; Chang, H.H. Extracellular vesicles: Function, resilience, biomarker, bioengineering, and clinical implications. Tzu Chi Med. J. 2024, 36, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Salmond, N.; Williams, K.C. Isolation and characterization of extracellular vesicles for clinical applications in cancer—time for standardization? Nanoscale Adv. 2021, 3, 1830–1852. [Google Scholar] [CrossRef] [PubMed]
- Hartjes, T.A.; Mytnyk, S.; Jenster, G.W.; van Steijn, V.; van Royen, M.E. Extracellular Vesicle Quantification and Characterization: Common Methods and Emerging Approaches. Bioengineering 2019, 6, 7. [Google Scholar] [CrossRef]
- Berggreen, A.H.; Petersen, J.L.; Lin, L.; Benabdellah, K.; Luo, Y. CRISPR delivery with extracellular vesicles: Promises and challenges. J. Extracell. Biol. 2023, 2, e111. [Google Scholar] [CrossRef]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Liu, M.; Wen, Z.; Zhang, T.; Zhang, L.; Liu, X.; Wang, M. The role of exosomal molecular cargo in exosome biogenesis and disease diagnosis. Front. Immunol. 2024, 15, 1417758. [Google Scholar] [CrossRef]
- Qiang, J.; Tao, Y.F.; Bao, J.W.; Chen, J.; Li, H.X.; He, J.; Xu, P. High Fat Diet-Induced miR-122 Regulates Lipid Metabolism and Fat Deposition in Genetically Improved Farmed Tilapia (GIFT, Oreochromis niloticus) Liver. Front. Physiol. 2018, 9, 1422. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Xie, L.; Wang, H.; Hu, J.; Liu, Z.; Hu, F. The role of novel adipokines and adipose-derived extracellular vesicles (ADEVs): Connections and interactions in liver diseases. Biochem. Pharmacol. 2024, 222, 116104. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Hall, C.; Glaser, S.; Francis, H.; Meng, F.; Alpini, G. Pathogenesis of Kupffer Cells in Cholestatic Liver Injury. Am. J. Pathol. 2016, 186, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.S.; Mustafa, T.; Connell, J.P.; Grande-Allen, K.J. Tumor necrosis factor alpha and interleukin 1 beta suppress myofibroblast activation via nuclear factor kappa B signaling in 3D-cultured mitral valve interstitial cells. Acta Biomater. 2021, 127, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef]
- Zhao, J.; Hu, Y.; Peng, J. Targeting programmed cell death in metabolic dysfunction-associated fatty liver disease (MAFLD): A promising new therapy. Cell. Mol. Biol. Lett. 2021, 26, 17. [Google Scholar] [CrossRef]
- Yu, J.; Sane, S.; Kim, J.E.; Yun, S.; Kim, H.J.; Jo, K.B.; Wright, J.P.; Khoshdoozmasouleh, N.; Lee, K.; Oh, H.T.; et al. Biogenesis and delivery of extracellular vesicles: Harnessing the power of EVs for diagnostics and therapeutics. Front. Mol. Biosci. 2023, 10, 1330400. [Google Scholar] [CrossRef]
- Zhang, Q.; Jeppesen, D.K.; Higginbotham, J.N.; Graves-Deal, R.; Trinh, V.Q.; Ramirez, M.A.; Sohn, Y.; Neininger, A.C.; Taneja, N.; McKinley, E.T.; et al. Supermeres are functional extracellular nanoparticles replete with disease biomarkers and therapeutic targets. Nat. Cell Biol. 2021, 23, 1240–1254. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Schneider, C.V. PNPLA3 I148M Interacts With Environmental Triggers to Cause Human Disease. Liver Int. 2025, 45, e16106. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Dong, B. Molecular mechanisms in MASLD/MASH-related HCC. Hepatology 2024. [Google Scholar] [CrossRef]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, E.A.; Yang, R.; Yerges-Armstrong, L.M.; Sreenivasan, U.; McFarland, R.; Leitch, C.C.; Wilson, M.H.; Narina, S.; Gorden, A.; Ryan, K.A.; et al. TM6SF2 rs58542926 impacts lipid processing in liver and small intestine. Hepatology 2017, 65, 1526–1542. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liu, S.; Zhao, Z.; Yu, X.; Liu, Q.; Xin, Y.; Xuan, S. Association of GCKR Gene Polymorphisms with the Risk of Nonalcoholic Fatty Liver Disease and Coronary Artery Disease in a Chinese Northern Han Population. J. Clin. Transl. Hepatol. 2019, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Li, J.; Zhang, J.; Yuan, F.; Yu, N.; Zhang, F.; Li, D.; Wang, J.; Zhang, L.; Shi, Y.; et al. Waist circumference mediates the association between rs1260326 in GCKR gene and the odds of lean NAFLD. Sci. Rep. 2023, 13, 6488. [Google Scholar] [CrossRef]
- Juanola, O.; Martínez-López, S.; Francés, R.; Gómez-Hurtado, I. Non-Alcoholic Fatty Liver Disease: Metabolic, Genetic, Epigenetic and Environmental Risk Factors. Int. J. Environ. Res. Public Health 2021, 18, 5227. [Google Scholar] [CrossRef]
- Romeo, S.; Sanyal, A.; Valenti, L. Leveraging Human Genetics to Identify Potential New Treatments for Fatty Liver Disease. Cell Metab. 2020, 31, 35–45. [Google Scholar] [CrossRef]
- Eslam, M.; Mangia, A.; Berg, T.; Chan, H.L.; Irving, W.L.; Dore, G.J.; Abate, M.L.; Bugianesi, E.; Adams, L.A.; Najim, M.A.; et al. Diverse impacts of the rs58542926 E167K variant in TM6SF2 on viral and metabolic liver disease phenotypes. Hepatology 2016, 64, 34–46. [Google Scholar] [CrossRef]
- Choudhary, N.S.; Duseja, A. Genetic and epigenetic disease modifiers: Non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD). Transl. Gastroenterol. Hepatol. 2021, 6, 2. [Google Scholar] [CrossRef]
- Katz, L.S.; Baumel-Alterzon, S.; Scott, D.K.; Herman, M.A. Adaptive and maladaptive roles for ChREBP in the liver and pancreatic islets. J. Biol. Chem. 2021, 296, 100623. [Google Scholar] [CrossRef]
- Geiger, M.; Gorica, E.; Mohammed, S.A.; Mongelli, A.; Mengozi, A.; Delfine, V.; Ruschitzka, F.; Costantino, S.; Paneni, F. Epigenetic Network in Immunometabolic Disease. Adv. Biol. 2024, 8, e2300211. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, Y.; Li, J.; Dang, Y.; Hu, D. Regulation of histone H3K27 methylation in inflammation and cancer. Mol. Biomed. 2025, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, C.M.; Frühbeck, G.; Escalada, J. Impact of Nutritional Changes on Nonalcoholic Fatty Liver Disease. Nutrients 2019, 11, 677. [Google Scholar] [CrossRef] [PubMed]
- Muriel, P.; López-Sánchez, P.; Ramos-Tovar, E. Fructose and the Liver. Int. J. Mol. Sci. 2021, 22, 6969. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Sargeant, J.A.; Gray, L.J.; Bodicoat, D.H.; Willis, S.A.; Stensel, D.J.; Nimmo, M.A.; Aithal, G.P.; King, J.A. The effect of exercise training on intrahepatic triglyceride and hepatic insulin sensitivity: A systematic review and meta-analysis. Obes. Rev. 2018, 19, 1446–1459. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, S.; Wu, J.; Wang, Y. Mitochondrial metabolic dysfunction and non-alcoholic fatty liver disease: New insights from pathogenic mechanisms to clinically targeted therapy. J. Transl. Med. 2023, 21, 510. [Google Scholar] [CrossRef]
- Cheng, W.-C.; Wong, P.-Y.; Wu, C.-D.; Cheng, P.-N.; Lee, P.-C.; Li, C.-Y. Non-linear association between long-term air pollution exposure and risk of metabolic dysfunction-associated steatotic liver disease. Environ. Health Prev. Med. 2024, 29, 7. [Google Scholar] [CrossRef]
- Guan, D.; Lazar, M.A. Circadian Regulation of Gene Expression and Metabolism in the Liver. Semin. Liver Dis. 2022, 42, 113–121. [Google Scholar] [CrossRef]
- Chung, H.; Chou, W.; Sears, D.D.; Patterson, R.E.; Webster, N.J.; Ellies, L.G. Time-restricted feeding improves insulin resistance and hepatic steatosis in a mouse model of postmenopausal obesity. Metabolism 2016, 65, 1743–1754. [Google Scholar] [CrossRef]
- Della Torre, S. Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology. Cells 2021, 10, 2502. [Google Scholar] [CrossRef]
- Kim, S.E.; Min, J.S.; Lee, S.; Lee, D.Y.; Choi, D. Different effects of menopausal hormone therapy on non-alcoholic fatty liver disease based on the route of estrogen administration. Sci. Rep. 2023, 13, 15461. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, M.; Xie, H.; Zhou, L.; Meng, X.; Shi, J.; Zheng, S. Role of reactive oxygen species in mediating hepatic ischemia-reperfusion injury and its therapeutic applications in liver transplantation. Transplant. Proc. 2007, 39, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Pagel-Langenickel, I.; Bao, J.; Pang, L.; Sack, M.N. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev. 2010, 31, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, Y.; Wang, D.; Huang, Z.; Xiao, X.; Zheng, Q.; Li, S.; Long, D.; Feng, L. Mitochondrial Dysfunction in Metabolic Dysfunction Fatty Liver Disease (MAFLD). Int. J. Mol. Sci. 2023, 24, 17514. [Google Scholar] [CrossRef]
- Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.F. PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients. Int. J. Mol. Sci. 2022, 23, 4305. [Google Scholar] [CrossRef]
- Chen, P.; Yao, L.; Yuan, M.; Wang, Z.; Zhang, Q.; Jiang, Y.; Li, L. Mitochondrial dysfunction: A promising therapeutic target for liver diseases. Genes Dis. 2024, 11, 101115. [Google Scholar] [CrossRef]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxid. Med. Cell. Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef]
- Cao, S.S.; Luo, K.L.; Shi, L. Endoplasmic Reticulum Stress Interacts With Inflammation in Human Diseases. J. Cell. Physiol. 2016, 231, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, N.; Doskey, L.C.; Malhi, H. The Role of Endoplasmic Reticulum in Lipotoxicity during Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) Pathogenesis. Am. J. Pathol. 2023, 193, 1887–1899. [Google Scholar] [CrossRef]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, M.-F.; Jiang, S.; Wu, J.; Liu, J.; Yuan, X.-W.; Shen, D.; Zhang, J.-Z.; Zhou, N.; He, J.; et al. Liver governs adipose remodelling via extracellular vesicles in response to lipid overload. Nat. Commun. 2020, 11, 719. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Huang, Y.-Q.; Da, M.-X.; Jin, W.-L.; Zhou, F.-H. Adipocyte-derived extracellular vesicles: Bridging the communications between obesity and tumor microenvironment. Discov. Oncol. 2023, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Dou, G.; Wang, L. MicroRNAs in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Int. J. Biol. Sci. 2021, 17, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Hochreuter, M.Y.; Dall, M.; Treebak, J.T.; Barrès, R. MicroRNAs in non-alcoholic fatty liver disease: Progress and perspectives. Mol. Metab. 2022, 65, 101581. [Google Scholar] [CrossRef]
- Heida, A.; Gruben, N.; Catrysse, L.; Koehorst, M.; Koster, M.; Kloosterhuis, N.J.; Gerding, A.; Havinga, R.; Bloks, V.W.; Bongiovanni, L.; et al. The hepatocyte IKK:NF-κB axis promotes liver steatosis by stimulating de novo lipogenesis and cholesterol synthesis. Mol. Metab. 2021, 54, 101349. [Google Scholar] [CrossRef]
- Hajduch, E.; Lachkar, F.; Ferré, P.; Foufelle, F. Roles of Ceramides in Non-Alcoholic Fatty Liver Disease. J. Clin. Med. 2021, 10, 792. [Google Scholar] [CrossRef]
- Cho, Y.K.; Lee, S.; Lee, J.; Doh, J.; Park, J.H.; Jung, Y.S.; Lee, Y.H. Lipid remodeling of adipose tissue in metabolic health and disease. Exp. Mol. Med. 2023, 55, 1955–1973. [Google Scholar] [CrossRef]
- Li, S.; Cheng, F.; Zhang, Z.; Xu, R.; Shi, H.; Yan, Y. The role of hepatocyte-derived extracellular vesicles in liver and extrahepatic diseases. Biomed. Pharmacother. 2024, 180, 117502. [Google Scholar] [CrossRef]
- Chen, Y.; Douanne, N.; Wu, T.; Kaur, I.; Tsering, T.; Erzingatzian, A.; Nadeau, A.; Juncker, D.; Nerguizian, V.; Burnier, J.V. Leveraging nature’s nanocarriers: Translating insights from extracellular vesicles to biomimetic synthetic vesicles for biomedical applications. Sci. Adv. 2025, 11, eads5249. [Google Scholar] [CrossRef]
- Yang, X.; Hao, J.; Luo, J.; Lu, X.; Kong, X. Adipose tissue-derived extracellular vesicles: Systemic messengers in health and disease (Review). Mol. Med. Rep. 2023, 28, 189. [Google Scholar] [CrossRef]
- Knight, B.L.; Hebbachi, A.; Hauton, D.; Brown, A.M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.-X.; Wei, S.; Yu, C.; Zhao, S.-Q.; Yang, W.-J.; Feng, Y.-H.; Pan, C.; Yang, K.-X.; Ma, Y. Activation of Kupffer cells in NAFLD and NASH: Mechanisms and therapeutic interventions. Front. Cell Dev. Biol. 2023, 11, 1199519. [Google Scholar] [CrossRef] [PubMed]
- Babuta, M.; Szabo, G. Extracellular vesicles in inflammation: Focus on the microRNA cargo of EVs in modulation of liver diseases. J. Leukoc. Biol. 2022, 111, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hu, W.; Lu, C.; Ma, Z.; Jiang, S.; Gu, C.; Acuña-Castroviejo, D.; Yang, Y. Targeting NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome in Cardiovascular Disorders. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2765–2779. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tao, Y.; Zhao, H.; Wang, Q. Adipose Extracellular Vesicles: Messengers From and to Macrophages in Regulating Immunometabolic Homeostasis or Disorders. Front. Immunol. 2021, 12, 666344. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.; Wang, Y.; Zhang, D.; Li, X.; Liu, S. The role of extracellular vesicles in non-alcoholic steatohepatitis: Emerging mechanisms, potential therapeutics and biomarkers. J. Adv. Res. 2025, 69, 157–168. [Google Scholar] [CrossRef]
- Catitti, G.; De Bellis, D.; Vespa, S.; Simeone, P.; Canonico, B.; Lanuti, P. Extracellular Vesicles as Players in the Anti-Inflammatory Inter-Cellular Crosstalk Induced by Exercise Training. Int. J. Mol. Sci. 2022, 23, 14098. [Google Scholar] [CrossRef]
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 2023, 23, 236–250. [Google Scholar] [CrossRef]
- Kolonics, F.; Szeifert, V.; Timár, C.I.; Ligeti, E.; Lőrincz, Á.M. The Functional Heterogeneity of Neutrophil-Derived Extracellular Vesicles Reflects the Status of the Parent Cell. Cells 2020, 9, 2718. [Google Scholar] [CrossRef]
- Li, J.; Yuan, Y.; Fu, Q.; Chen, M.; Liang, H.; Chen, X.; Long, X.; Zhang, B.; Zhao, J.; Chen, Q. Novel insights into the role of immunomodulatory extracellular vesicles in the pathogenesis of liver fibrosis. Biomark. Res. 2024, 12, 119. [Google Scholar] [CrossRef]
- Povero, D.; Panera, N.; Eguchi, A.; Johnson, C.D.; Papouchado, B.G.; de Araujo Horcel, L.; Pinatel, E.M.; Alisi, A.; Nobili, V.; Feldstein, A.E. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-γ. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 646–663.e4. [Google Scholar] [CrossRef] [PubMed]
- Martín-Taboada, M.; Corrales, P.; Medina-Gómez, G.; Vila-Bedmar, R. Tackling the effects of extracellular vesicles in fibrosis. Eur. J. Cell Biol. 2022, 101, 151221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xiao, X.; Wang, L.; Shi, X.; Fu, N.; Wang, S.; Zhao, R.C. Human adipose and umbilical cord mesenchymal stem cell-derived extracellular vesicles mitigate photoaging via TIMP1/Notch1. Signal Transduct. Target. Ther. 2024, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-González, S.; Marín-Royo, G.; Jurado-López, R.; Bartolomé, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef]
- Berdiaki, A.; Neagu, M.; Tzanakakis, P.; Spyridaki, I.; Pérez, S.; Nikitovic, D. Extracellular Matrix Components and Mechanosensing Pathways in Health and Disease. Biomolecules 2024, 14, 1186. [Google Scholar] [CrossRef]
- Zhang, B.; Zhao, J.; Jiang, M.; Peng, D.; Dou, X.; Song, Y.; Shi, J. The Potential Role of Gut Microbial-Derived Exosomes in Metabolic-Associated Fatty Liver Disease: Implications for Treatment. Front. Immunol. 2022, 13, 893617. [Google Scholar] [CrossRef]
- Jain, H.; Kumar, A.; Almousa, S.; Mishra, S.; Langsten, K.L.; Kim, S.; Sharma, M.; Su, Y.; Singh, S.; Kerr, B.A.; et al. Characterisation of LPS+ bacterial extracellular vesicles along the gut-hepatic portal vein-liver axis. J. Extracell. Vesicles 2024, 13, e12474. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Arvanitakis, K.; Stergiou, I.E.; Vallilas, C.; Sougioultzis, S.; Germanidis, G.; Theocharis, S. Interplay of Extracellular Vesicles and TLR4 Signaling in Hepatocellular Carcinoma Pathophysiology and Therapeutics. Pharmaceutics 2023, 15, 2460. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Q.; Tan, D.E.L.; Sikka, V.; Ng, C.H.; Xian, Y.; Li, D.; Muthiah, M.; Chew, N.W.S.; Storm, G.; et al. Gut-liver axis: Potential mechanisms of action of food-derived extracellular vesicles. J. Extracell. Vesicles 2024, 13, e12466. [Google Scholar] [CrossRef]
- Chiang, J.Y.; Pathak, P.; Liu, H.; Donepudi, A.; Ferrell, J.; Boehme, S. Intestinal Farnesoid X Receptor and Takeda G Protein Couple Receptor 5 Signaling in Metabolic Regulation. Dig. Dis. 2017, 35, 241–245. [Google Scholar] [CrossRef]
- Díaz-Garrido, N.; Badia, J.; Baldomà, L. Microbiota-derived extracellular vesicles in interkingdom communication in the gut. J. Extracell. Vesicles 2021, 10, e12161. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C. Energetic Stress-Induced Metabolic Regulation by Extracellular Vesicles. Compr. Physiol. 2023, 13, 5051–5068. [Google Scholar] [CrossRef]
- Gurjar, S.; Bhat, A.R.; Upadhya, R.; Shenoy, R.P. Extracellular vesicle-mediated approaches for the diagnosis and therapy of MASLD: Current advances and future prospective. Lipids Health Dis. 2025, 24, 5. [Google Scholar] [CrossRef] [PubMed]
- Rome, S.; Tacconi, S. High-fat diets: You are what you eat….your extracellular vesicles too! J. Extracell. Vesicles 2024, 13, e12382. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.-Y.; Liu, Y.-F.; Zeng, Z.-L.; Zhao, Z.-B.; Yan, X.-L.; Zheng, J.; Chen, W.-H.; Wang, Z.-X.; Xie, H.; Liu, J.-H. Antibiotic-induced gut microbiota disruption promotes vascular calcification by reducing short-chain fatty acid acetate. Mol. Med. 2024, 30, 130. [Google Scholar] [CrossRef]
- González-Lozano, E.; García-García, J.; Gálvez, J.; Hidalgo-García, L.; Rodríguez-Nogales, A.; Rodríguez-Cabezas, M.E.; Sánchez, M. Novel Horizons in Postbiotics: Lactobacillaceae Extracellular Vesicles and Their Applications in Health and Disease. Nutrients 2022, 14, 5296. [Google Scholar] [CrossRef]
- Li, J.; Shi, M.; Wang, Y.; Liu, J.; Liu, S.; Kang, W.; Liu, X.; Chen, X.; Huang, K.; Liu, Y. Probiotic-derived extracellular vesicles alleviate AFB1-induced intestinal injury by modulating the gut microbiota and AHR activation. J. Nanobiotechnol. 2024, 22, 697. [Google Scholar] [CrossRef]
- Mann, E.R.; Lam, Y.K.; Uhlig, H.H. Short-chain fatty acids: Linking diet, the microbiome and immunity. Nat. Rev. Immunol. 2024, 24, 577–595. [Google Scholar] [CrossRef]
- Li, C.; Yao, J.; Yang, C.; Yu, S.; Yang, Z.; Wang, L.; Li, S.; He, N. Gut microbiota-derived short chain fatty acids act as mediators of the gut-liver-brain axis. Metab. Brain Dis. 2025, 40, 122. [Google Scholar] [CrossRef]
- Zhou, W.; Pal, A.S.; Hsu, A.Y.; Gurol, T.; Zhu, X.; Wirbisky-Hershberger, S.E.; Freeman, J.L.; Kasinski, A.L.; Deng, Q. MicroRNA-223 Suppresses the Canonical NF-κB Pathway in Basal Keratinocytes to Dampen Neutrophilic Inflammation. Cell Rep. 2018, 22, 1810–1823. [Google Scholar] [CrossRef]
- Jiang, J.; Gao, Y.; Wang, J.; Huang, Y.; Yang, R.; Zhang, Y.; Ma, Y.; Wen, Y.; Luo, G.; Zhang, S.; et al. Hepatic sphingomyelin phosphodiesterase 3 promotes steatohepatitis by disrupting membrane sphingolipid metabolism. Cell Metab. 2025, 37, 1119–1136.e13. [Google Scholar] [CrossRef]
- Kouroumalis, E.; Tsomidis, I.; Voumvouraki, A. Extracellular Vesicles in Viral Liver Diseases. Viruses 2024, 16, 1785. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Niu, J.; Shi, Y. Exosomes target HBV-host interactions to remodel the hepatic immune microenvironment. J. Nanobiotechnol. 2024, 22, 315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ou, M.; Yang, P.; Ning, M. The role of extracellular vesicle immune checkpoints in cancer. Clin. Exp. Immunol. 2024, 216, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Romano, R.; Bucci, C.; Marzetti, E. Extracellular Vesicles and Damage-Associated Molecular Patterns: A Pandora’s Box in Health and Disease. Front. Immunol. 2020, 11, 601740. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, J.H. The emerging roles of extracellular vesicles as intercellular messengers in liver physiology and pathology. Clin. Mol. Hepatol. 2022, 28, 706–724. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, S.; Yang, D.; Xu, W.; Qian, H. Extracellular vesicles: Emerging roles, biomarkers and therapeutic strategies in fibrotic diseases. J. Nanobiotechnol. 2023, 21, 164. [Google Scholar] [CrossRef]
- Cho, Y.E.; Song, B.J.; Akbar, M.; Baek, M.C. Extracellular vesicles as potential biomarkers for alcohol- and drug-induced liver injury and their therapeutic applications. Pharmacol. Ther. 2018, 187, 180–194. [Google Scholar] [CrossRef]
- Eguchi, A.; Yan, R.; Pan, S.Q.; Wu, R.; Kim, J.; Chen, Y.; Ansong, C.; Smith, R.D.; Tempaku, M.; Ohno-Machado, L.; et al. Comprehensive characterization of hepatocyte-derived extracellular vesicles identifies direct miRNA-based regulation of hepatic stellate cells and DAMP-based hepatic macrophage IL-1β and IL-17 upregulation in alcoholic hepatitis mice. J. Mol. Med. 2020, 98, 1021–1034. [Google Scholar] [CrossRef]
- Rahman, M.A.; Patters, B.J.; Kodidela, S.; Kumar, S. Extracellular Vesicles: Intercellular Mediators in Alcohol-Induced Pathologies. J. Neuroimmune Pharmacol. 2020, 15, 409–421. [Google Scholar] [CrossRef]
- Hu, Q.; Lyon, C.J.; Fletcher, J.K.; Tang, W.; Wan, M.; Hu, T.Y. Extracellular vesicle activities regulating macrophage- and tissue-mediated injury and repair responses. Acta Pharm. Sin. B 2021, 11, 1493–1512. [Google Scholar] [CrossRef]
- Sosnowski, K.; Przybyłkowski, A. Ethanol-induced changes to the gut microbiome compromise the intestinal homeostasis: A review. Gut Microbes 2024, 16, 2393272. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Yang, L.; Chu, H. The role of gut microbiota, exosomes, and their interaction in the pathogenesis of ALD. J. Adv. Res. 2025, 72, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.A.; Baba, S.K.; Sadida, H.Q.; Marzooqi, S.A.; Jerobin, J.; Altemani, F.H.; Algehainy, N.; Alanazi, M.A.; Abou-Samra, A.B.; Kumar, R.; et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduct. Target. Ther. 2024, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Momen-Heravi, F.; Furi, I.; Kodys, K.; Catalano, D.; Gangopadhyay, A.; Haraszti, R.; Satishchandran, A.; Iracheta-Vellve, A.; Adejumo, A.; et al. Extracellular vesicles from mice with alcoholic liver disease carry a distinct protein cargo and induce macrophage activation through heat shock protein 90. Hepatology 2018, 67, 1986–2000. [Google Scholar] [CrossRef]
- Umbaugh, D.S.; Jaeschke, H. Extracellular vesicles: Roles and applications in drug-induced liver injury. Adv. Clin. Chem. 2021, 102, 63–125. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, S.; Marzolf, B.; Troisch, P.; Brightman, A.; Hu, Z.; Hood, L.E.; Galas, D.J. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc. Natl. Acad. Sci. USA 2009, 106, 4402–4407. [Google Scholar] [CrossRef]
- Andrade, R.J.; Chalasani, N.; Björnsson, E.S.; Suzuki, A.; Kullak-Ublick, G.A.; Watkins, P.B.; Devarbhavi, H.; Merz, M.; Lucena, M.I.; Kaplowitz, N.; et al. Drug-induced liver injury. Nat. Rev. Dis. Primers 2019, 5, 58. [Google Scholar] [CrossRef]
- Royo, F.; Palomo, L.; Mleczko, J.; Gonzalez, E.; Alonso, C.; Martínez, I.; Pérez-Cormenzana, M.; Castro, A.; Falcon-Perez, J.M. Metabolically active extracellular vesicles released from hepatocytes under drug-induced liver-damaging conditions modify serum metabolome and might affect different pathophysiological processes. Eur. J. Pharm. Sci. 2017, 98, 51–57. [Google Scholar] [CrossRef]
- Seay, T.W.; Suo, Z. Roles of Extracellular Vesicles on the Progression and Metastasis of Hepatocellular Carcinoma. Cells 2023, 12, 1879. [Google Scholar] [CrossRef]
- Olejarz, W.; Kubiak-Tomaszewska, G.; Chrzanowska, A.; Lorenc, T. Exosomes in Angiogenesis and Anti-angiogenic Therapy in Cancers. Int. J. Mol. Sci. 2020, 21, 5840. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, L.; Wang, X.; Wang, Y.; Yu, J.; Li, M.; Ma, Z.; Chi-Lui Ho, P.; Chen, X.; Wang, L.; et al. Extracellular vesicles in the HCC microenvironment: Implications for therapy and biomarkers. Pharmacol. Res. 2024, 209, 107419. [Google Scholar] [CrossRef] [PubMed]
- Belhabib, I.; Zaghdoudi, S.; Lac, C.; Bousquet, C.; Jean, C. Extracellular Matrices and Cancer-Associated Fibroblasts: Targets for Cancer Diagnosis and Therapy? Cancers 2021, 13, 3466. [Google Scholar] [CrossRef]
- Kuang, L.; Wu, L.; Li, Y. Extracellular vesicles in tumor immunity: Mechanisms and novel insights. Mol. Cancer 2025, 24, 45. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Zhang, S.; He, G.; Zheng, H.; Fan, Q.; et al. Extracellular vesicle PD-L1 in reshaping tumor immune microenvironment: Biological function and potential therapy strategies. Cell Commun. Signal 2022, 20, 14. [Google Scholar] [CrossRef]
- Cheng, H.-Y.; Su, G.-L.; Wu, Y.-X.; Chen, G.; Yu, Z.-L. Extracellular vesicles in anti-tumor drug resistance: Mechanisms and therapeutic prospects. J. Pharm. Anal. 2024, 14, 100920. [Google Scholar] [CrossRef]
- Zhou, Y.; Ren, H.; Dai, B.; Li, J.; Shang, L.; Huang, J.; Shi, X. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J. Exp. Clin. Cancer Res. 2018, 37, 324. [Google Scholar] [CrossRef]
- Zeng, Y.; Hu, S.; Luo, Y.; He, K. Exosome Cargos as Biomarkers for Diagnosis and Prognosis of Hepatocellular Carcinoma. Pharmaceutics 2023, 15, 2365. [Google Scholar] [CrossRef]
- Mauro, M.; Ugo, P.; Walton, Z.; Ali, S.; Rastellini, C.; Cicalese, L. Glypican-3 (GPC-3) Structural Analysis and Cargo in Serum Small Extracellular Vesicles of Hepatocellular Carcinoma Patients. Int. J. Mol. Sci. 2023, 24, 10922. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, D.; Li, Y. Extracellular Vesicles in Pathogenesis and Treatment of Metabolic Associated Fatty Liver Disease. Front. Physiol. 2022, 13, 909518. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, S.Y.; Ko, E.; Lee, J.H.; Yi, H.S.; Yoo, Y.J.; Je, J.; Suh, S.J.; Jung, Y.K.; Kim, J.H.; et al. Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci. Rep. 2017, 7, 3710. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Luo, S.-Z.; Xu, Z.-X.; Zhou, C.; Li, Z.-H.; Zhou, X.-Y.; Xu, M.-Y. Lipotoxic hepatocyte-derived exosomal miR-1297 promotes hepatic stellate cell activation through the PTEN signaling pathway in metabolic-associated fatty liver disease. World J. Gastroenterol. 2021, 27, 1419–1434. [Google Scholar] [CrossRef]
- Shi, Y.; Du, L.; Lv, D.; Li, Y.; Zhang, Z.; Huang, X.; Tang, H. Emerging role and therapeutic application of exosome in hepatitis virus infection and associated diseases. J. Gastroenterol. 2021, 56, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, S.; Gaal, K.; Britton, R.S.; Bacon, B.R.; Triadafilopoulos, G.; Tsukamoto, H. Increased 4-hydroxynonenal levels in experimental alcoholic liver disease: Association of lipid peroxidation with liver fibrogenesis. Hepatology 1992, 16, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Abdelhameed, F.; Kite, C.; Lagojda, L.; Dallaway, A.; Chatha, K.K.; Chaggar, S.S.; Dalamaga, M.; Kassi, E.; Kyrou, I.; Randeva, H.S. Non-invasive Scores and Serum Biomarkers for Fatty Liver in the Era of Metabolic Dysfunction-associated Steatotic Liver Disease (MASLD): A Comprehensive Review From NAFLD to MAFLD and MASLD. Curr. Obes. Rep. 2024, 13, 510–531. [Google Scholar] [CrossRef]
- Dilsiz, N. A comprehensive review on recent advances in exosome isolation and characterization: Toward clinical applications. Transl. Oncol. 2024, 50, 102121. [Google Scholar] [CrossRef]
- Rodrigues-Junior, D.M.; Tsirigoti, C.; Lim, S.K.; Heldin, C.H.; Moustakas, A. Extracellular Vesicles and Transforming Growth Factor β Signaling in Cancer. Front. Cell Dev. Biol. 2022, 10, 849938. [Google Scholar] [CrossRef]
- Sun, M.; Tang, M.; Qian, Y.; Zong, G.; Zhu, G.; Jiang, Y.; Mu, Y.; Zhou, M.; Ding, Q.; Wang, H.; et al. Extracellular vesicles-derived ferritin from lipid-induced hepatocytes regulates activation of hepatic stellate cells. Heliyon 2024, 10, e33741. [Google Scholar] [CrossRef]
- Yoo, D.; Jung, S.Y.; Go, D.; Park, J.Y.; You, D.G.; Jung, W.K.; Li, Y.; Ding, J.; Park, J.H.; Um, W. Functionalized extracellular vesicles of mesenchymal stem cells for regenerative medicine. J. Nanobiotechnol. 2025, 23, 219. [Google Scholar] [CrossRef]
- Gong, Y.; You, Q.; Yuan, X.; Zeng, F.; Zhang, F.; Xiao, J.; Chen, H.; Liu, Y.; Wang, T.; Yan, X.; et al. Mesenchymal stem cell-derived extracellular vesicles attenuate ferroptosis in aged hepatic ischemia/reperfusion injury by transferring miR-1275. Redox Biol. 2025, 81, 103556. [Google Scholar] [CrossRef]
- Owliaee, I.; Khaledian, M.; Boroujeni, A.K.; Shojaeian, A. Engineered small extracellular vesicles as a novel platform to suppress human oncovirus-associated cancers. Infect. Agent. Cancer 2023, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Zheng, L.; Li, Y.; Yang, J.; Mao, T.; Zhang, J.; Liu, Y.; Ning, J.; Zhang, T.; Huang, H.; et al. Engineered extracellular vesicles for delivering functional Cas9/gRNA to eliminate hepatitis B virus cccDNA and integration. Emerg. Microbes. Infect. 2024, 13, 2284286. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, J.; Yang, Y.; Chen, Y. PD-1/PD-L1 checkpoint inhibitors in advanced hepatocellular carcinoma immunotherapy. Front. Immunol. 2022, 13, 1070961. [Google Scholar] [CrossRef]
- Newman, L.A.; Muller, K.; Rowland, A. Circulating cell-specific extracellular vesicles as biomarkers for the diagnosis and monitoring of chronic liver diseases. Cell. Mol. Life Sci. 2022, 79, 232. [Google Scholar] [CrossRef]
- Lim, H.K.; Jeffrey, G.P.; Ramm, G.A.; Soekmadji, C. Pathogenesis of Viral Hepatitis-Induced Chronic Liver Disease: Role of Extracellular Vesicles. Front. Cell. Infect. Microbiol. 2020, 10, 587628. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, T.D.; Buonocore, L.; Rose, N.F.; Rose, J.K.; Robek, M.D. Virus-Like Vesicle-Based Therapeutic Vaccine Vectors for Chronic Hepatitis B Virus Infection. J. Virol. 2015, 89, 10407–10415. [Google Scholar] [CrossRef]
- De Sousa, K.P.; Rossi, I.; Abdullahi, M.; Ramirez, M.I.; Stratton, D.; Inal, J.M. Isolation and characterization of extracellular vesicles and future directions in diagnosis and therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2023, 15, e1835. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Jakhmola, V.R.; Baweja, S. Bioengineered extracellular vesicles: The path to precision medicine in liver diseases. Liver Res. 2025, 9, 17–28. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Hirsova, P.; Kostallari, E.; Sidhu, G.S.; Ibrahim, S.H.; Malhi, H. Extracellular Vesicles in Hepatobiliary Health and Disease. Compr. Physiol. 2023, 13, 4631–4658. [Google Scholar] [CrossRef]
- Shaba, E.; Vantaggiato, L.; Governini, L.; Haxhiu, A.; Sebastiani, G.; Fignani, D.; Grieco, G.E.; Bergantini, L.; Bini, L.; Landi, C. Multi-Omics Integrative Approach of Extracellular Vesicles: A Future Challenging Milestone. Proteomes 2022, 10, 12. [Google Scholar] [CrossRef]
- Qian, F.; Huang, Z.; Zhong, H.; Lei, Q.; Ai, Y.; Xie, Z.; Zhang, T.; Jiang, B.; Zhu, W.; Sheng, Y.; et al. Analysis and Biomedical Applications of Functional Cargo in Extracellular Vesicles. ACS Nano 2022, 16, 19980–20001. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, Z.F.; Graim, K.S.; He, M. Towards artificial intelligence-enabled extracellular vesicle precision drug delivery. Adv. Drug Deliv. Rev. 2023, 199, 114974. [Google Scholar] [CrossRef] [PubMed]
- Bordanaba-Florit, G.; Royo, F.; Kruglik, S.G.; Falcón-Pérez, J.M. Using single-vesicle technologies to unravel the heterogeneity of extracellular vesicles. Nat. Protoc. 2021, 16, 3163–3185. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.T.; Ko, J. Future of Digital Assays to Resolve Clinical Heterogeneity of Single Extracellular Vesicles. ACS Nano 2022, 16, 11619–11645. [Google Scholar] [CrossRef]
- Lawrence, S.R.; Shah, K.M. Prospects and Current Challenges of Extracellular Vesicle-Based Biomarkers in Cancer. Biology 2024, 13, 694. [Google Scholar] [CrossRef]
- Ghosh, S.; Zhao, X.; Alim, M.; Brudno, M.; Bhat, M. Artificial intelligence applied to ‘omics data in liver disease: Towards a personalised approach for diagnosis, prognosis and treatment. Gut 2025, 74, 295–311. [Google Scholar] [CrossRef]
- Wu, Z.; Xia, M.; Salas, S.S.; Trillos-Almanza, M.C.; Aguilar, M.M.; Arroyave-Ospina, J.C.; Wang, J.; Arrese, M.; Sydor, S.; Bechmann, L.P.; et al. Extracellular vesicles in metabolic dysfunction associated fatty liver disease: Mechanisms, diagnostic and therapeutic implications. Explor. Dig. Dis. 2022, 1, 4–20. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EVs Subtype | Size (Diameter) | Key Markers | Biogenesis Pathways | Cargo Types | References |
|---|---|---|---|---|---|
| Exosomes | 30–150 nanometers | CD63, CD81, CD9; nucleic acids (e.g., miRNAs, mRNAs) | Inward budding of endosomal membranes forming intraluminal vesicles in multivesicular bodies; regulated by ESCRT complex, Alix, and Tsg101 | Proteins (e.g., tetraspanins), lipids, nucleic acids, metabolites. | [26,27,28,29] |
| MVs | 100–1000 nanometers | Influenced by ADAM10 and ADAM17 enzymes | Direct outward budding from plasma membrane; driven by cytoskeletal dynamics, calcium levels, and enzymatic activities | Larger cytoplasmic fragments, organelles, lipids (e.g., ceramides), pro-inflammatory cytokines (e.g., TNF-α, IL-1β) | [30,31,32,33] |
| Apoptotic Bodies | 500–4000 nanometers | Phosphatidylserine, annexin V, thrombospondin, complement protein C3b | Formed during apoptosis; outward blebbing of cell membrane driven by actin-myosin interactions and cellular fragmentation | Cellular debris, nuclear chromatin, proteins; associated with cell death and inflammation | [34,35,36] |
| Ectosomes | 100–1000 nanometers | Not specified | Outward budding of plasma membrane; involves reorganization of membrane and cytoskeletal components | Implied to be distinct from exosomes or microvesicles, potentially including unique proteins and lipids | [37] |
| Exomeres | <50 nanometers | Unique proteomic profiles | Nonvesicular nanoparticles; biogenesis not fully described, but distinct from vesicular EVs | Proteins with specialized profiles; involved in intercellular communication and disease progression | [38] |
| Supermeres | <50 nanometers | Enriched with RNAs | Nonvesicular nanoparticles; biogenesis mechanisms unclear, but associated with enhanced tissue accumulation | RNAs (highly enriched), disease biomarkers, therapeutic targets; exhibits biodistribution patterns for intercellular signalling | [38] |
| Gene | Variant | Prevalence Rate | Functional Consequences | Clinical Implications for MASLD Prevention/Control | Reference |
|---|---|---|---|---|---|
| PNPLA3 | I148M (rs738409) | ~20–40% globally | Reduces triacylglycerol hydrolysis, leading to lipid accumulation, inflammation, and fibrosis progression. | Carriers are at higher risk for severe liver disease; lifestyle modifications (e.g., calorie restriction, exercise) may mitigate risk. | [39] |
| TM6SF2 | E167K (rs58542926) | ~5–10% globally | Impairs very-low-density lipoprotein secretion, increasing hepatic fat content but reducing circulating lipids and cardiovascular risk. | Personalized dietary and pharmacological interventions targeting lipid export pathways could reduce hepatic steatosis. | [42] |
| GCKR | P446L (rs1260326) | ~20–30% globally | Enhances glucokinase activity, promoting insulin sensitivity but upregulating de novo lipogenesis. | Genotype-guided dietary strategies (e.g., low-carbohydrate diets) may help manage lipid synthesis and reduce hepatic fat accumulation. | [43] |
| MBOAT7 | rs641738 | ~10–20% globally | Alters phospholipid remodelling, leading to altered lipid composition and increased inflammation. | Targeting phospholipid metabolism pathways may offer therapeutic potential to reduce inflammation and fibrosis risk. | [45] |
| HSD17B13 | rs72613567 | ~5–15% globally | Protective effects against MASLD progression by reducing inflammation and fibrosis risk. | Understanding protective mechanisms could guide the development of anti-inflammatory therapies for MASLD. | [45] |
| Liver Disease | EV-Associated Biomarker | Significance | Reference |
|---|---|---|---|
| MASLD | miR-128-3p | Promotes fibrogenesis by targeting PPARγ in HSCs | [91] |
| MASLD | miR-122 | Early detection, disease staging, correlates with liver histology scores | [128] |
| MASLD | Glypican-3 | Reflects hepatocyte injury levels, predicts fibrosis stages | [140] |
| MASLD | miR-192 | Upregulates fibrosis markers, enhances profibrogenic activity of HSCs | [141] |
| MASLD | miRNA-1297 | Activates HSCs via the PTEN pathway | [143] |
| Viral Hepatitis | HBV DNA/HCV RNA | Indicates viral load, reflects antiviral therapy response, aids in personalized treatment adjustments | [144] |
| ALD | Oxidative Stress Markers (e.g., Malondialdehyde, 4-Hydroxynonenal) | Provides insights into ethanol-induced liver damage and fibrosis development | [145] |
| HCC | Alpha-fetoprotein, Des-gamma-carboxy prothrombin | Enables early detection of HCC in high-risk populations | [146] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grossini, E.; Ola Pour, M.M.; Venkatesan, S. The Role of Extracellular Vesicles in the Pathogenesis of Metabolic Dysfunction-Associated Steatotic Liver Disease and Other Liver Diseases. Int. J. Mol. Sci. 2025, 26, 5033. https://doi.org/10.3390/ijms26115033
Grossini E, Ola Pour MM, Venkatesan S. The Role of Extracellular Vesicles in the Pathogenesis of Metabolic Dysfunction-Associated Steatotic Liver Disease and Other Liver Diseases. International Journal of Molecular Sciences. 2025; 26(11):5033. https://doi.org/10.3390/ijms26115033
Chicago/Turabian StyleGrossini, Elena, Mohammad Mostafa Ola Pour, and Sakthipriyan Venkatesan. 2025. "The Role of Extracellular Vesicles in the Pathogenesis of Metabolic Dysfunction-Associated Steatotic Liver Disease and Other Liver Diseases" International Journal of Molecular Sciences 26, no. 11: 5033. https://doi.org/10.3390/ijms26115033
APA StyleGrossini, E., Ola Pour, M. M., & Venkatesan, S. (2025). The Role of Extracellular Vesicles in the Pathogenesis of Metabolic Dysfunction-Associated Steatotic Liver Disease and Other Liver Diseases. International Journal of Molecular Sciences, 26(11), 5033. https://doi.org/10.3390/ijms26115033