
Cannabidiolic Acid Rescues Deficits in Hippocampal Long-Term Potentiation in Models of Alzheimer’s Disease: An Electrophysiological and Proteomic Analysis

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
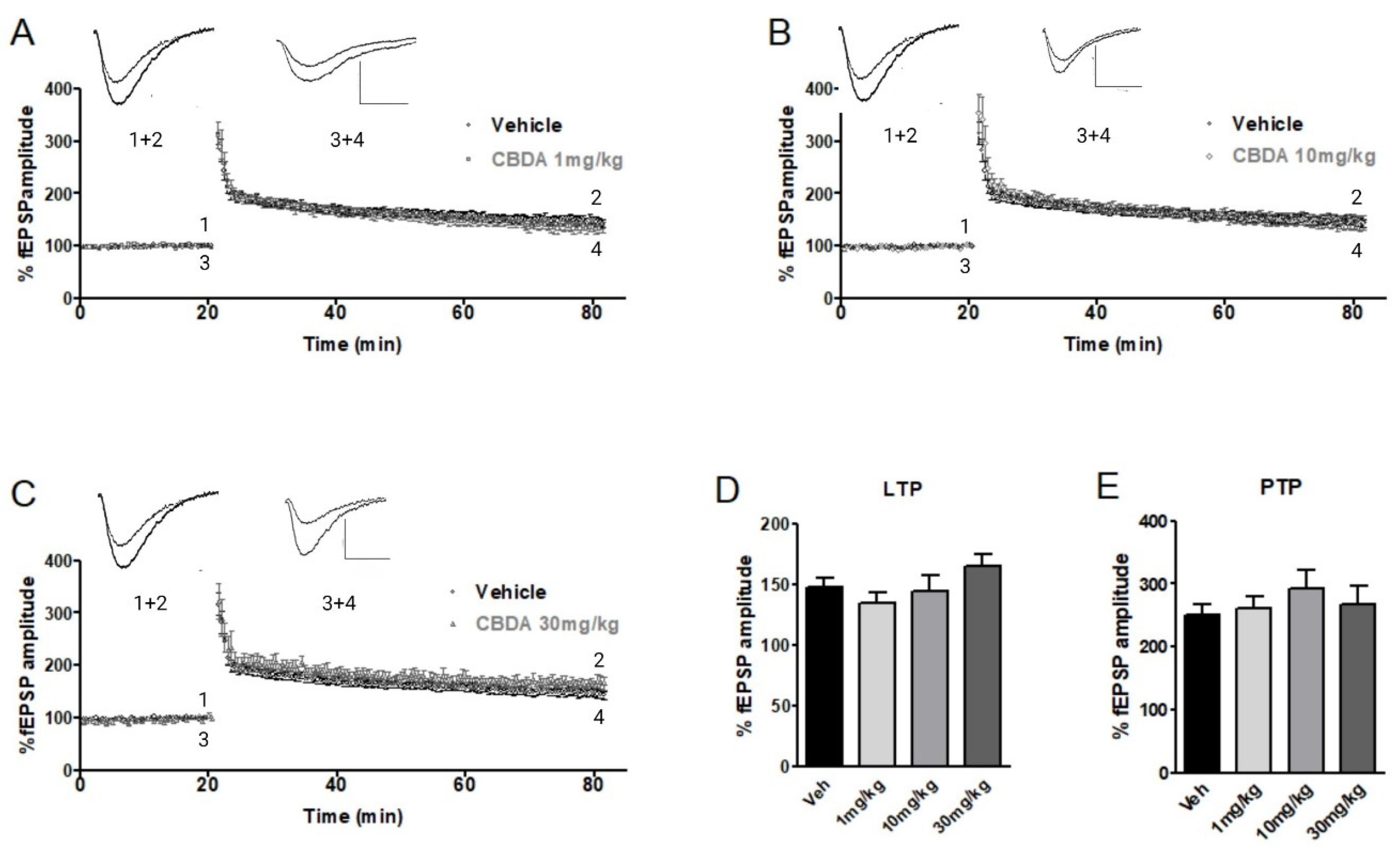
2.1. LTP in Hippocampal Slices from C57B6 Mice
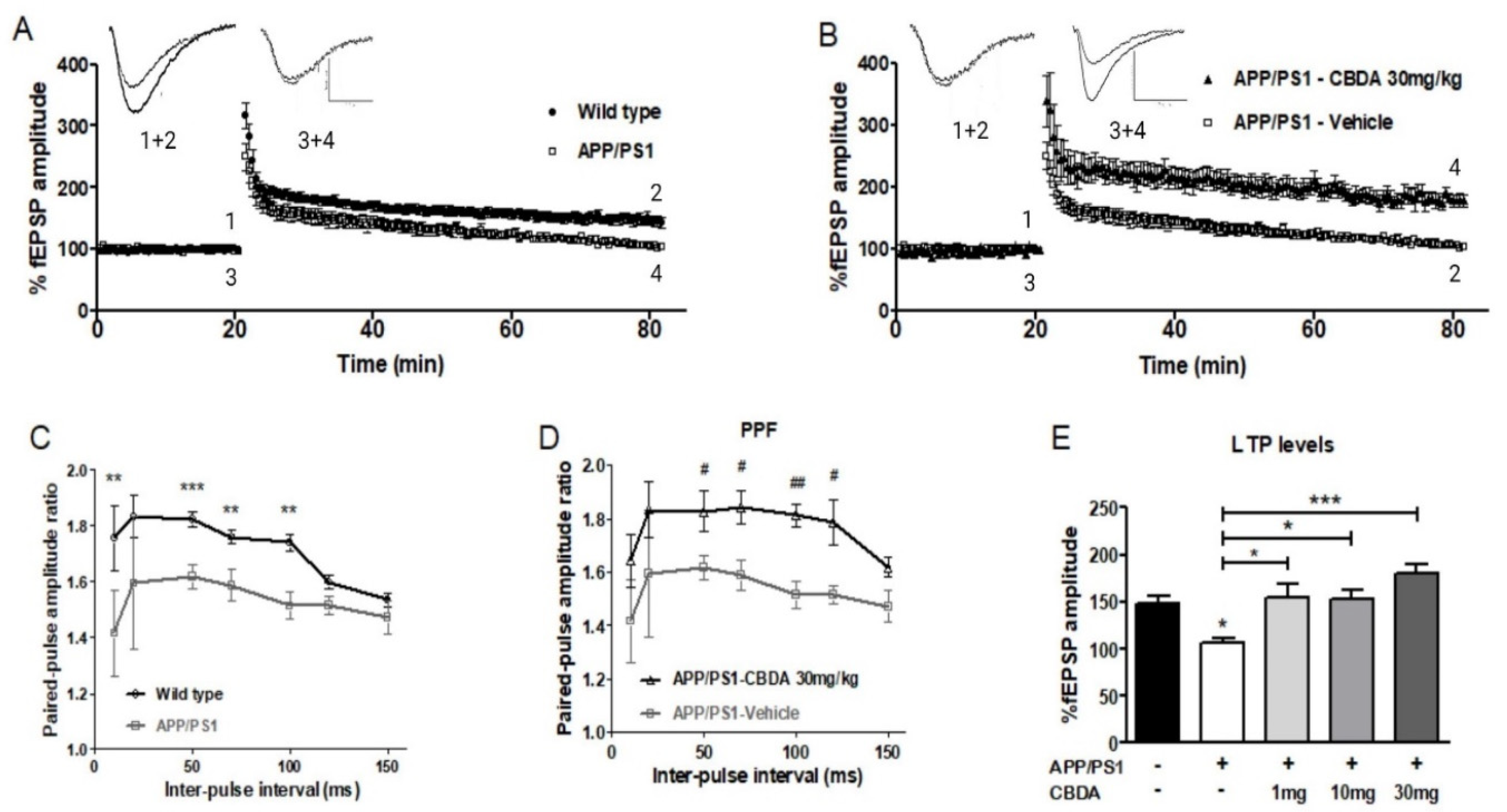
2.2. LTP in Hippocampal Slices from APP/PS1 Mice and Wild-Type Littermates Chronically Treated with CBDA or Vehicle
2.3. LTP in Vehicle-Treated and CBDA-Treated APP/PS1 Mice
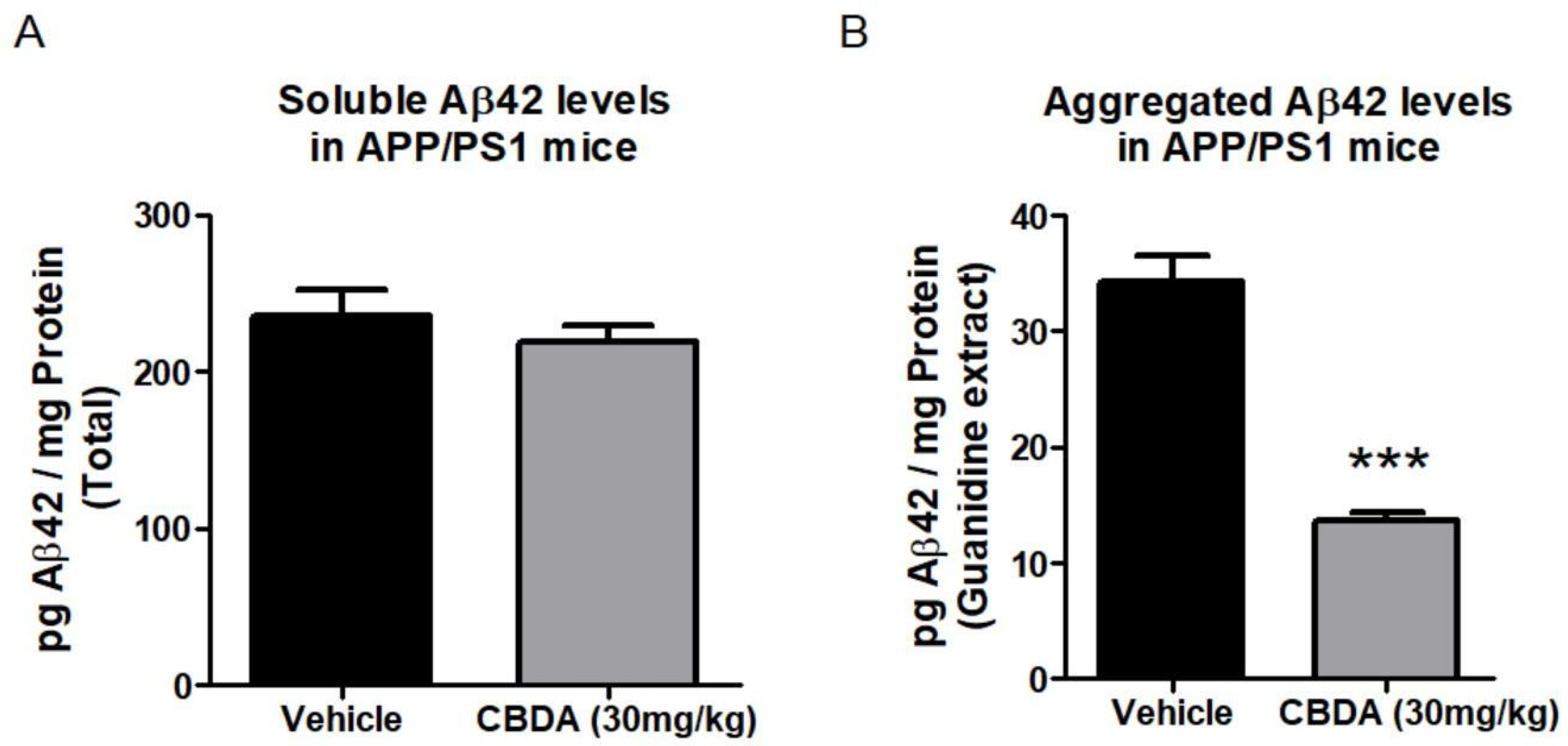
2.4. Levels of Beta Amyloid Peptide (Aβ)
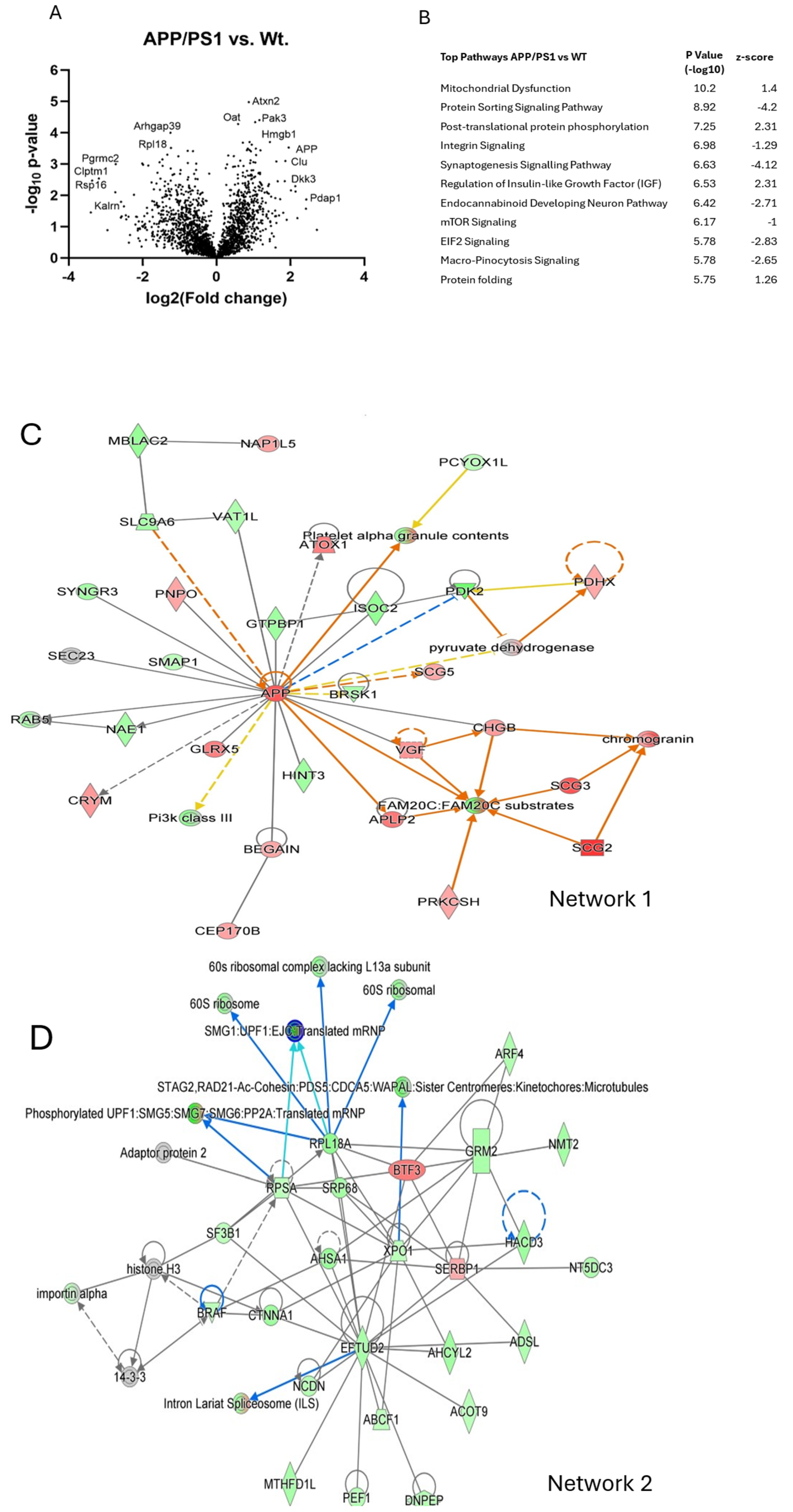
2.5. Mass Spectrometry and Proteomic Analysis
APP/PS1 Compared to Wild-Type Cortex
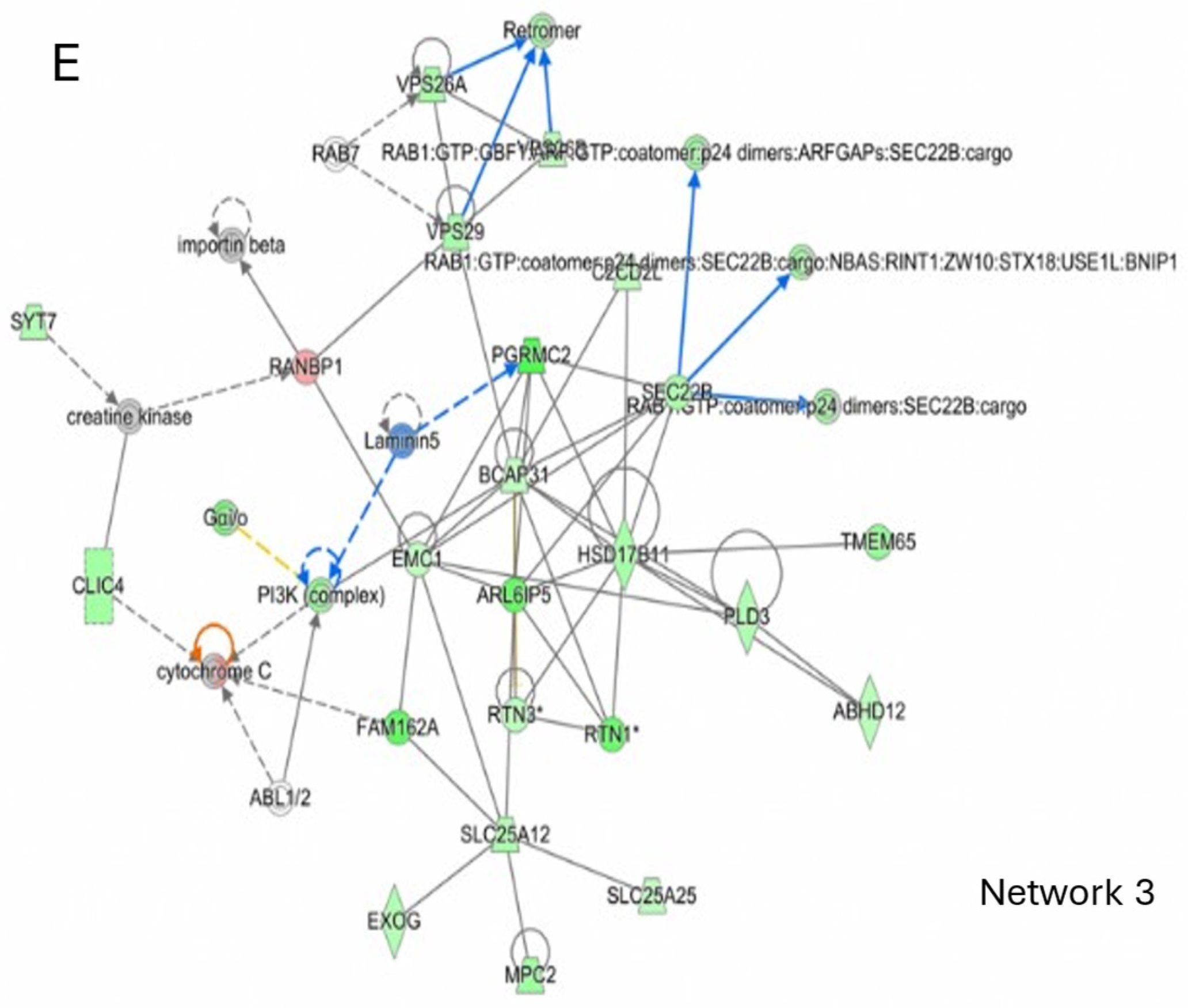
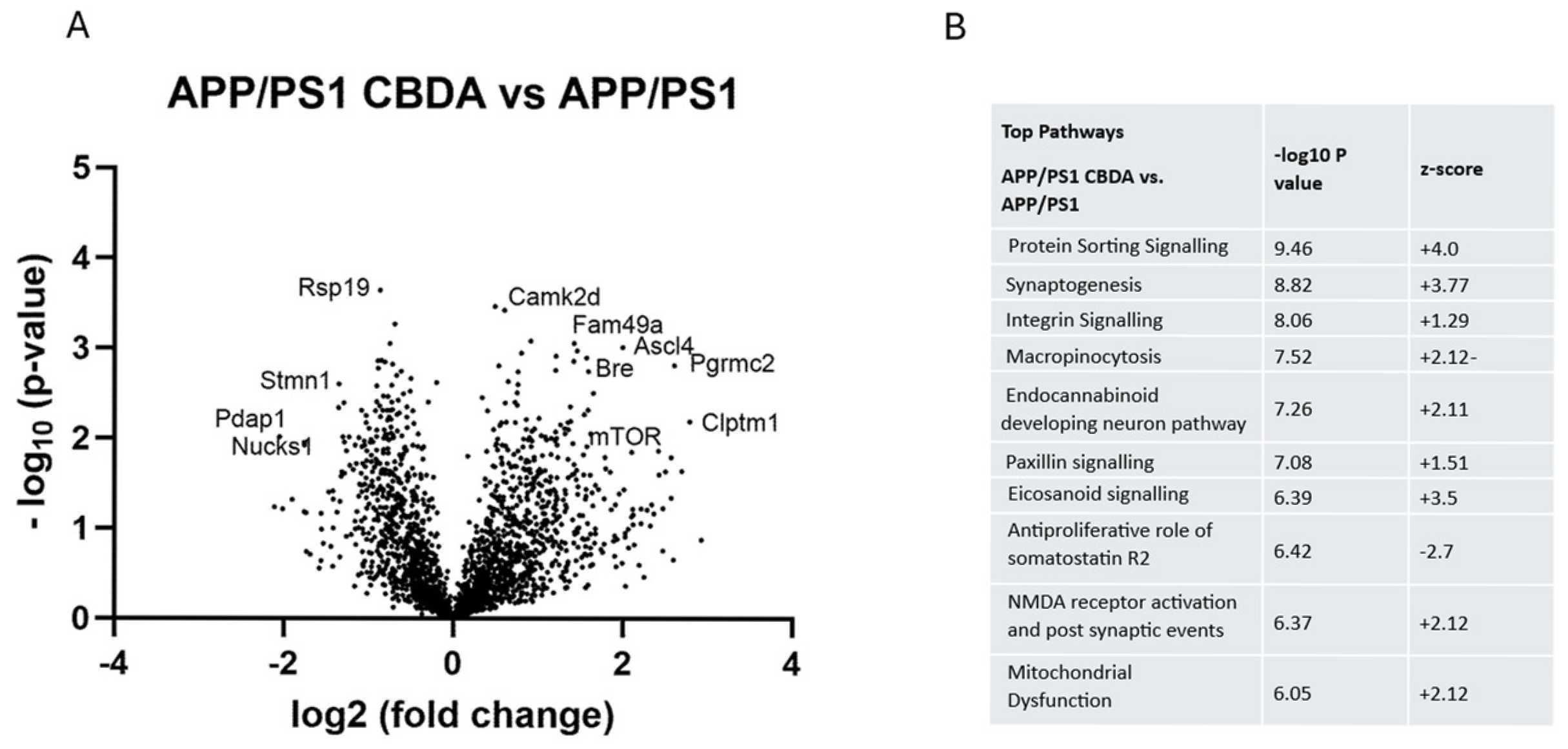
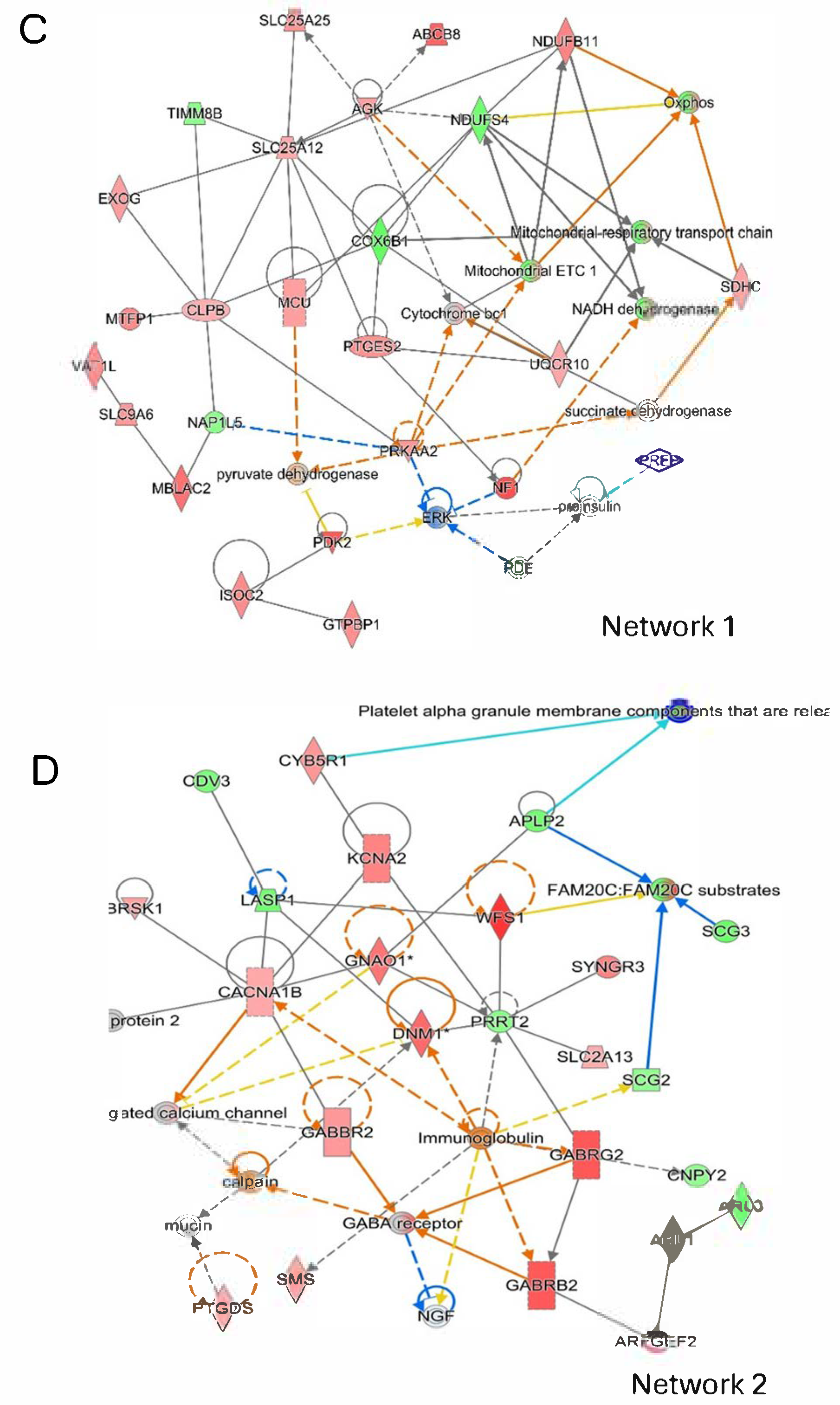
2.6. CBDA Versus Vehicle Treatment in APP/PS1 Cortex
2.7. CBDA-Treated APP/PS1 Mice Versus Vehicle-Treated Wild-Type Control Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Genotyping Procedures
4.3. Hippocampal Slice Electrophysiology
4.4. Chronic Treatment with CBDA
4.5. Protein Extraction for Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Aβ ELISA
4.7. Mass Spectrometry Tissue Preparation
4.8. Sample Preparation
4.9. Mass Spectrometry and nLC
4.10. Data Analysis
4.11. Functional Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. APPswe/PSN1dE9 (APP/PS1)
Appendix A.2. For Detection of Control DNA (324 bb)
Appendix A.3. Preparation of Amyloid-Derived Diffusible Ligands (ADDLs) Aβ1−42
References
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Mc Donald, J.M.; Savva, G.M.; Brayne, C.; Welzel, A.T.; Forster, G.; Shankar, G.M.; Selkoe, D.J.; Ince, P.G.; Walsh, D.M.; Medical Research Council Cognitive Function and Ageing Study. The presence of sodium dodecyl sulphate-stable Aβ dimers is strongly associated with Alzheimer-type dementia. Brain 2010, 133, 1328–1341. [Google Scholar] [CrossRef]
- Sebastian Monasor, L.; Muller, S.A.; Colombo, A.V.; Tanrioever, G.; Konig, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar Abeta triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. eLife 2020, 9, e54083. [Google Scholar] [CrossRef]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement. 2023, 19, 333–342. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- O’Nuallain, B.; Freir, D.B.; Nicoll, A.J.; Risse, E.; Ferguson, N.; Herron, C.E.; Collinge, J.; Walsh, D.M. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J. Neurosci. 2010, 30, 14411–14419. [Google Scholar] [CrossRef]
- Freir, D.B.; Nicoll, A.J.; Klyubin, I.; Panico, S.; Mc Donald, J.M.; Risse, E.; Asante, E.A.; Farrow, M.A.; Sessions, R.B.; Saibil, H.R.; et al. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2011, 2, 336. [Google Scholar] [CrossRef]
- Hughes, B.; Herron, C.E. Cannabidiol Reverses Deficits in Hippocampal LTP in a Model of Alzheimer’s Disease. Neurochem. Res. 2018, 9, e54083. [Google Scholar] [CrossRef] [PubMed]
- Costello, D.A.; O’Leary, D.M.; Herron, C.E. Agonists of peroxisome proliferator-activated receptor-gamma attenuate the Abeta-mediated impairment of LTP in the hippocampus in vitro. Neuropharmacology 2005, 49, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Metais, C.; Brennan, K.; Mably, A.J.; Scott, M.; Walsh, D.M.; Herron, C.E. Simvastatin treatment preserves synaptic plasticity in AbetaPPswe/PS1dE9 mice. J. Alzheimers Dis. 2014, 39, 315–329. [Google Scholar] [CrossRef]
- Palmeri, A.; Ricciarelli, R.; Gulisano, W.; Rivera, D.; Rebosio, C.; Calcagno, E.; Tropea, M.R.; Conti, S.; Das, U.; Roy, S.; et al. Amyloid-beta Peptide Is Needed for cGMP-Induced Long-Term Potentiation and Memory. J. Neurosci. 2017, 37, 6926–6937. [Google Scholar] [CrossRef]
- Kang, C. Donanemab: First Approval. Drugs 2024, 84, 1313–1318. [Google Scholar] [CrossRef]
- Hampson, A.J.; Grimaldi, M.; Lolic, M.; Wink, D.; Rosenthal, R.; Axelrod, J. Neuroprotective antioxidants from marijuana. Ann. N. Y Acad. Sci. 2000, 899, 274–282. [Google Scholar] [CrossRef]
- Hamelink, C.; Hampson, A.; Wink, D.A.; Eiden, L.E.; Eskay, R.L. Comparison of cannabidiol, antioxidants, and diuretics in reversing binge ethanol-induced neurotoxicity. J. Pharmacol. Exp. Ther. 2005, 314, 780–788. [Google Scholar] [CrossRef]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5YAPP+ cells through PPARγ involvement. Phytother. Res. 2014, 28, 1007–1013. [Google Scholar] [CrossRef]
- Rock, E.M.; Limebeer, C.L.; Parker, L.A. Effect of cannabidiolic acid and ∆9-tetrahydrocannabinol on carrageenan-induced hyperalgesia and edema in a rodent model of inflammatory pain. Psychopharmacology 2018, 235, 3259–3271. [Google Scholar] [CrossRef]
- Vigli, D.; Cosentino, L.; Pellas, M.; De Filippis, B. Chronic Treatment with Cannabidiolic Acid (CBDA) Reduces Thermal Pain Sensitivity in Male Mice and Rescues the Hyperalgesia in a Mouse Model of Rett Syndrome. Neuroscience 2021, 453, 113–123. [Google Scholar] [CrossRef]
- Goerl, B.; Watkins, S.; Metcalf, C.; Smith, M.; Beenhakker, M. Cannabidiolic acid exhibits entourage-like improvements of anticonvulsant activity in an acute rat model of seizures. Epilepsy Res. 2021, 169, 106525. [Google Scholar] [CrossRef] [PubMed]
- Ruhaak, L.R.; Felth, J.; Karlsson, P.C.; Rafter, J.J.; Verpoorte, R.; Bohlin, L. Evaluation of the cyclooxygenase inhibiting effects of six major cannabinoids isolated from Cannabis sativa. Biol. Pharm. Bull. 2011, 34, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Ben-Cnaan, E.; Permyakova, A.; Azar, S.; Hirsch, S.; Baraghithy, S.; Hinden, L.; Tam, J. The Metabolic Efficacy of a Cannabidiolic Acid (CBDA) Derivative in Treating Diet- and Genetic-Induced Obesity. Int. J. Mol. Sci. 2022, 23, 5610. [Google Scholar] [CrossRef]
- Dawidowicz, A.L.; Olszowy-Tomczyk, M.; Typek, R. CBG, CBD, Delta9-THC, CBN, CBGA, CBDA and Delta9-THCA as antioxidant agents and their intervention abilities in antioxidant action. Fitoterapia 2021, 152, 104915. [Google Scholar] [CrossRef]
- Hirao-Suzuki, M.; Takeda, S.; Koga, T.; Takiguchi, M.; Toda, A. Cannabidiolic acid dampens the expression of cyclooxygenase-2 in MDA-MB-231 breast cancer cells: Possible implication of the peroxisome proliferator-activated receptor β/δ abrogation. J. Toxicol. Sci. 2020, 45, 227–236. [Google Scholar] [CrossRef]
- Kim, J.; Choi, P.; Park, Y.T.; Kim, T.; Ham, J.; Kim, J.C. The Cannabinoids, CBDA and THCA, Rescue Memory Deficits and Reduce Amyloid-Beta and Tau Pathology in an Alzheimer’s Disease-like Mouse Model. Int. J. Mol. Sci. 2023, 24, 6827. [Google Scholar] [CrossRef]
- Pellati, F.; Borgonetti, V.; Brighenti, V.; Biagi, M.; Benvenuti, S.; Corsi, L. Cannabis sativa L and Nonpsychoactive Cannabinoids: Their Chemistry and Role against Oxidative Stress, Inflammation, and Cancer. BioMed Res. Int. 2018, 2018, 1691428. [Google Scholar] [CrossRef]
- Pellati, F.; Brighenti, V.; Sperlea, J.; Marchetti, L.; Bertelli, D.; Benvenuti, S. New methods for the comprehensive analysis of bioactive compounds in Cannabis sativa L. (hemp). Molecules 2018, 23, 2639. [Google Scholar] [CrossRef]
- Formato, M.; Crescente, G.; Scognamiglio, M.; Fiorentino, A.; Pecoraro, M.T.; Piccolella, S.; Catauro, M.; Pacifico, S. (−)-Cannabidiolic Acid, a Still Overlooked Bioactive Compound: An Introductory Review and Preliminary Research. Molecules 2020, 25, 2638. [Google Scholar] [CrossRef]
- Savonenko, A.; Xu, G.M.; Melnikova, T.; Morton, J.L.; Gonzales, V.; Wong, M.P.; Price, D.L.; Tang, F.; Markowska, A.L.; Borchelt, D.R. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: Relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 2005, 18, 602–617. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Priller, C.; Bauer, T.; Mitteregger, G.; Krebs, B.; Kretzschmar, H.A.; Herms, J. Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 2006, 26, 7212–7221. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Berman, P.; Futoran, K.; Lewitus, G.M.; Mukha, D.; Benami, M.; Shlomi, T.; Meiri, D. A new ESI-LC/MS approach for comprehensive metabolic profiling of phytocannabinoids in Cannabis. Sci. Rep. 2018, 8, 14280. [Google Scholar] [CrossRef]
- Anderson, L.L.; Low, I.K.; Banister, S.D.; McGregor, I.S.; Arnold, J.C. Pharmacokinetics of Phytocannabinoid Acids and Anticonvulsant Effect of Cannabidiolic Acid in a Mouse Model of Dravet Syndrome. J. Nat. Prod. 2019, 82, 3047–3055. [Google Scholar] [CrossRef]
- Bolognini, D.; Rock, E.M.; Cluny, N.L.; Cascio, M.G.; Limebeer, C.L.; Duncan, M.; Stott, C.G.; Javid, F.A.; Parker, L.A.; Pertwee, R.G. Cannabidiolic acid prevents vomiting in Suncus murinus and nausea-induced behaviour in rats by enhancing 5-HT1A receptor activation. Br. J. Pharmacol. 2013, 168, 1456–1470. [Google Scholar] [CrossRef]
- Cheng, D.; Low, J.K.; Logge, W.; Garner, B.; Karl, T. Chronic cannabidiol treatment improves social and object recognition in double transgenic APPswe/PS1E9 mice. Psychopharmacology 2014, 231, 3009–3017. [Google Scholar] [CrossRef]
- Cheng, D.; Spiro, A.S.; Jenner, A.M.; Garner, B.; Karl, T. Long-term cannabidiol treatment prevents the development of social recognition memory deficits in Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 2014, 42, 1383–1396. [Google Scholar] [CrossRef]
- Johns, T.N.; Wakshlag, J.J.; Lyubimov, A.V.; Zakharov, A.; Burnside, W.M. Pharmacokinetics of cannabidiol-/cannabidiolic acid-rich hemp oil in juvenile cynomolgus macaques (Macaca fascicularis). Front. Vet. Sci. 2023, 10, 1286158. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular Prion Protein Mediates Impairment of Synaptic Plasticity by Amyloid-β Oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- D’Aniello, E.; Fellous, T.; Iannotti, F.A.; Gentile, A.; Allara, M.; Balestrieri, F.; Gray, R.; Amodeo, P.; Vitale, R.M.; Di Marzo, V. Identification and characterization of phytocannabinoids as novel dual PPARalpha/gamma agonists by a computational and in vitro experimental approach. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 586–597. [Google Scholar] [CrossRef]
- Heneka, M.T.; Landreth, G.E. PPARs in the brain. Biochim. Et Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 1031–1045. [Google Scholar] [CrossRef]
- Takeda, S.; Okazaki, H.; Ikeda, E.; Abe, S.; Yoshioka, Y.; Watanabe, K.; Aramaki, H. Down-regulation of cyclooxygenase-2 (cox-2) by cannabidiolic acid in human breast cancer cells. J. Toxicol. Sci. 2014, 39, 711–716. [Google Scholar] [CrossRef]
- Gelman, S.; Palma, J.; Tombaugh, G.; Ghavami, A. Differences in Synaptic Dysfunction Between rTg4510 and APP/PS1 Mouse Models of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 195–208. [Google Scholar] [CrossRef]
- Pereda, D.; Al-Osta, I.; Okorocha, A.E.; Easton, A.; Hartell, N.A. Changes in presynaptic calcium signalling accompany age-related deficits in hippocampal LTP and cognitive impairment. Aging Cell 2019, 18, e13008. [Google Scholar] [CrossRef]
- Mirlohi, S.; Bladen, C.; Santiago, M.J.; Arnold, J.C.; McGregor, I.; Connor, M. Inhibition of human recombinant T-type calcium channels by phytocannabinoids in vitro. Br. J. Pharmacol. 2022, 179, 4031–4043. [Google Scholar] [CrossRef]
- Thibault, O.; Pancani, T.; Landfield, P.W.; Norris, C.M. Reduction in neuronal L-type calcium channel activity in a double knock-in mouse model of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 546–549. [Google Scholar] [CrossRef]
- Straiker, A.; Wilson, S.; Corey, W.; Dvorakova, M.; Bosquez, T.; Tracey, J.; Wilkowski, C.; Ho, K.; Wager-Miller, J.; Mackie, K. An Evaluation of Understudied Phytocannabinoids and Their Effects in Two Neuronal Models. Molecules 2021, 26, 5352. [Google Scholar] [CrossRef]
- Straiker, A.; Dvorakova, M.; Zimmowitch, A.; MacKie, K. Cannabidiol inhibits endocannabinoid signaling in autaptic hippocampal neurons. Mol. Pharmacol. 2018, 94, 743–748. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef]
- Navarro, G.; Varani, K.; Lillo, A.; Vincenzi, F.; Rivas-Santisteban, R.; Raïch, I.; Reyes-Resina, I.; Ferreiro-Vera, C.; Borea, P.A.; Sánchez de Medina, V.; et al. Pharmacological data of cannabidiol- and cannabigerol-type phytocannabinoids acting on cannabinoid CB1, CB2 and CB1/CB2 heteromer receptors. Pharmacol. Res. 2020, 159, 104940. [Google Scholar] [CrossRef]
- Filer, C.N. Acidic Cannabinoid Decarboxylation. Cannabis Cannabinoid Res. 2022, 7, 262–273. [Google Scholar] [CrossRef]
- de Petrocellis, L.; Orlando, P.; Moriello, A.S.; Aviello, G.; Stott, C.; Izzo, A.A.; di Marzo, V. Cannabinoid actions at TRPV channels: Effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol. 2012, 204, 255–266. [Google Scholar] [CrossRef]
- Kedziora, M.; Boccella, S.; Marabese, I.; Mlost, J.; Infantino, R.; Maione, S.; Starowicz, K. Inhibition of anandamide breakdown reduces pain and restores LTP and monoamine levels in the rat hippocampus via the CB(1) receptor following osteoarthritis. Neuropharmacology 2023, 222, 109304. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Nagre, N.N.; Xie, S.; Subbanna, S. Elevation of endogenous anandamide impairs LTP, learning, and memory through CB1 receptor signaling in mice. Hippocampus 2014, 24, 808–818. [Google Scholar] [CrossRef]
- Izco, M.; Martinez, P.; Corrales, A.; Fandos, N.; Garcia, S.; Insua, D.; Montanes, M.; Perez-Grijalba, V.; Rueda, N.; Vidal, V.; et al. Changes in the brain and plasma Abeta peptide levels with age and its relationship with cognitive impairment in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neuroscience 2014, 263, 269–279. [Google Scholar] [CrossRef]
- Timmer, N.M.; van Dijk, L.; van der Zee, C.E.; Kiliaan, A.; de Waal, R.M.; Verbeek, M.M. Enoxaparin treatment administered at both early and late stages of amyloid beta deposition improves cognition of APPswe/PS1dE9 mice with differential effects on brain Abeta levels. Neurobiol. Dis. 2010, 40, 340–347. [Google Scholar] [CrossRef]
- Cao, C.; Li, Y.; Liu, H.; Bai, G.; Mayl, J.; Lin, X.; Sutherland, K.; Nabar, N.; Cai, J. The potential therapeutic effects of THC on Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Du, Y.; Zhao, X.; Tang, Q.; Su, W.; Hu, Y.; Yu, P. Cannabidiol Enhances Microglial Beta-Amyloid Peptide Phagocytosis and Clearance via Vanilloid Family Type 2 Channel Activation. Int. J. Mol. Sci. 2022, 23, 5367. [Google Scholar] [CrossRef] [PubMed]
- Todorova, V.; Blokland, A. Mitochondria and Synaptic Plasticity in the Mature and Aging Nervous System. Curr. Neuropharmacol. 2017, 15, 166–173. [Google Scholar] [CrossRef]
- Entezari, M.; Hashemi, D.; Taheriazam, A.; Zabolian, A.; Mohammadi, S.; Fakhri, F.; Hashemi, M.; Hushmandi, K.; Ashrafizadeh, M.; Zarrabi, A.; et al. AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: A pre-clinical and clinical investigation. Biomed. Pharmacother. 2022, 146, 112563. [Google Scholar] [CrossRef]
- He, K.; Nie, L.; Zhou, Q.; Rahman, S.U.; Liu, J.; Yang, X.; Li, S. Proteomic Profiles of the Early Mitochondrial Changes in APP/PS1 and ApoE4 Transgenic Mice Models of Alzheimer’s Disease. J. Proteome Res. 2019, 18, 2632–2642. [Google Scholar] [CrossRef]
- Elder, M.K.; Erdjument-Bromage, H.; Oliveira, M.M.; Mamcarz, M.; Neubert, T.A.; Klann, E. Age-dependent shift in the de novo proteome accompanies pathogenesis in an Alzheimer’s disease mouse model. Commun. Biol. 2021, 4, 823. [Google Scholar] [CrossRef]
- Xiyang, Y.B.; Liu, R.; Wang, X.Y.; Li, S.; Zhao, Y.; Lu, B.T.; Xiao, Z.C.; Zhang, L.F.; Wang, T.H.; Zhang, J. COX5A Plays a Vital Role in Memory Impairment Associated With Brain Aging via the BDNF/ERK1/2 Signaling Pathway. Front. Aging Neurosci. 2020, 12, 215. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Murphy, A.N. Do Two Mitochondrial Wrongs Help Make Cells Right? Trends Mol. Med. 2020, 26, 3–6. [Google Scholar] [CrossRef]
- Qi, X.; Disatnik, M.H.; Shen, N.; Sobel, R.A.; Mochly-Rosen, D. Aberrant mitochondrial fission in neurons induced by protein kinase Cdelta under oxidative stress conditions in vivo. Mol. Biol. Cell 2011, 22, 256–265. [Google Scholar] [CrossRef]
- Usmani, M.A.; Ahmed, Z.M.; Magini, P.; Pienkowski, V.M.; Rasmussen, K.J.; Hernan, R.; Rasheed, F.; Hussain, M.; Shahzad, M.; Lanpher, B.C.; et al. De novo and bi-allelic variants in AP1G1 cause neurodevelopmental disorder with developmental delay, intellectual disability, and epilepsy. Am. J. Hum. Genet. 2021, 108, 1330–1341. [Google Scholar] [CrossRef]
- Guerrini, R.; Parrini, E. Neuronal migration disorders. Neurobiol. Dis. 2010, 38, 154–166. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, X.; Ju, W.; Sun, L.; Zhang, H. Genetic and Expression Analysis of COPI Genes and Alzheimer’s Disease Susceptibility. Front. Genet. 2019, 10, 866. [Google Scholar] [CrossRef]
- Holm, J.E.J.; Soares, S.G.; Symmons, M.F.; Huddin, A.S.; Moncrieffe, M.C.; Gay, N.J. Anterograde trafficking of Toll-like receptors requires the cargo sorting adaptors TMED-2 and 7. Traffic 2023, 24, 508–521. [Google Scholar] [CrossRef]
- Altas, B.; Romanowski, A.J.; Bunce, G.W.; Poulopoulos, A. Neuronal mTOR Outposts: Implications for Translation, Signaling, and Plasticity. Front. Cell Neurosci. 2022, 16, 853634. [Google Scholar] [CrossRef]
- Vevea, J.D.; Kusick, G.F.; Courtney, K.C.; Chen, E.; Watanabe, S.; Chapman, E.R. Synaptotagmin 7 is targeted to the axonal plasma membrane through gamma-secretase processing to promote synaptic vesicle docking in mouse hippocampal neurons. eLife 2021, 10, e67261. [Google Scholar] [CrossRef]
- Nikonenko, I.; Toni, N.; Moosmayer, M.; Shigeri, Y.; Muller, D.; Sargent Jones, L. Integrins are involved in synaptogenesis, cell spreading, and adhesion in the postnatal brain. Brain Res. Dev. Brain Res. 2003, 140, 185–194. [Google Scholar] [CrossRef]
- Parnell, E.; Shapiro, L.P.; Voorn, R.A.; Forrest, M.P.; Jalloul, H.A.; Loizzo, D.D.; Penzes, P. KALRN: A central regulator of synaptic function and synaptopathies. Gene 2021, 768, 145306. [Google Scholar] [CrossRef]
- Youn, H.; Jeoung, M.; Koo, Y.; Ji, H.; Markesbery, W.R.; Ji, I.; Ji, T.H. Kalirin is under-expressed in Alzheimer’s disease hippocampus. J. Alzheimers Dis. 2007, 11, 385–397. [Google Scholar] [CrossRef]
- Pang, S.; Luo, Z.; Dong, W.; Gao, S.; Chen, W.; Liu, N.; Zhang, X.; Gao, X.; Li, J.; Gao, K.; et al. Integrin beta1/FAK/SRC signal pathway is involved in autism spectrum disorder in Tspan7 knockout rats. Life Sci. Alliance 2023, 6, e202201616. [Google Scholar] [CrossRef]
- Gupton, S.L.; Gertler, F.B. Integrin signaling switches the cytoskeletal and exocytic machinery that drives neuritogenesis. Dev. Cell 2010, 18, 725–736. [Google Scholar] [CrossRef]
- Jain, S.; Yoon, S.Y.; Zhu, L.; Brodbeck, J.; Dai, J.; Walker, D.; Huang, Y. Arf4 determines dentate gyrus-mediated pattern separation by regulating dendritic spine development. PLoS ONE 2012, 7, e46340. [Google Scholar] [CrossRef] [PubMed]
- Bassani, S.; Cingolani, L.A.; Valnegri, P.; Folci, A.; Zapata, J.; Gianfelice, A.; Sala, C.; Goda, Y.; Passafaro, M. The X-linked intellectual disability protein TSPAN7 regulates excitatory synapse development and AMPAR trafficking. Neuron 2012, 73, 1143–1158. [Google Scholar] [CrossRef]
- Seipold, L.; Saftig, P. The Emerging Role of Tetraspanins in the Proteolytic Processing of the Amyloid Precursor Protein. Front. Mol. Neurosci. 2016, 9, 149. [Google Scholar] [CrossRef]
- Gulati, P.; Gaspers, L.D.; Dann, S.G.; Joaquin, M.; Nobukuni, T.; Natt, F.; Kozma, S.C.; Thomas, A.P.; Thomas, G. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008, 7, 456–465. [Google Scholar] [CrossRef]
- Wang, L.; Budolfson, K.; Wang, F. Pik3c3 deletion in pyramidal neurons results in loss of synapses, extensive gliosis and progressive neurodegeneration. Neuroscience 2011, 172, 427–442. [Google Scholar] [CrossRef]
- McPhie, D.L.; Coopersmith, R.; Hines-Peralta, A.; Chen, Y.; Ivins, K.J.; Manly, S.P.; Kozlowski, M.R.; Neve, K.A.; Neve, R.L. DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J. Neurosci. 2003, 23, 6914–6927. [Google Scholar] [CrossRef]
- Park, Y.K.; Goda, Y. Integrins in synapse regulation. Nat. Rev. Neurosci. 2016, 17, 745–756. [Google Scholar] [CrossRef]
- Beauchamp, M.C.; Jerome-Majewska, L.A. A protective role for EFTUD2 in the brain. Neuron 2024, 112, 3378–3380. [Google Scholar] [CrossRef]
- Sun, W.; Tian, B.X.; Wang, S.H.; Liu, P.J.; Wang, Y.C. The function of SEC22B and its role in human diseases. Cytoskeleton 2020, 77, 303–312. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. Implication of HMGB1 signaling pathways in Amyotrophic lateral sclerosis (ALS): From molecular mechanisms to pre-clinical results. Pharmacol. Res. 2020, 156, 104792. [Google Scholar] [CrossRef]
- Barbash, S.; Garfinkel, B.P.; Maoz, R.; Simchovitz, A.; Nadorp, B.; Guffanti, A.; Bennett, E.R.; Nadeau, C.; Turk, A.; Paul, L.; et al. Alzheimer’s brains show inter-related changes in RNA and lipid metabolism. Neurobiol. Dis. 2017, 106, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for Mitochondrial UPR Gene Activation in Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Vasak, M.; Knipp, M.; Castellani, R.J.; Perry, G. Dimethylargininase, a nitric oxide regulatory protein, in Alzheimer disease. Free Radic. Biol. Med. 1998, 25, 898–902. [Google Scholar] [CrossRef]
- Ruiz-Perez, G.; Ruiz de Martin Esteban, S.; Marques, S.; Aparicio, N.; Grande, M.T.; Benito-Cuesta, I.; Martinez-Relimpio, A.M.; Arnanz, M.A.; Tolon, R.M.; Posada-Ayala, M.; et al. Potentiation of amyloid beta phagocytosis and amelioration of synaptic dysfunction upon FAAH deletion in a mouse model of Alzheimer’s disease. J. Neuroinflammation 2021, 18, 223. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, W.; Cao, Y.; Tan, W. Hippocampus Proteomics and Brain Lipidomics Reveal Network Dysfunction and Lipid Molecular Abnormalities in APP/PS1 Mouse Model of Alzheimer’s Disease. J. Proteome Res. 2020, 19, 3427–3437. [Google Scholar] [CrossRef]
- Zhang, W.; Bai, M.; Xi, Y.; Hao, J.; Liu, L.; Mao, N.; Su, C.; Miao, J.; Li, Z. Early memory deficits precede plaque deposition in APPswe/PS1dE9 mice: Involvement of oxidative stress and cholinergic dysfunction. Free Radic. Biol. Med. 2012, 52, 1443–1452. [Google Scholar] [CrossRef]
- Xu, L.L.; Shen, Y.; Wang, X.; Wei, L.F.; Wang, P.; Yang, H.; Wang, C.F.; Xie, Z.H.; Bi, J.Z. Mitochondrial dynamics changes with age in an APPsw/PS1dE9 mouse model of Alzheimer’s disease. Neuroreport 2017, 28, 222–228. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Ramagli, L.S.; Rodriguez, L.V. Quantitation of Microgram Amounts of Protein in Two-Dimensional Polyacrylamide-Gel Electrophoresis Sample Buffer. Electrophoresis 1985, 6, 559–563. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil, B.; Sullivan, M.; Scaife, C.; Glennon, J.C.; Herron, C. Cannabidiolic Acid Rescues Deficits in Hippocampal Long-Term Potentiation in Models of Alzheimer’s Disease: An Electrophysiological and Proteomic Analysis. Int. J. Mol. Sci. 2025, 26, 4944. https://doi.org/10.3390/ijms26104944
Gil B, Sullivan M, Scaife C, Glennon JC, Herron C. Cannabidiolic Acid Rescues Deficits in Hippocampal Long-Term Potentiation in Models of Alzheimer’s Disease: An Electrophysiological and Proteomic Analysis. International Journal of Molecular Sciences. 2025; 26(10):4944. https://doi.org/10.3390/ijms26104944
Chicago/Turabian StyleGil, Beatriz, Mairéad Sullivan, Caitriona Scaife, Jeffrey C. Glennon, and Caroline Herron. 2025. "Cannabidiolic Acid Rescues Deficits in Hippocampal Long-Term Potentiation in Models of Alzheimer’s Disease: An Electrophysiological and Proteomic Analysis" International Journal of Molecular Sciences 26, no. 10: 4944. https://doi.org/10.3390/ijms26104944
APA StyleGil, B., Sullivan, M., Scaife, C., Glennon, J. C., & Herron, C. (2025). Cannabidiolic Acid Rescues Deficits in Hippocampal Long-Term Potentiation in Models of Alzheimer’s Disease: An Electrophysiological and Proteomic Analysis. International Journal of Molecular Sciences, 26(10), 4944. https://doi.org/10.3390/ijms26104944