3. Materials and Methods
3.1. General Methods
Commercially available solvents were used as obtained from suppliers (Molar Chemicals Ltd., Halásztelek, Hungary; Merck Ltd., Budapest, Hungary and VWR International Ltd., Debrecen, Hungary), while applied solvents were dried according to standard procedures. Optical rotations were measured in MeOH at 20 °C with a Perkin-Elmer 341 polarimeter (PerkinElmer Inc., Shelton, CT, USA). Chromatographic separations and monitoring of reactions were carried out on a Merck Kieselgel 60 (Merck Ltd., Budapest, Hungary). HRMS flow injection analysis was performed with a Thermo Scientific Orbitrap Exploris 240 hybrid quadrupole-Orbitrap (Thermo Fischer Scientific, Waltham, MA, USA) mass spectrometer coupled to a Waters Acquity I-Class UPLC TM (Waters, Manchester, UK). GC measurements for direct separation of commercially available enantiomers of isopulegol, to determine the enantiomeric purity of the starting material (−)-
21 and its enantiomer (+)-
21, were performed on a Chirasil-DEX CB column (2500 × 0.25 mm I.D.) on a Perkin-Elmer Autosystem XL GC consisting of a Flame Ionization Detector (Perkin-Elmer Corp., Norwalk, CT, USA) and a Turbochrom Workstation data system (Perkin-Elmer Corp., Norwalk, CT, USA). Melting points were determined on a Kofler apparatus (Nagema, Dresden, Germany) and they are uncorrected.
1H NMR,
13C NMR, and
19F NMR were recorded on a Brucker Avance DRX 500 spectrometer [500 MHz (
1H), 471 MHz (
19F), and 125 MHz (
13C)]. The
1H and
13C chemical shifts are given relative to TMS and
19F to CFCl
3 (0.00 ppm). J values are given in Hz. Images of NMR spectra are found in
Figures S1–S86 in the Supplementary Materials.
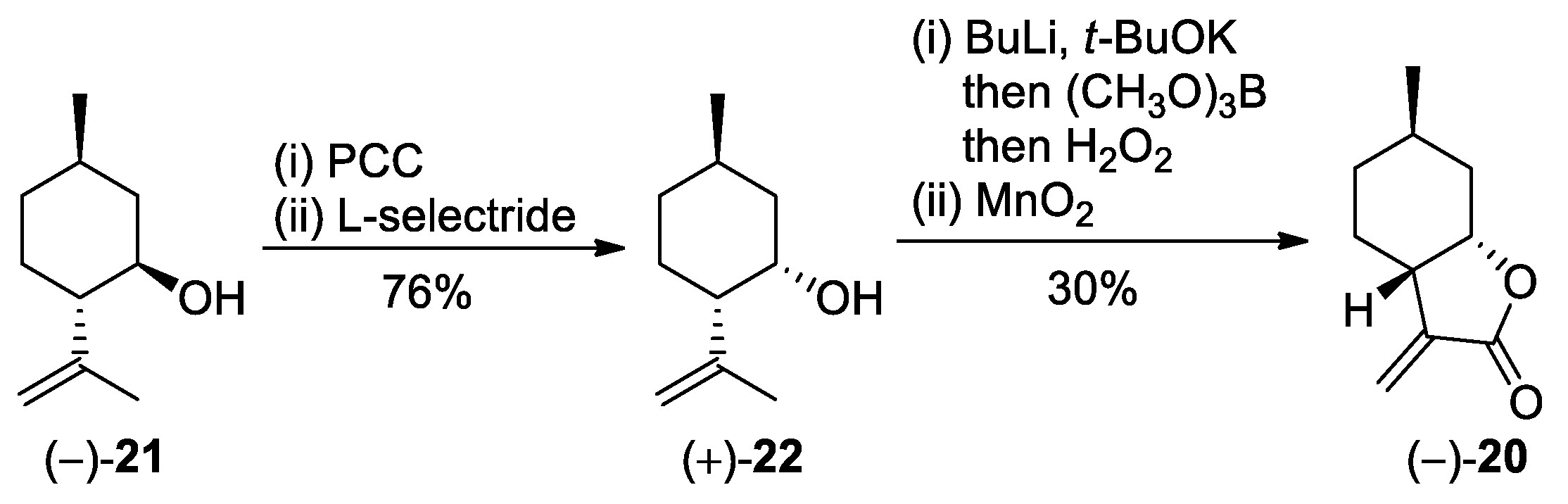
(−)-Isopulegol (−)-
21 is available commercially from Merck Co with
ee% = 95%. (+)-Neoisopulegol (+)-
22 [
36,
38], (−)-
α-methylene-
γ-butyrolactone (–)-
20 [
36,
37], and cysteine derivatives (WO 2015148880 A1) were prepared according to modified literature procedures.
3.2. Preparation of (+)-Neoisopulegol (+)-22
Oxidation of (−)-isopulegol was based on Ref. [
38]. To a slurry of PCC (27.6 g, 2 equiv.) and silica gel (55 g) in DCM (200 mL), (−)-isopulegol
9 (10 g, 64 mmol) was added. The reaction mixture was stirred at room temperature for 48 h. Upon completion of the reaction (as monitored by TLC), the mixture was filtered and the filtrate was evaporated to dryness. The crude product was purified by column chromatography on silica gel using
n-hexane/EtOAc 9:1 as the eluent. This provided (–)-isopulegone as a pale yellow oil (yield: 80%).
Reduction of (–)-isopulegone was based on Ref. [
36]. (–)-Isopulegone (2 g, 12.5 mmol) was dissolved in dry THF (40 mL). L-Selectride (15 mL, 1.2 equiv.) was dissolved in dry THF (20 mL), and added dropwise to the previous solution under an argon atmosphere at −78 °C. After 1–2 h, the reaction mixture was allowed to warm up to RT, and then it was treated with H
2O
2 in 5% NaOH (40 mL) and extracted with Et
2O (3 × 100 mL). The organic layer was dried over Na
2SO
4 and evaporated to dryness. The crude product was purified by column chromatography on silica gel using
n-hexane/EtOAc 9:1 as the eluent. This provided (+)-neoisopulegol (+)-
22 as a pale yellow oil (yield: 95%).
3.3. Preparation of (–)-α-Methylene-γ-Butyrolactone (−)-20
Hydroxylation of (+)-neoisopulegol was based on Ref. [
37].
t-BuOK (2.2 g, 1.5 equiv.) was dissolved in dry
n-hexane (15 mL). Then,
n-BuLi (2 g, 2 equiv.) was added dropwise under an argon atmosphere at 0 °C. The mixture was stirred for one hour, during which the color changed from white to light orange. After the dropwise addition of (+)-neoisopulegol (+)-
22 (2 g, 13 mmol), the reaction mixture was stirred under an argon atmosphere at RT overnight. B(OMe)
3 (4.5 mL, 3 equiv.) was added dropwise at −78 °C and stirred with the mixture for one hour. Upon reaching −20 °C, 30% aqueous H
2O
2 (7 mL) was introduced into the mixture and stirred for one hour. Water was then added to quench the reaction, and the mixture was extracted with Et
2O (3 × 100 mL). The organic layer was dried over Na
2SO
4, filtered, and evaporated to dryness. The crude product was crystallized from
n-hexane to give (1
S,2
S,5
R)-2-(3-hydroxyprop-1-en-2-yl)-5-methylcyclohexanol as white crystals (yield: 50%).
Oxidation of (1
S,2
S,5
R)-2-(3-hydroxyprop-1-en-2-yl)-5-methylcyclohexanol is based on Ref. [
36]. To a suspension of MnO
2 (29.6 g, 20 equiv.) in DCM (50 mL), a solution of (1
S,2
S,5
R)-2-(3-hydroxyprop-1-en-2-yl)-5-methylcyclohexanol (3 g, 17 mmol) in DCM (50 mL) was added. The reaction mixture was vigorously stirred at 50–60 °C under reflux for 48 h. Upon completion of the reaction (monitored by TLC), the mixture was filtered through a Celite pad, and the filtrate was concentrated to afford the corresponding lactone (−)-
20 as a yellow oil (yield: 60%).
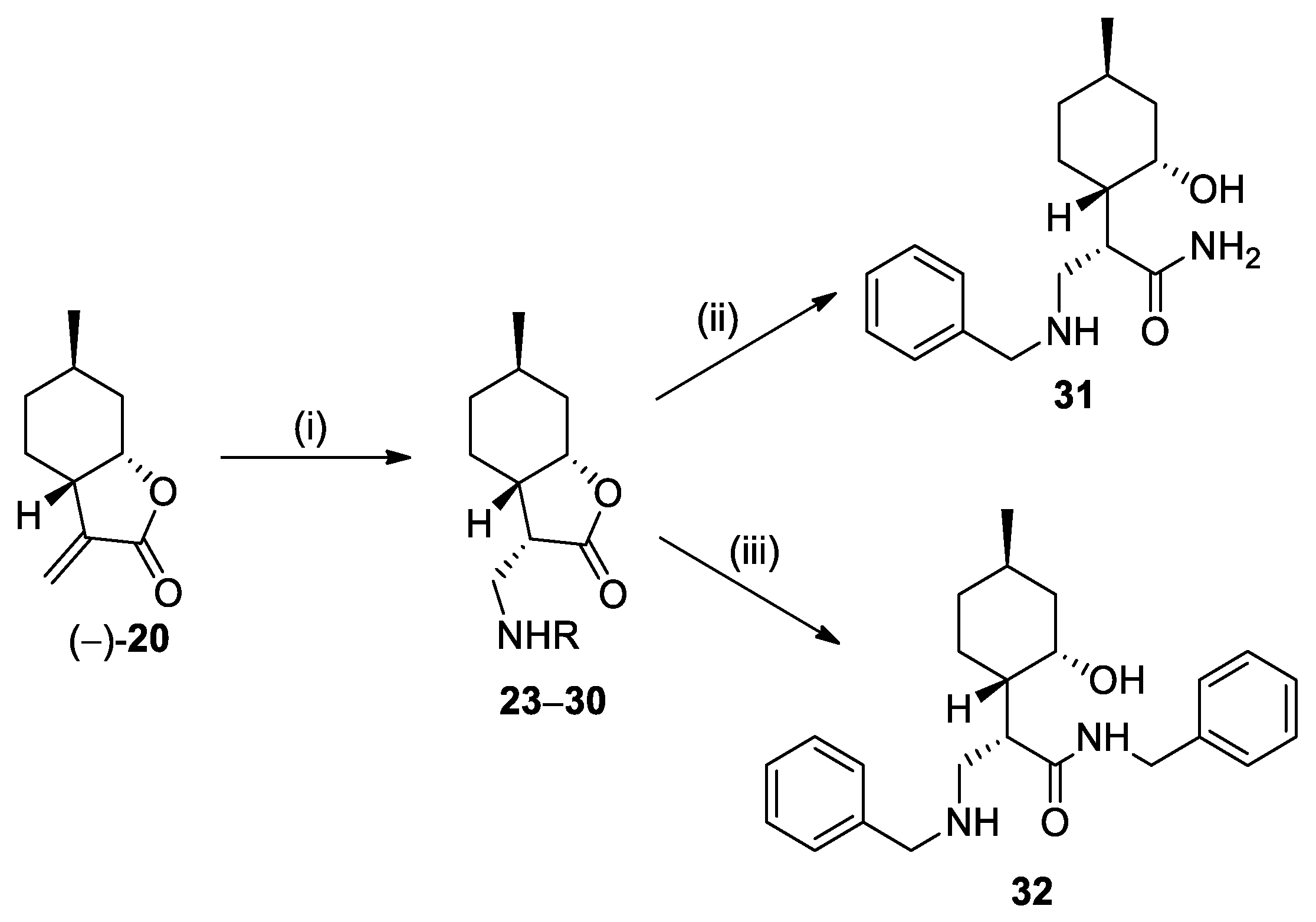
3.4. General Procedure for the Preparation of (+)-Neoisopulegol-Based β-Aminolactone Compounds 23–30
To a solution of compound (−)-20 (50 mg, 0.3 mmol) in dry EtOH (1.0 mL), the appropriate amines (0.45 mmol, 1.5 equiv.) were added. The reaction mixture was stirred at room temperature for 24 h. After the completion of the reaction (as monitored by TLC), the mixture was evaporated to dryness. The crude product was purified by column chromatography on silica gel using CHCl3:MeOH = 39:1 as the eluent.
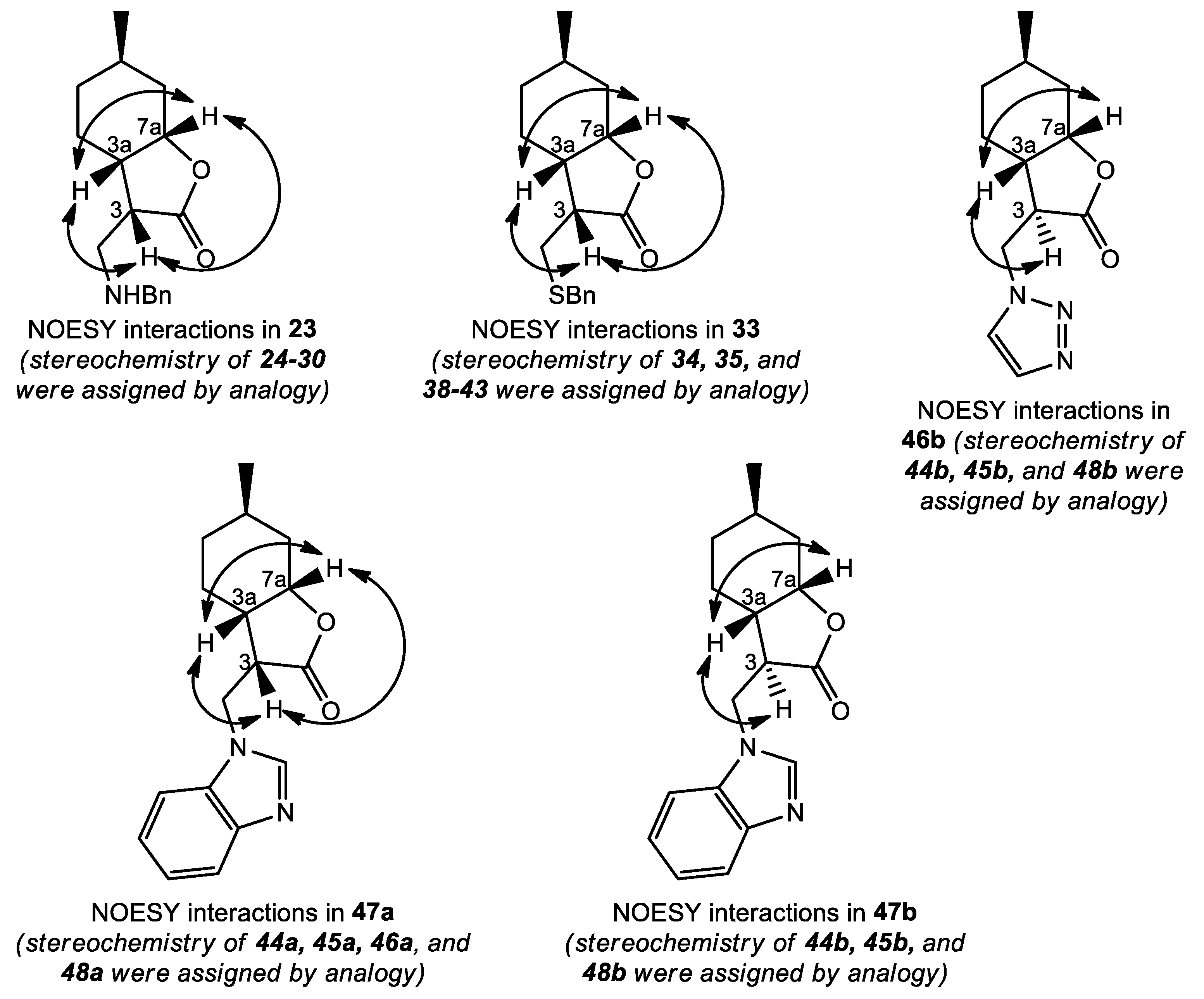
(3S,3aS,6R,7aS)-3-((Benzylamino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (23): The reaction was accomplished with benzylamine. Yield: 89%; yellow oil; = −44.45 (c = 0.15, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.34–7.30 (m, 4H), 7.28–7.22 (m, 1H), 4.45 (d, 1H, J = 2.7 Hz), 3.87–3. 76 (m, 2H), 2.99 (dd, J = 11.8, 7.1 Hz, 1H), 2.94–2.87 (1H, m), 2.69 (dd, J = 11.8, 7.4 Hz, 1H), 2.32 (ddd, J = 11.6, 9.9, 5.7 Hz, 1H), 2.22 (1H, d, J = 15 Hz), 1.66–1.60 (m, 2H), 1.60–1.51 (m, 1H), 1.08–1.28 (m, 2H), 0.94–0.81 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 178.6, 140.2, 128.6, 128.3, 127.2, 78.8, 54.4, 48.3, 44.9, 38.0, 36.2, 32.1, 26.4, 23.1, 22.1. HRMS (ESI): m/z calcd for C17H24NO2 [M+H]+: 274.1807; found: 274.1798.
(3S,3aS,6R,7aS)-3-(((2-Fluorobenzyl)amino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (24): The reaction was accomplished with 2-fluorobenzylamine. Yield: 42%; yellow oil; = −33.80 (c = 0.17, MeOH). ¹H NMR (500 MHz, CDCl3): δ = 7.34 (t, J = 7.5 Hz, 1H), 7.25–7.21 (m, 1H), 7.11 (t, J = 7.4 Hz, 1H), 7.07–7.01 (m, 1H), 4.45 (d, J = 2.6 Hz, 1H), 3.92–3.82 (m, 2H), 3.00 (dd, J = 11.8, 6.9 Hz, 1H), 2.91 (dd, J = 13.7, 6.8 Hz, 1H), 2.68 (dd, J = 11.7, 7.7 Hz, 1H), 2.33 (ddd, J = 11.4, 10.0, 5.7 Hz, 1H), 2.22 (d, J = 14.9 Hz, 1H), 1.69–1.61 (m, 2H), 1.61–1.51 (m, 1H), 1.23–1.08 (m, 2H), 0.94–0.81 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 178.5, 161.4 (d, 1JC-F = 245.1 Hz), 130.5 (d, 3JC-F = 4.6 Hz), 129 (d, 3JC-F = 8.1 Hz), 127 (d, 2JC-F = 14.5 Hz), 124.3 (d, 4JC-F = 3.6 Hz), 115.5 (d, 2JC-F = 21.8 Hz), 78.8, 48.2, 47.6 (d, 3JC-F = 2.9 Hz), 44.7, 37.9, 36.2, 32.1, 26.4, 23.1, 22.1. 19F NMR (470 MHz, CDCl3) δ = −119.46. HRMS (ESI): m/z calcd for C17H23FNO2 [M+H]+: 292.1713; found: 292.1701.
(3S,3aS,6R,7aS)-3-(((3-Fluorobenzyl)amino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (25): The reaction was accomplished with 3-fluorobenzylamine. Yield: 39%; yellow oil; = −36.11 (c = 0.18, MeOH). ¹H NMR (500 MHz, CDCl3): δ = 7.31–7.26 (m, 1H), 7.12–7.03 (m, 2H), 6.94 (td, J = 8.5, 2.1 Hz, 1H), 4.46 (d, J = 2.6 Hz, 1H), 3.87–3.75 (m, 2H), 2.98 (dd, J = 11.6, 7.3 Hz, 1H), 2.90 (dd, J = 13.6, 6.8 Hz, 1H), 2.67 (dd, J = 11.6, 7.1 Hz, 1H), 2.32 (ddd, J = 11.4, 10.0, 5.7 Hz, 1H), 2.22 (d, J = 14.9 Hz, 1H), 1.70–1.63 (m, 2H), 1.59–1.51 (m, 1H), 1.25–1.09 (m, 2H), 0.95–0.81 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 178.5, 163.0 (d, 1JC-F = 245.7 Hz), 142.7 (d, 3JC-F = 6.9 Hz), 129.9 (d, 3JC-F = 8.2 Hz), 123.5 (d, 4JC-F = 2.7 Hz), 114.8 (d, 2JC-F = 21.2 Hz), 113.9 (d, 2JC-F = 21.1 Hz), 78.7, 53.6 (d, 4JC-F = 1.4 Hz), 48.1, 44.7, 37.9, 36.0, 31.9, 26.2, 23.0, 21.9. 19F NMR (470 MHz, CDCl3) δ = −113.46. HRMS (ESI): m/z calcd for C17H23FNO2 [M+H]+: 292.1713; found: 292.1703.
(3S,3aS,6R,7aS)-3-(((4-Fluorobenzyl)amino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (26): The reaction was accomplished with 4-fluorobenzylamine. Yield: 52%; yellow oil; = −44.45 (c = 0.16, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.29 (dd, J = 8.3, 5.6 Hz, 2H), 7.01 (t, J = 8.7 Hz, 2H), 4.46 (d, J = 2.7 Hz, 1H), 3.84–3.73 (m, 2H), 2.97 (dd, J = 11.5, 7.3 Hz, 1H), 2.91 (dd, J = 13.4, 6.7 Hz, 1H), 2.68 (dd, J = 11.5, 6.9 Hz, 1H), 2.32 (ddd, J = 11.5, 9.8, 5.7 Hz, 1H), 2.22 (d, J = 15.2 Hz, 1H), 1.69–1.59 (m, 2H), 1.59–1.51 (m, 1H), 1.28–1.07 (m, 2H), 0.94–0.81 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 178.5, 162.0 (d, 1JC-F = 245.7 Hz), 135.5 (d, 4JC-F = 3.2 Hz), 129.7 (d, 3JC-F = 8.4 Hz), 115.3 (d, 2JC-F = 21.3 Hz), 78.7, 53.4, 47.9, 44.6, 37.9, 36.0, 31.9, 26.2, 23.0, 21.9. 19F NMR (470 MHz, CDCl3) δ = −115.78. HRMS (ESI): m/z calcd for C17H23FNO2 [M+H]+: 292.1713; found: 292.1703.
(3S,3aS,6R,7aS)-3-(((2-Methoxybenzyl)amino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (27): The reaction was accomplished with 2-methoxybenzylamine. Yield: 48% (35 mg); white powder, mp. 95.8–97.1 °C; = −31.60 (c = 0.19, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.25–7.21 (m, 2H), 6.91 (t, J = 7.4 Hz, 1H), 6.88–6.85 (m, 1H), 4.44 (d, J = 2.6 Hz, 1H), 3.84 (s, 3H), 3.81 (s, 2H), 2.98 (dd, J = 11.7, 6.5 Hz, 1H), 2.92 (dd, J = 6.5, 13.9 Hz, 1H), 2.66 (dd, J = 11.7, 7.9 Hz, 1H), 2.33 (ddd, J = 11.4, 9.9, 5.6 Hz, 1H), 2.21 (d, J = 13.3 Hz, 1H), 1.67–1.64 (m, 3H), 1.23–1.16 (m, 1H), 1.15–1.07 (m, 1H), 0.93–0.82 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 178.3, 129.8, 128.4, 128.0, 120.5, 110.3, 78.6, 55.3, 49.6, 48.1, 44.5, 37.8, 36.1, 32.0, 26.2, 22.8, 21.9. HRMS (ESI): m/z calcd for C18H26NO3 [M+H]+: 304.1913; found: 304.1901.
(3S,3aS,6R,7aS)-3-(((3-Methoxybenzyl)amino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (28): The reaction was accomplished with 3-methoxybenzylamine. Yield: 66%; yellow oil; = −37.92 (c = 0.19, MeOH). ¹H NMR (500 MHz, CDCl3): δ = 7.23 (t, J = 7.9 Hz, 1H), 6.92–6.88 (m, 2H), 6.81–6.78 (m, 1H), 4.45 (d, J = 2.7 Hz, 1H), 3.84–3.74 (m, 5H), 2.99 (dd, J = 11.8, 7.1 Hz, 1H), 2.90 (q, J = 6.8 Hz, 1H), 2.68 (dd, J = 11.8, 7.3 Hz, 1H), 2.35–2.28 (m, 1H), 2.21 (d, J = 15.1 Hz, 1H), 1.69–1.63 (m, 2H), 1.58–1.51 (m, 1H), 1.24–1.09 (m, 2H), 0.91 (d, J = 6.6 Hz, 3H), 0.89–0.82 (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 178.6, 160.0, 141.9, 129.6, 120.6, 113.7, 112.7, 78.8, 55.4, 54.3, 48.3, 44.9, 38.1, 36.2, 32.1, 26.4, 23.2, 22.1. HRMS (ESI): m/z calcd for C18H26NO3 [M+H]+: 304.1913; found: 304.1902.
(3S,3aS,6R,7aS)-3-(((4-Methoxybenzyl)amino)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (29): The reaction was accomplished with 4-methoxybenzylamine. Yield: 69%; white powder, mp. 48.5–50.5 °C; = −10.96 (c = 0.17, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.23 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 4.45 (d, J = 2.6 Hz, 1H), 3.80 (s, 3H), 3.79–3.70 (m, 2H), 2.97 (dd, J = 11.7, 7.0 Hz, 1H), 2.89 (dd, J = 13.5, 6.9Hz, 1H), 2.67 (dd, J = 11.7, 7.4Hz, 1H), 2.34–2.27 (m, 1H), 2.21 (d, J = 14.9 Hz, 1H), 1.68–1.63 (m, 2H), 1.58–1.50 (m, 1H), 1.23–1.08 (m, 2H), 0.91 (d, J = 6.5 Hz, 3H), 0.88–0.81 (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 178.6, 158.9, 132.3, 129.4, 114.0, 78.8, 55.4, 53.8, 48.3, 44.7, 38.0, 36.2, 32.1, 26.4, 23.1, 22.1. HRMS (ESI): m/z calcd for C18H26NO3 [M+H]+: 304.1913; found: 304.1902.
(3S,3aS,6R,7aS)-6-Methyl-3-(((naphthalen-1-ylmethyl)amino)methyl)hexahydro-benzofuran-2(3H)-one (30): The reaction was accomplished with 1-naphthylmethylamine. Yield: 90%; white crystals, mp. 62.1–64.5 °C; = −40.88 (c = 0.18, MeOH). 1H NMR (500 MHz, CDCl3): δ = 8.13 (d, J = 8.3 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.56–7.45 (m, 3H), 7.45–7.39 (m, 1H), 4.46–4.41 (m, 1H), 4.33–4.19 (m, 2H), 3.11 (dd, J = 11.8, 6.8Hz, 1H), 2.93 (dd, J = 13.8, 6.8 Hz, 1H), 2.80 (dd, J = 11.8, 7.8 Hz, 1H), 2.33–2.26 (m, 1H), 2.21 (d, J = 14.8 Hz, 1H), 1.65–1.48 (m, 3H), 1.23–1.10 (m, 2H), 0.93–0.80 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 178.6, 135.6, 134.1, 131.9, 128.9, 128.0, 126.3, 126.2, 125.8, 125.5, 123.8, 78.8, 52.1, 48.3, 45.3, 38.0, 36.2, 32.1, 26.4, 23.1, 22.1. HRMS (ESI): m/z calcd for C21H26NO2 [M+H]+: 324.1964; found: 324.1953.
3.5. General Procedure for the Preparation of (+)-Neoisopulegol-Based Thiol Adducts 33–35 and 38–43
To a solution of compound (−)-20 (50 mg, 0.3 mmol) in dry EtOH (1.0 mL), the appropriate benzyl mercaptan derivatives or cysteine derivatives (0.45 mmol, 1.5 equiv.) and Et3N (0.84 mmol, 2.8 equiv.) were added. After stirring for 24 h at room temperature (as monitored by TLC), the mixture was evaporated to dryness. The crude product was purified by column chromatography on silica gel, eluted with appropriate solvent.
(3R,3aS,6R,7aS)-3-((Benzylthio)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (33): The reaction was accomplished with benzyl mercaptan and chromatographed by n-hexane/EtOAc 9:1. Yield: 80%, white powder, mp. 88.7–90.1 °C; = −116.51 (c = 0.17, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.31 (d, J = 4.3 Hz, 4H), 7.27–7.22 (m, 1H), 4.37 (dd, J = 6.0, 3.2 Hz, 1H), 3.75 (s, 2H), 2.91 (dd, J = 13.0, 4.1 Hz, 1H), 2.78–2.71 (m, 1H), 2.46 (t, J = 12.3 Hz, 1H), 2.35–2.27 (m, 1H), 2.19 (d, J = 15.0 Hz, 1H), 1.68–1.59 (m, 2H), 1.56–1.48 (m, 1H), 1.22–1.14 (m, 1H), 1.02–0.92 (m, 1H), 0.91–0.80 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 177.3, 138.2, 128.9, 128.8, 127.4, 78.6, 47.7, 37.6, 37.2, 36.2, 31.9, 26.8, 26.3, 22.4, 22.0. HRMS (ESI): m/z calcd for C17H23O2S [M+H]+: 291.1419; found: 291.1410.
(3R,3aS,6R,7aS)-3-(((4-Fluorobenzyl)thio)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (34): The reaction was accomplished with 4-fluorobenzyl mercaptan and chromatographed by n-hexane/EtOAc 9:1. Yield: 76%; white powder, mp. 58.4–61.2 °C; = −107.25 (c = 0.16, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.31–7.26 (m, 2H), 7.00 (t, J = 8.6 Hz, 2H), 4.40 (d, J = 2.5 Hz, 1H), 3.72 (s, 2H), 2.89 (dd, J = 12.9, 4.1 Hz, 1H),), 2.82–2.73 (m, 1H), 2.45 (t, J = 12.2 Hz, 1H), 2.32 (ddd, J = 11.5, 9.9, 5.5 Hz, 1H), 2.20 (d, J = 14.9 Hz, 1H), 1.70–1.61 (m, 2H), 1.54–1.49 (m, 1H), 1.24–1.14 (m, 1H), 1.05–0.93 (m, 1H), 0.92–0.83 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 177.2, 162.1 (d, 1JC-F = 245.8 Hz), 133.9 (d, 4JC-F = 3.0 Hz), 130.5 (d, 3JC-F = 8.2 Hz), 115.6 (d, 2JC-F = 21.5 Hz), 78.7, 47.7, 37.6, 36.5, 36.2, 31.9, 26.8, 26.3, 22.4, 22.0. 19F NMR (470 MHz, CDCl3) δ = −115.20. HRMS (ESI): m/z calcd for C17H22FO2S [M+H]+: 309.1325; found: 309.1317.
(3R,3aS,6R,7aS)-3-(((4-Methoxybenzyl)thio)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (35): The reaction was accomplished with 4-methoxybenzyl mercaptan and chromatographed by n-hexane/EtOAc 9:1. Yield: 73%; white powder, mp. 60.7–62.3 °C; = −114.78 (c = 0.15, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.22 (d, J = 8.5 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 4.39 (d, J = 2.6 Hz, 1H), 3.80 (s, 3H), 3.70 (s, 2H), 2.90 (dd, J = 13.0, 4.1 Hz, 1H), 2.80–2.72 (m, 1H), 2.45 (t, J = 12.3 Hz, 1H), 2.37–2.28 (m, 1H), 2.20 (d, J = 15.1 Hz, 1H), 1.69–1.63 (m, 2H), 1.53–1.50 (m, 1H), 1.23–1.14 (m, 1H), 1.02–0.94 (m, 1H), 0.93–0.82 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 177.3, 159.0, 130.1, 130.0, 114.2, 78.7, 55.5, 47.8, 37.6, 36.6, 36.2, 31.9, 26.7, 26.3, 22.4, 22.0. HRMS (ESI): m/z calcd for C18H25O3S [M+H]+: 321.1524; found: 321.1514.
(R)-2-Acetamido-3-((((3R,3aS,6R,7aS)-6-methyl-2-oxooctahydrobenzofuran-3-yl)-methyl)-thio)propanoic acid (38): The reaction was accomplished with N-acetyl-L-cysteine and chromatographed by CHCl3/CH3OH 19:1. Yield: 71%; white powder, mp. 136.2–138.3 °C; = −51.80 (c = 0.17, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 7.63 (d, J = 6.5 Hz, 1H), 4.51 (s, 1H), 4.12 (d, J = 3.8 Hz, 1H), 3.13–3.06 (m, 1H), 3.04 (dd, J = 13.3, 3.7 Hz, 1H), 2.79 (dd, J =13.1, 3.9 Hz, 1H), 2.71 (dd, J = 13.2, 7.2 Hz, 1H),), 2.42 (t, J = 12.3 Hz, 1H), 2.34 (d, J = 4.6 Hz, 1H), 2.04 (d, J = 14.5 Hz, 1H), 1.84 (s, 3H), 1.67 (s, 1H), 1.58 (s, 1H), 1.38 (s, 1H), 1.23 (t, J = 12.1 Hz, 1H), 1.09 (t, J = 6.9 Hz, 1H), 0.87 (d, J = 6.3 Hz, 5H). 13C NMR (125 MHz, DMSO-d6): δ = 177.7, 173.4, 169.1, 78.1, 54.3, 47.2, 37.2, 35.8, 35.3, 31.8, 27.3, 26.5, 23.3, 22.5, 22.3. HRMS (ESI): m/z calcd for C15H24NO5S [M+H]+: 330.1375; found: 330.1367.
(R)-Methyl 2-amino-3-((((3R,3aS,6R,7aS)-6-methyl-2-oxooctahydrobenzofuran-3-yl)-methyl)thio)propanoate (39): The reaction was accomplished with L-cysteine methyl ester and chromatographed by CHCl3/CH3OH 19:1. Yield: 50%; orange powder, mp. 78.4–80.3 °C; = −134.73 (c = 0.15, MeOH). 1H NMR (500 MHz, CDCl3): δ =4.46 (dd, J = 5.9, 3.1 Hz, 1H), 3.75 (s, 3H), 3.68 (dd, J = 7.4, 4.6 Hz, 1H), 2.98 (ddd, J = 13.5, 6.7, 4.4 Hz, 2H), 2.94–2.88 (m, 1H), 2.80 (dd, J = 13.5, 7.4 Hz, 1H), 2.60 (dd, J = 12.8, 11.5 Hz, 1H), 2.40 (ddd, J = 11.9, 9.8, 5.9 Hz, 1H), 2.26–2.19 (m, 1H), 1.82–1.74 (m, 1H), 1.74–1.65 (m, 1H), 1.61–1.51 (m, 1H), 1.21 (ddd, J = 15.6, 12.5, 3.5 Hz, 1H), 1.06 (ddd, J = 25.6, 13.3, 3.3 Hz, 1H), 0.95–0.84 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 177.1, 174.5, 78.7, 54.3, 52.5, 48.0, 38.0, 37.5, 36.2, 31.9, 27.8, 26.3, 22.5, 22.0. HRMS (ESI): m/z calcd for C14H24NO4S [M+H]+: 302.1426; found: 302.1420.
(R)-Methyl 2-acetamido-3-((((3R,3aS,6R,7aS)-6-methyl-2-oxooctahydrobenzofuran-3-yl)methyl)thio)propanoate (40): The reaction was accomplished with N-acetyl-L-cysteine methyl ester and chromatographed by n-hexane/EtOAc 1:2. Yield: 62%; white powder, mp. 105.4–107.6 °C; = −104.50 (c = 0.16, MeOH). 1H NMR (500 MHz, CDCl3): δ = 6.32 (d, J = 6.9 Hz, 1H), 4.85 (dt, J = 7.4, 4.9 Hz, 1H), 4.46 (dd, J1 = 5.8, 3.1 Hz, 1H), 3.79 (s, 3H), 3.13 (dd, J = 13.9, 4.8 Hz, 1H), 3.03–2.92 (m, 2H), 2.87 (ddd, J = 10.7, 5.9, 4.5 Hz, 1H), 2.58 (dd, J = 12.9, 11.1 Hz, 1H), 2.37 (ddd, J = 11.9, 9.8, 5.9 Hz, 1H), 2.26–2.18 (m, 1H), 2.06 (s, 3H), 1.78–1.66 (m, 2H), 1.60–1.51 (m, 1H), 1.25–1.17 (m, 1H), 1.06 (ddd, J = 25.4, 13.2, 3.2 Hz, 1H), 0.95–0.82 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 177.0, 171.3, 170.0, 78.6, 53.0, 52.4, 48.0, 37.6, 36.1, 34.9, 31.9, 28.1, 26.3, 23.3, 22.5, 22.0. HRMS (ESI): m/z calcd for C16H26NO5S [M+H]+: 344.1532; found: 344.1522.
(R)-2-Acetamido-3-((((3R,3aS,6R,7aS)-6-methyl-2-oxooctahydrobenzofuran-3-yl)-methyl)thio)propanamide (41):The reaction was accomplished with N-acetyl-L-cysteine amide and chromatographed by n-hexane/EtOAc 1:2. Yield: 41%; white powder, mp. 99.3–101.2 °C; = −116.83 (c = 0.12, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 8.04 (d, J = 8.3 Hz, 1H), 7.52 (s, 1H), 7.12 (s, 1H), 4.51 (s, 1H), 4.38 (dd, J = 12.6, 6.8 Hz, 1H), 3.15–3.08 (m, 1H), 2.88 (ddd, J = 17.5, 13.4, 4.7 Hz, 2H), 2.61 (dd, J = 13.6, 8.8 Hz, 1H), 2.46 (d, J = 12.4 Hz, 1H), 2.39–2.30 (m, 1H), 2.05 (d, J = 14.6 Hz, 1H), 1.85 (s, 3H), 1.74–1.62 (m, 1H), 1.58 (d, J = 7.9 Hz, 1H), 1.39 (s, 1H), 1.29–1.19 (m, 1H), 0.95–0.82 (m, 5H). 13C NMR (125 MHz, DMSO-d6): δ = 177.5, 172.7, 169.7, 78.1, 52.3, 47.0, 37.2, 35.8, 34.4, 31.8, 27.0, 26.5, 23.0, 22.5, 22.3. HRMS (ESI): m/z calcd for C15H25N2O4S [M+H]+: 329.1535; found: 329.1525.
(R)-Ethyl 2-acetamido-3-((((3R,3aS,6R,7aS)-6-methyl-2-oxooctahydrobenzofuran-3-yl)-methyl)thio)propanoate (42): The reaction was accomplished with N-acetyl-L-cysteine ethyl ester and chromatographed by n-hexane/EtOAc 1:2. Yield: 93%; white powder, mp. 98.3–100.7 °C; = −95.53 (c = 0.17, MeOH). 1H NMR (500 MHz, CDCl3): δ = 6.31 (d, J = 6.9 Hz, 1H), 4.82 (dt, J = 7.3, 4.9 Hz,1H), 4.46 (d, J = 2.8 Hz, 1H), 4.24 (q, J = 7.1 Hz, 2H), 3.13 (dd, J = 13.9, 4.7 Hz, 1H), 2.97 (dd, J1 = 13.2, 4.5 Hz, 2H), 2.88–2.84 (m, 1H), 2.58 (dd, J = 12.9, 11.3 Hz, 1H), 2.37 (ddd, J = 11.9, 9.8, 5.8 Hz, 1H), 2.22 (dd, J = 15.0, 3.0 Hz, 1H), 2.05 (s, 3H), 1.77–1.72 (m, 1H), 1.72–1.65 (m, 1H), 1.60–1.50 (m, 1H), 1.31 (t, J = 7.1 Hz, 3H), 1.24–1.18 (m, 1H), 1.05 (qd, J = 13.2, 3.1 Hz, 1H), 0.96–0.84 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 177.0, 170.8, 170.0, 78.6, 62.2, 52.5, 48.0, 37.6, 36.2, 34.8, 31.9, 28.1, 26.3, 23.3, 22.5, 22.0, 14.3. HRMS (ESI): m/z calcd for C17H28NO5S [M+H]+: 358.1688; found: 358.1677.
(R)-Methyl 2-((tert-butoxycarbonyl)amino)-3-((((3R,3aS,6R,7aS)-6-methyl-2-oxooctahydrobenzofuran-3-yl)methyl)thio)propanoate (43): The reaction was accomplished with N-Boc-L-cysteine methyl ester and chromatographed by n-hexane/EtOAc 2:1. Yield: 54%; white powder, mp. 88.3–90.3 °C; = −82.13 (c = 0.19, MeOH). 1H NMR (500 MHz, CDCl3): δ = 5.33 (d, J = 6.2 Hz, 1H), 4.56 (d, J = 3.2 Hz, 1H), 4.45 (d, J = 2.7 Hz, 1H), 3.78 (s, 3H), 3.08 (dd, J = 13.8, 4.2 Hz, 1H), 3.01–2.92 (m, 2H), 2.91–2.84 (m, 1H), 2.65–2.58 (m, 1H), 2.37 (ddd, J = 11.8, 9.8, 5.8 Hz, 1H), 2.25–2.19 (m, 1H), 1.81–1.74 (m, 1H), 1.72–1.64 (m, 1H), 1.61–1.51 (m, 1H), 1.45 (s, 9H), 1.25–1.16 (m, 1H), p1.06 (ddd, J =25.7, 13.2, 3.2 Hz, 1H), 0.97–0.84 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 176.8, 171.3, 155.1, 80.3, 78.5, 53.5, 52.7, 47.9, 37.4, 36.0, 35.3, 31.8, 28.3, 28.0, 26.2, 22.3, 21. 9. HRMS (ESI): m/z calcd for C19H32NO6S [M+H]+: 402.1950; found: 402.1941.
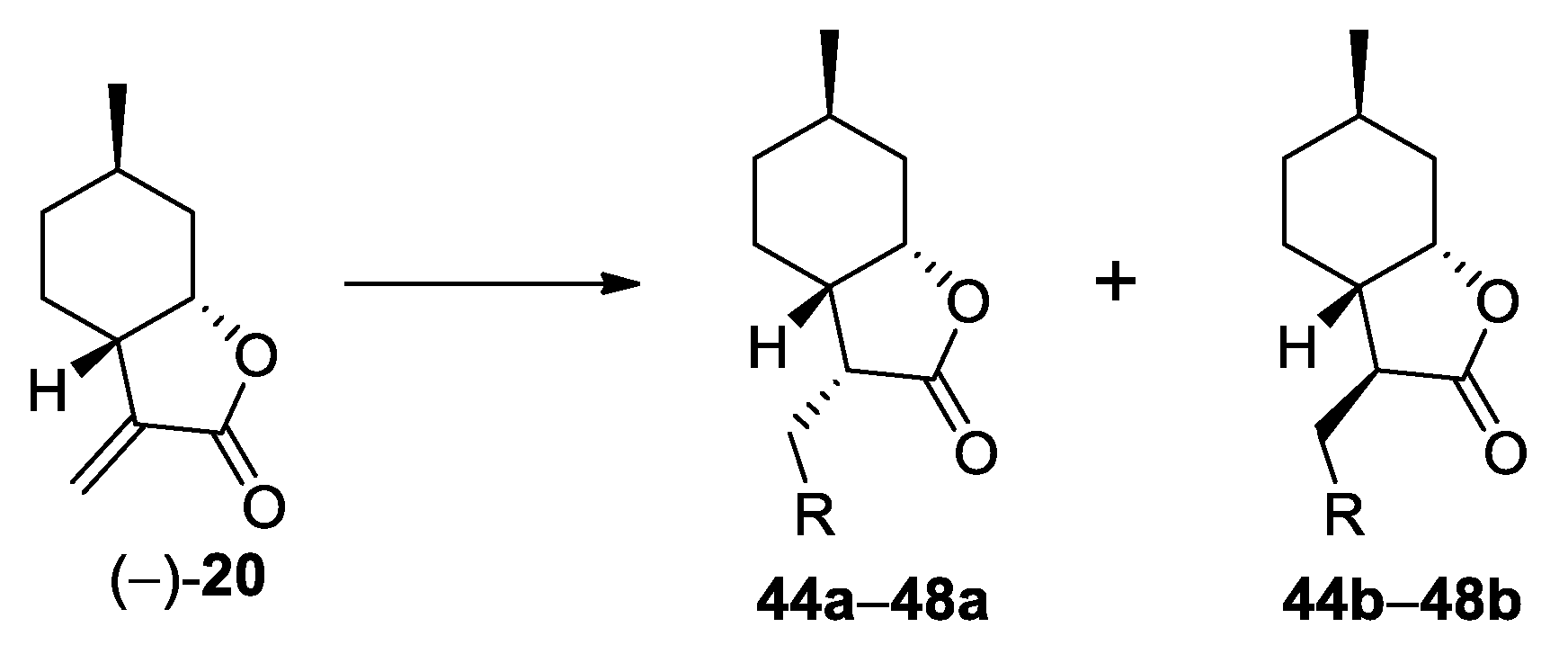
3.6. General Procedure for the Preparation of (+)-Neoisopulegol-Based Azole Adducts 44–48
Compound (−)-20 (50 mg, 0.3 mmol) and the azole derivative (0.6 mmol, 2 equiv.) were dissolved in acetonitrile (5.0 mL), and then DBU (0.3 mmol, 1 equiv.) was added. The mixture was stirred at 70 °C overnight. Then, it was evaporated to dryness, and the crude product was purified by column chromatography on silica gel with an appropriate solvent mixture.
(3S,3aS,6R,7aS)-3-((1H-Imidazol-1-yl)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (44a): The reaction was accomplished with imidazole and chromatographed by CHCl3/isopropanol 19:1. Yield: 50%; white crystals, mp. 43.5–44.3 °C; = −73.36 (c = 0.15, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.52 (s, 1H), 7.10 (s, 1H), 6.98 (s, 1H), 4.48 (d, J =2.7 Hz, 1H), 4.41 (dd, J = 14.7, 4.4 Hz, 1H), 4.07 (dd, J = 14.7, 9.8 Hz, 1H), 3.14–3.08 (m, 1H), 2.33–2.22 (m, 2H), 1.83–1.71 (m, 2H), 1.63–1.61 (m, 1H), 1.28–1.18 (m, 2H), 0.98–0.86 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 130.4, 78.7, 49.9, 42.5, 37.4, 36.0, 31.8, 26.1, 23.2, 21.9. HRMS (ESI): m/z calcd for C13H19N2O2 [M+H]+: 235.1447; found: 235.1438.
(3R,3aS,6R,7aS)-3-((1H-Imidazol-1-yl)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (44b): The reaction was accomplished with imidazole and chromatographed by CHCl3/isopropanol 19:1. Yield: 14%; white crystals, mp. 150.2–152.3 °C; = −45.30 (c = 0.20, MeOH). 1H NMR (500 MHz, CDCl3): δ =7.5 (s, 1H), 7.11 (s, 1H), 6.96 (s, 1H), 4.31–4.21 (m, 2H), 4.18 (q, J= 4.2 Hz, 1H), 2.71–2.65 (m, 1H), 2.09–2.02 (m, 2H), 1.82–1.71 (m, 1H), 1.71–1.56 (m, 2H), 1.33–1.19 (m, 2H), 1.00–0.87 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 176.5, 130.8, 78.0, 50.8, 45.5, 37.3, 35.7, 31.0, 27.0, 25.9, 21.2. HRMS (ESI): m/z calcd for C13H19N2O2 [M+H]+: 235.1447; found: 235.1438.
(3S,3aS,6R,7aS)-3-((1H-1,2,4-Triazol-1-yl)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (45a): The reaction was accomplished with 1,2,4-triazole and chromatographed by CHCl3/isopropanol 19:1. Yield: 57%; colorless oil; = −75.33 (c = 0.18, MeOH). 1H NMR (500 MHz, CDCl3): δ = 8.17 (s, 1H), 7.95 (s, 1H), 4.58 (dd, J = 14.3, 4.8 Hz, 1H), 4.51 (d, J=15.0 Hz, 1H), 4.28 (dd, J = 14.3, 9.4 Hz, 1H), 3.34–3.30 (m, 1H), 2.43–2.34 (m, 1H), 2.25 (d, J = 15.0 Hz, 1H), 1.92–1.87 (m, 1H), 1.77–1.70 (m, 1H), 1.64–1.56 (m, 1H), 1.26 (s, 1H), 1.24–1.18 (m, 1H), 0.98–0.86 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 175.9, 152.6, 78.9, 48.5, 45.0, 37.6, 36.0, 31.8, 26.2, 23.1, 21.9. HRMS (ESI): m/z calcd for C12H18N3O2 [M+H]+: 236.1399; found: 236.1390.
(3R,3aS,6R,7aS)-3-((1H-1,2,4-Triazol-1-yl)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (45b): The reaction was accomplished with 1,2,4-triazole and chromatographed by CHCl3/isopropanol 19:1. Yield: 18%; white crystals, mp. 102.5–103.1 °C; = −42.11 (c =0.17, MeOH). 1H NMR (500 MHz, CDCl3): δ = 8.12 (s, 1H), 7.96 (s, 1H), 4.52–4.41 (m, 2H), 4.38 (q, J= 4.3 Hz, 1H), 2.86–2.80 (m, 1H), 2.35–2.30 (m, 1H), 2.07 (d, J= 14.7 Hz, 1H), 1.81–1.75 (m, 1H), 1.72–1.60 (m, 2H), 1.36–1.22 (m, 2H), 1.01–0.96 (m, 1H), 0.93 (d, J= 6.58 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 176.2, 152.6, 143.7, 78.1, 50.0, 47.7, 37.2, 35.6, 30.9, 26.8, 26.0, 21.2. HRMS (ESI): m/z calcd for C12H18N3O2 [M+H]+: 236.1399; found: 236.1390.
(3S,3aS,6R,7aS)-3-((1H-1,2,3-Triazol-1-yl)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (46a): The reaction was accomplished with 1,2,3-triazole and chromatographed by CHCl3/isopropanol 19:1. Yield: 53%; white crystals, mp. 99.3–101.2 °C; = −82.49 (c =0.18, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.71 (d, J= 11.5 Hz, 2H), 4.69 (dd, J= 14.4, 4.9 Hz, 1H), 4.57 (dd, J= 14.4, 9.1 Hz, 1H), 4.51 (d, J= 2.7 Hz, 1H), 3.34 (dt, J= 9.2, 5.5 Hz, 1H), 2.45 (ddd, J = 11.9, 9.9, 5.9 Hz, 1H), 2.26–2.25 (d, J= 15.2 Hz, 1H), 1.98–1.93 (m, 1H), 1.77–1.70 (m, 1H), 1.65–1.57 (m, 1H), 1.29–1.16 (m, 2H), 0.98–0.87 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 175.9, 134.1, 124.4, 79.0, 49.02, 45.7, 37.9, 36.0, 31.8, 26.2, 23.1, 21.9. HRMS (ESI): m/z calcd for C12H18N3O2 [M+H]+: 236.1399; found: 236.1390.
(3R,3aS,6R,7aS)-3-((1H-1,2,3-Triazol-1-yl)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (46b): The reaction was accomplished with 1,2,3-triazole and chromatographed by CHCl3/isopropanol 19:1. Yield: 28%; white crystals, mp. 101.8–104.3 °C; = −46.11 (c = 0.15, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.73 (s, 1H), 7.64 (s, 1H), 4.75–4.67 (m, 2H), 4.23 (dd, J = 8.7, 4.3 Hz, 1H), 2.80 (td, J = 6.2, 3.0 Hz, 1H), 2.36–2.31 (m, 1H), 2.03 (d, J= 15.0 Hz, 1H), 1.81–1.75 (m, 1H), 1.70–1.58 (m, 2H), 1.35–1.22 (m, 2H), 1.02–0.94 (m, 1H), 0.91 (d, J= 6.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 176.3, 134.7, 124.1, 78.3, 50.3, 48.2, 37.1, 35.6, 30.8, 26.8, 26.0, 21.1. HRMS (ESI): m/z calcd for C12H18N3O2 [M+H]+: 236.1399; found: 236.1390.
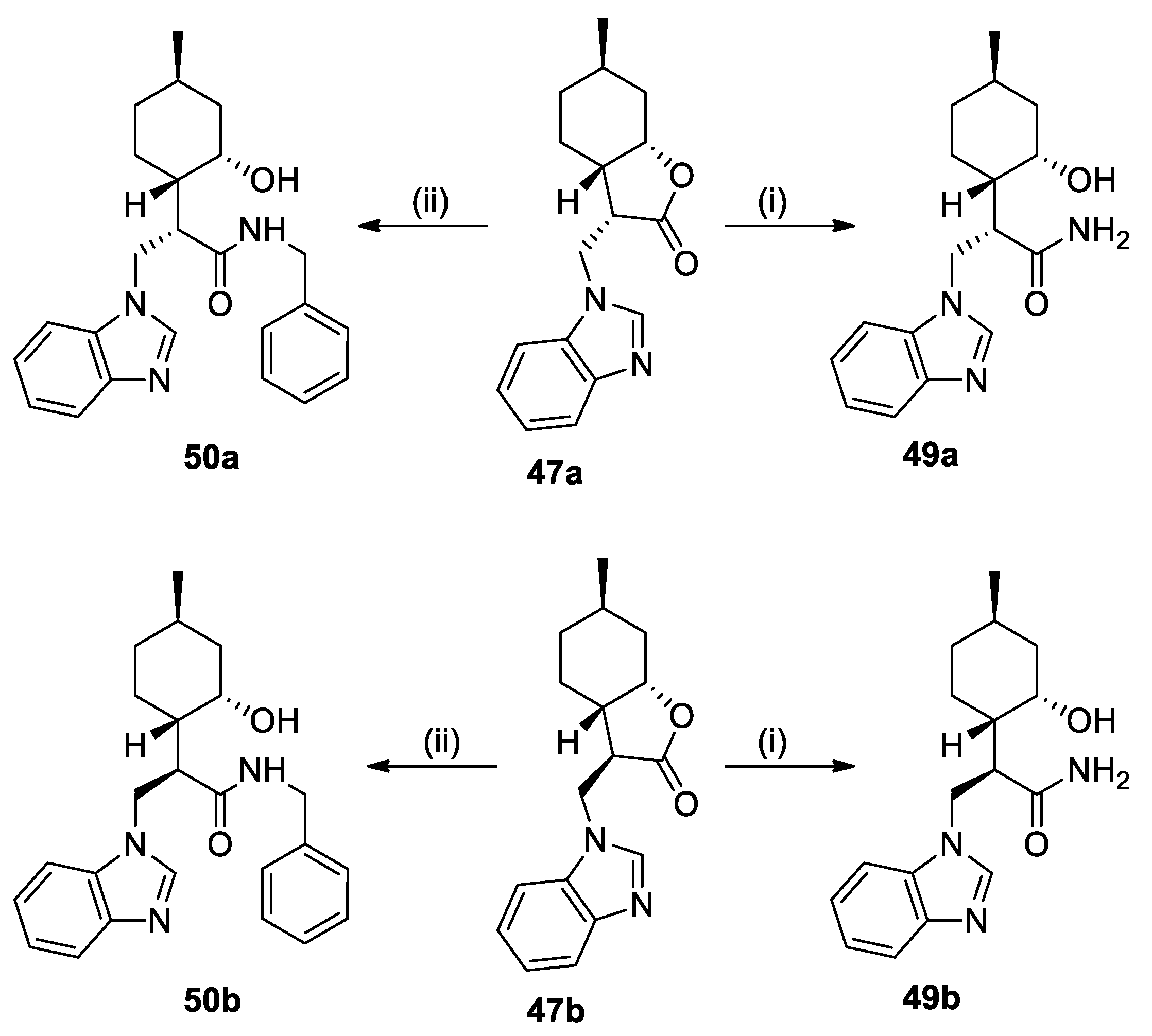
(3S,3aS,6R,7aS)-3-((1H-Benzo[d]imidazol-1-yl)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (47a): The reaction was accomplished with benzimidazole and chromatographed by CHCl3/t-BuOH 19:1. Yield: 20%; white crystals, mp. 120.1–122.9 °C; = −74.60 (c = 0.22, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.97 (s, 1H), 7.83 (d, J = 7.3 Hz, 1H), 7.42 (d, J = 7.9 Hz, 1H), 7.36–7.29 (m, 2H), 4.64 (dd, J = 15.0, 4.6 Hz, 1H), 4.45 (d, J = 2.65 Hz, 1H), 4.31 (dd, J = 15.0, 9.3 Hz, 1H), 3.31–3.27 (m, 1H), 2.28–2.23 (m, 2H), 1.89–1.86 (m, 1H), 1.77–1.75 (m, 1H), 1.67–1.60 (m, 1H), 1.37–1.16 (m, 2H), 0.94–0.87 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 176.0, 144.1, 143.0, 133.4, 123.5, 122.7, 121.0, 109.4, 78.7, 48.1, 40.6, 37.4, 36.0, 31.8, 26.1, 23.3, 21.9. HRMS (ESI): m/z calcd for C17H21N2O2 [M+H]+: 285.1603; found: 285.1592.
(3R,3aS,6R,7aS)-3-((1H-Benzo[d]imidazol-1-yl)methyl)-6-methylhexahydrobenzofuran-2(3H)-one (47b): The reaction was accomplished with benzimidazole and chromatographed by CHCl3/t-BuOH 19:1. Yield: 12%; white powder, mp. 125.3–126.5 °C; = −22.77 (c = 0.21, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.92 (s, 1H), 7.83 (d, J = 7.5 Hz, 1H), 7.44 (d, J = 7.8 Hz, 1H), 7.36–7.31 (m, 2H), 4.54–4.40 (m, 3H), 2.87 (ddd, J = 7.9, 5.1, 2.7 Hz, 1H), 2.14–2.02 (m, 2H), 1.71–1.62 (m, 2H), 1.58 (d, J = 3.4 Hz, 1H), 1.32–1.17 (m, 2H), 0.95–0.86 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 142.8, 141.7, 123.6, 122.7, 120.8, 109.2, 77.8, 49.7, 43.1, 37.3, 35.5, 30.7, 26.7, 25.7, 20.9. HRMS (ESI): m/z calcd for C17H21N2O2 [M+H]+: 285.1603; found: 285.1592.
(3S,3aS,6R,7aS)-3-((1H-Benzo[d][1,2,3]triazol-1-yl)methyl)-6-methylhexahydrobenzo-furan-2(3H)-one (48a): The reaction was accomplished with benzotriazole and chromatographed by CHCl3/t-BuOH 19:1. Yield: 21%; white powder, mp. 129.8–131.7 °C; = −91.04 (c = 0.15, MeOH). 1H NMR (500 MHz, CDCl3): δ = 8.09 (d, J = 8.35 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 4.94 (dd, J = 14.6, 3.8 Hz, 1H), 4.77 (dd, J = 14.6, 10.7 Hz, 1H), 4.52 (d, J = 2.6 Hz, 1H), 3.58–3.52 (m, 1H), 2.41 (ddd, J = 11.8, 9.9, 5.9 Hz, 1H), 2.26 (d, J = 15.2 Hz, 1H), 2.08–2.02 (m, 1H), 1.78–1.70 (m, 1H), 1.68–1.58 (m, 1H), 1.34–1.18 (m, 2H), 0.97–0.86 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 176.0, 127.9, 124.4, 120.4, 109.2, 79.1, 48.3, 43.4, 37.6, 36.0, 31.8, 26.2, 23.1, 22.0. HRMS (ESI): m/z calcd for C16H20N3O2 [M+H]+: 286.1556; found: 286.1546.
(3R,3aS,6R,7aS)-3-((1H-Benzo[d][1,2,3]triazol-1-yl)methyl)-6-methylhexahydrobenzo-furan-2(3H)-one (48b): The reaction was accomplished with benzotriazole and chromatographed by CHCl3/t-BuOH 19:1. Yield: 12%; white crystals, mp. 135.9–137.5 °C; = −38.03 (c = 0.19, MeOH). 1H NMR (500 MHz, CDCl3): δ = 8.08 (d, J= 8.4 Hz, 1H), 7.61 (d, J = 8.3 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 4.94 (qd, J = 14.6, 6.7 Hz, 2H), 4.36 (q, J = 3.9 Hz, 1H), 2.94–2.92 (m, 1H), 2.4–2.36 (m, 1H), 2.08 (d, J = 15.5 Hz, 1H), 1.69–1.58 (m, 2H), 1.28–1.16 (m, 3H), 0.94–0.86 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 128.3, 124.6, 120.4, 109.4, 78.5, 50.3, 46.2, 37.2, 35.7, 31.3, 27.3, 25.9, 21.4. HRMS (ESI): m/z calcd for C16H20N3O2 [M+H]+: 286.1556; found: 286.1545.
3.7. General Procedure for the Ring Opening of Lactones with NH3 (Synthesis of 31, 36, and 49a–b)
To a solution of lactone 23, 33, or 47a–b (0.36 mmol) in MeOH (2.0 mL), a solution of 25% NH3 in MeOH (5.0 mL) was added. The mixture was stirred at room temperature for 12 h. After the completion of the reaction (as monitored by TLC), the mixture was evaporated to dryness. The crude product was purified by column chromatography on silica gel then recrystallized in Et2O, resulting in compounds 31, 36, and 49a–b, respectively.
(S)-3-(Benzylamino)-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)propanamide (31): Recrystallized in Et2O. Yield: 94%; white powder; mp. 140.3–142.5 °C; = +27.52 (c = 0.20, MeOH). ¹H NMR (500 MHz, CD3OD): δ = 7.31 (dd, J = 7.3, 5.9 Hz, 4H), 7.27–7.22 (m, 1H), 3.81–3.70 (m, 2H), 3.40 (td, J = 10.6, 4.2 Hz, 1H), 2.98–2. 89 (m, 2H), 2.71–2.65 (m, 1H), 1.95 (d, J = 12.2 Hz, 1H), 1.71–1.55 (m, 3H), 1.46–1.34 (m, 1H), 1.15–1.04 (m, 1H), 0.96 (dd, J = 23.4, 12.0 Hz, 1H), 0.91 (d, J = 6.5 Hz, 3H), 0.89–0.79 (m, 1H).¹³C NMR (125 MHz, CD3OD) δ = 129.5, 129.5, 128.2, 71.6, 54.6, 48.3, 47.1, 46.7, 45.7, 35.7, 32.8, 27.8, 22.5. HRMS (ESI) m/z: calcd for C17H27N2O2[M+H]+: 291.2073; found: 291.2063.
(R)-3-(Benzylthio)-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)propanamide (36): Chromatographed by CHCl3/MeOH 19:1. Yield: 95%; white powder, mp. 156.3–158.6 °C; = +86.75 (c = 0.16, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.36–7.29 (m, 4H), 7.27–7.23 (m, 1H), 5.62 (s, 1H), 5.39 (s, 1H), 3.94 (d, J = 1.8 Hz, 1H), 3.78–3.69 (m, 2H), 2.71 (d, J = 7.1 Hz, 2H), 2.32 (dd, J = 15.5, 7.1, 1H), 1.79 (ddd, J = 13.8, 5.9, 3.3 Hz, 1H), 1.75–1.63 (m, 2H), 1.59–1.51 (m, 1H), 1.51–1.44 (m, 1H), 1.44–1.33 (m, 1H), 1.17–1.10 (m, 1H), 0.94–0.83 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 176.6, 138.7, 129.2, 128.7, 127.3, 67.5, 49.4, 43.3, 42.5, 37.3, 34.6, 31.9, 25.9, 24.6, 22.3. HRMS (ESI) m/z: calcd for C17H26NO2S [M+H]+: 308.1684; found: 308.1675.
(S)-3-(1H-Benzo[d]imidazol-1-yl)-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)-propanamide (49a): Chromatographed by CHCl3/acetone 9:1. Yield: 59%; mp. 263.3–265.0 °C; = +89.60 (c = 0.20, MeOH). 1H NMR (500 MHz, CD3OD): δ = 8.00 (s, 1H), 7.64 (t, J = 8.2 Hz, 2H), 7.31 (t, J = 7.6 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 4.58 (dd, J = 13.9, 3.8 Hz, 1H), 4.47 (dd, J = 22.0, 10.3 Hz, 2H), 4.22 (s, 1H), 2.99 (td, J1 = 10.9, 3.7 Hz, 1H), 1.94 (dd, J = 13.9, 2.4 Hz, 1H), 1.89–1.79 (m, 1H), 1.76–1.67 (m, 1H), 1.60–1.52 (m, 2H), 1.52–1.45 (m, 1H), 1.32–1.20 (m, 1H), 0.98 (td, J = 12.6, 3.9 Hz, 1H),
0.95–0.90 (m, 3H). 13C NMR (125 MHz, CD3OD): δ = 124.2, 123.5, 120.0, 111.8, 66.9, 50.3, 43.7, 42.6, 35.7, 26.9, 26.4, 22.7. HRMS (ESI) m/z: calcd for C17H24N3O2 [M+H]+: 302.1868; found: 302.1859.
(R)-3-(1H-Benzo[d]imidazol-1-yl)-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)-propanamide (49b): Chromatographed by CHCl3/acetone 9:1. Yield: 69%; white crystals, mp. 240.4–243.3 °C; = −48.20 (c = 0.20, MeOH). 1H NMR (500 MHz, CD3OD): δ = 0.92 (d, J = 6.3 Hz, 3H), 0.99–1.11 (m, 1H), 1.15–1.21 (m, 1H), 1.66–1.80 (m, 2H), 1.80–1.95 (m, 4H), 3.03 (ddd, J1 = 4.5 Hz, J2 = 8.0 Hz, J3 = 12.0 Hz, 1H), 4.01 (s, 1H), 4.50–4.62 (m, 2H), 7.28 (t, J = 7.6 Hz, 1H), 7.34 (t, J = 7.6 Hz, 1H), 7.60 (d, J = 8.1 Hz, 1H), 7.66 (d, J = 8.0 Hz, 1H), 8.04 (s, 1H). 13C NMR (125 MHz, CD3OD): δ = 124.3, 123.5, 120.1, 111.6, 68.9, 50.8, 46.3, 43.7, 43.5, 35.9, 26.8, 24.8, 22.6. HRMS (ESI) m/z: calcd for C17H24N3O2 [M+H]+: 302.1868; found: 302.1859.
3.8. General Procedure for the Ring Opening of Lactones with Benzylamine (Synthesis of 32, 37, and 50a–b)
To a solution of lactone 23, 33, or 47a–b (0.36 mmol) in dry THF (5.0 mL), benzylamine (0.72 mmol) was added. The mixture was stirred at 70 °C for 48 h. When the reaction was completed, the mixture was evaporated to dryness. The crude product was purified by column chromatography on silica gel mixture, then recrystallized in n-hexane/Et2O, resulting in compounds 32, 37, and 50a–b, respectively.
(S)-N-Benzyl-3-(benzylamino)-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)-propanamide (32): Chromatographed by CHCl3/CH3OH 19:1. Yield: 70%; white powder, mp. 211.0–212.7 °C; = +21.05 (c = 0.20, MeOH). 1H NMR (500 MHz, DMSO-d6): δ = 9.18 (s, 1H), 9.06 (s, 1H), 8.75 (t, J = 5.7 Hz, 1H), 7.54 (d, J = 4.0 Hz, 2H), 7.42 (d, J = 4.9 Hz, 3H), 7.35–7.20 (m, 5H), 4.74 (s, 1H), 4.43–4.15 (m, 2H), 4.11 (s, 2H), 3.80 (s, 1H), 3.23–3.12 (m, 1H), 3.05 (d, J = 11.8 Hz, 1H), 2.79–2.71 (m, 1H), 1.75–1.63 (d, J = 11.2 Hz, 2H), 1.60–1.50 (m, 2H), 1.47–1.36 (m, 1H), 1.18 (d, J = 10.2 Hz, 1H), 1.01 (t, J = 13.0 Hz, 1H), 0.80–0.74 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ = 172.3, 139.0, 131.8, 130.1, 128.9, 128.6, 128.2, 127.5, 126.9, 64.0, 50.1, 45.3, 44.3, 42.9, 41.9, 41.3, 34.3, 25.1, 24.4, 22.2. HRMS (ESI) m/z: calcd for C24H33N2O2 [M+H]+: 381.2542; found: 381.2530.
(R)-N-Benzyl-3-(benzylthio)-2-((1S,2S,4R)-2-hydroxy-4-methylcyclohexyl)propanamide (37): Chromatographed by n-hexane/EtOAc 2:1. Yield: 54%; white powder, mp. 172.1–175.4 °C; = +84.00 (c = 0.17, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.38–7.16 (m, 10H), 5.77 (t, J = 4.8 Hz, 1H), 4.48 (dd, J = 14.7, 5.7 Hz, 1H), 4.39 (dd, J = 14.7, 5.5 Hz, 1H), 3.94 (s, 1H), 3.74–3.64 (m, 2H), 2.74 (d, J = 7.2 Hz, 2H), 2.20 (dd, J = 15.9, 7.2 Hz, 1H), 1.78 (ddd, J = 13.8, 5.7, 3.2 Hz, 1H), 1.72–1.58 (m, 3H), 1.44–1.37 (m, 1H), 1.36–1.27 (m, 1H), 1.18–1.06 (m, 1H), 0.93–0.82 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 174.0, 138.9, 138.3, 129.1, 128.8, 128.7, 128.2, 127.6, 127.3, 67.2, 50.3, 43.8, 43.5, 42.5, 37.4, 34.6, 32.2, 25.9, 24.8, 22.3. HRMS (ESI) m/z: calcd for C24H32NO2S [M+H]+: 398.2154; found: 398.2144.
(S)-3-(1H-Benzo[d]imidazol-1-yl)-N-benzyl-2-((1S,2S,4R)-2-hydroxy-4-methylcyclo-hexyl)propanamide (50a): Chromatographed by CHCl3/t-BuOH 19:1. Yield: 39%; white powder, mp. 181.0–184.2 °C; = −52.53 (c = 0.23, MeOH). 1H NMR (500 MHz, CDCl3): δ = 7.86 (s, 1H), 7.84–7.77 (m, 1H), 7.42–7.36 (m, 1H), 7.28 (d, J = 4.5 Hz, 2H), 7.18 (d, J = 3.3 Hz, 3H), 6.89–6.82 (m, 2H), 5.78 (s, 1H), 4.65 (dd, J = 13.5, 11.9 Hz, 1H), 4.35 (dd, J = 14.0, 3.1 Hz, 1H), 4.26 (dd, J = 14.7, 5.6 Hz, 1H), 4.15 (dd, J = 14.7, 5.6 Hz, 1H), 3.96 (s, 1H), 2.80–2.70 (m, 1H), 2.05 (s, 1H), 1.86–1.73 (m, 5H), 1.72–1.60 (m, 1H), 1.19 (t, J = 13.6 Hz, 1H), 1.06–0.95 (m, 1H), 0.91 (d, J = 6.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 173.0, 144.0, 143.9, 137.4, 133.6, 128.8, 127.7, 127.6, 123.2, 122.4, 120.7, 109.6, 67.6, 51.3, 45.6, 43.9, 42.4, 42.5, 34.7, 25.8, 24.8, 22.2. HRMS (ESI) m/z: calcd for C24H30N3O2 [M+H]+: 392.2338; found: 392.2326.
(R)-3-(1H-Benzo[d]imidazol-1-yl)-N-benzyl-2-((1S,2S,4R)-2-hydroxy-4-methylcyclo-hexyl)propanamide (50b): Chromatographed by CHCl3/t-BuOH 19:1. Yield: 60%; white powder, mp. 258.2–260.2 °C; = +41.23 (c = 0.21, MeOH). 1H NMR (500 MHz, CD3OD): δ = 7.97 (s, 1H), 7.69 (d, J = 7.5 Hz, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.34–7.26 (m, 2H), 7.14–7.06 (m, 3H), 6.71–6.66 (m, 2H), 4.60 (dd, J = 13.8, 3.9 Hz, 1H), 4.50 (t, J = 12.7 Hz, 1H), 4.26–4.15 (m, 2H), 3.95 (d, J = 14.9 Hz, 1H), 3.01 (td, J = 11.0, 3.9 Hz, 1H), 1.93 (dd, J = 13.8, 1.9 Hz, 1H), 1.88–1.80 (m, 1H), 1.80–1.72 (m, 1H), 1.69 (d, J = 12.9 Hz, 1H), 1.50 (qd, J = 13.0, 3.2 Hz, 1H), 1.37 (dd, J = 13.0, 3.2 Hz, 1H), 1.39–1.21 (m, 1H), 1.00–0.88 (m, 4H). 13C NMR (125 MHz, CD3OD): δ = 175.4, 145.0, 144.0, 139.1, 134.9, 129.3, 128.2, 128.0, 124.3, 123.5, 120.1, 111.9, 66.9, 50.8, 46.0, 43.9, 43.6, 42.9, 35.6, 26.9, 26.4, 22.7. HRMS (ESI) m/z: calcd for C24H30N3O2 [M+H]+: 392.2338; found: 392.2327.
3.9. Determination of the Antimicrobial Effect
3.9.1. Reagents and Media
DMSO (Sigma-Aldrich, St Louis, MO, USA), phosphate-buffered saline (PBS; pH 7.4), Mueller–Hinton (MH) broth, tryptic soy broth (TSB), tryptic soy agar (TSA), Luria–Bertani broth (LBB), and Luria–Bertani agar (LBA) were used. All reagents were purchased from Sigma.
3.9.2. Bacterial Strains
Escherichia coli ATCC 25922 was used as the Gram-negative strain. Gram-positive strains investigated in this study included the following: Staphylococcus aureus American Type Culture Collection (ATCC) 25923 as a methicillin-susceptible reference strain, and the methicillin- and oxacillin-resistant S. aureus MRSA ATCC 43300 strain.
3.9.3. Antibacterial Activity
The minimum inhibitory concentrations (MICs) of the compounds were determined according to the Clinical and Laboratory Standard Institute guidelines [
41]. Two-fold serial dilutions of the compounds were prepared in Mueller–Hinton broth in 96-well plates, and the starting concentration of the compounds was 100 µM (Sigma-Aldrich, St. Louis, MO, USA). The MIC values of the compounds were determined by visual inspection. DMSO as a solvent was also assayed to ensure that it had no antibacterial effect.
3.10. Computational Studies
The crystal structures of the protein templates were obtained from PDB (protein data bank), and the tested structures were drawn by ChemBioDraw Ultra 12.0. The Accelrys Discovery Studio 3.5 software was used to perform the docking study and the in silico ADMET prediction.
3.10.1. Preparation of the Crystal Structures of the Protein Targets
The structures were prepared by using the Accelrys Discovery Studio 3.5 software’s Clean Geometry option after eliminating the water molecules. Then, the absent hydrogen atoms were furnished by applying the CHARMm force field. Adopted Basis minimization was utilized to minimize the complex energy and to get the most stable structure without altering the basic protein skeleton. Thereafter, a 10 Å radius sphere was created to define the active site residues [
46].
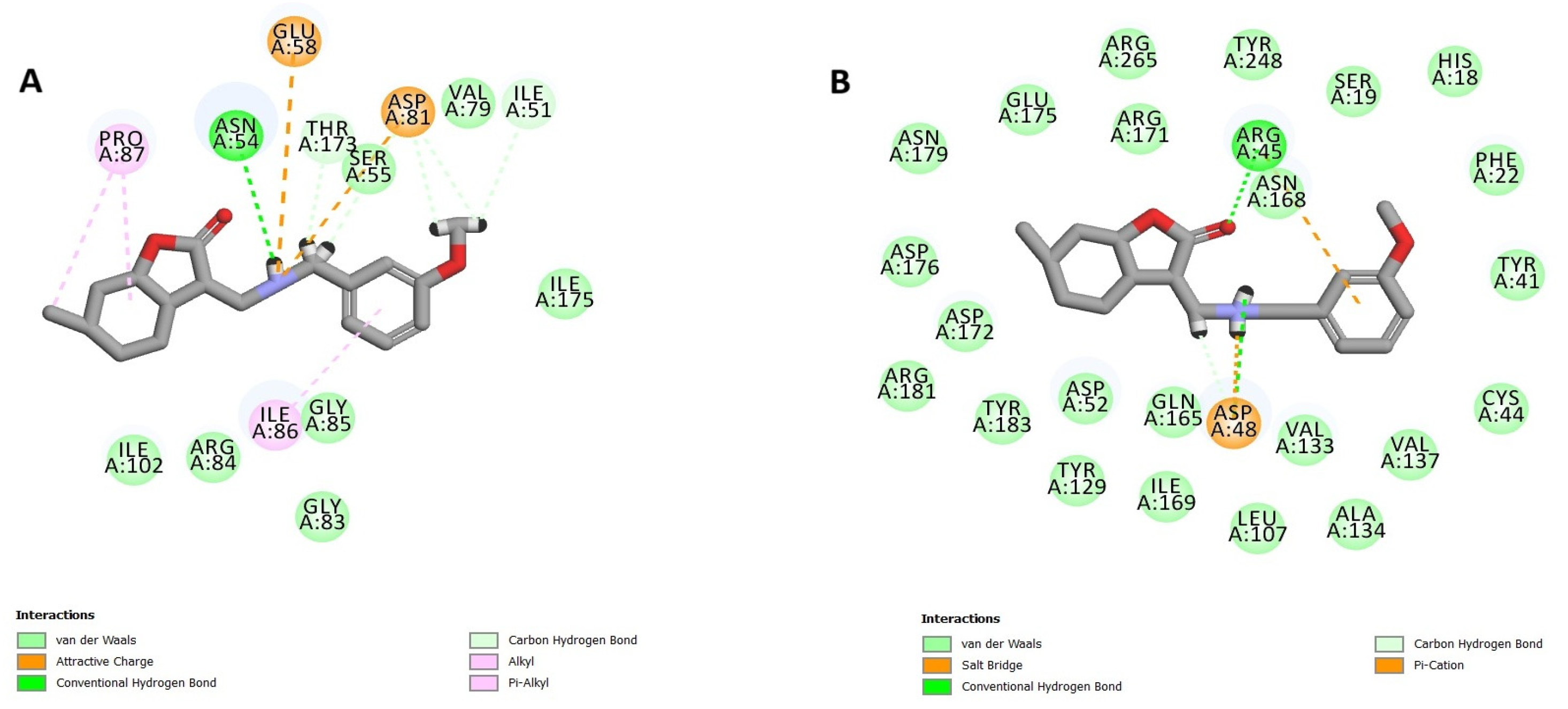
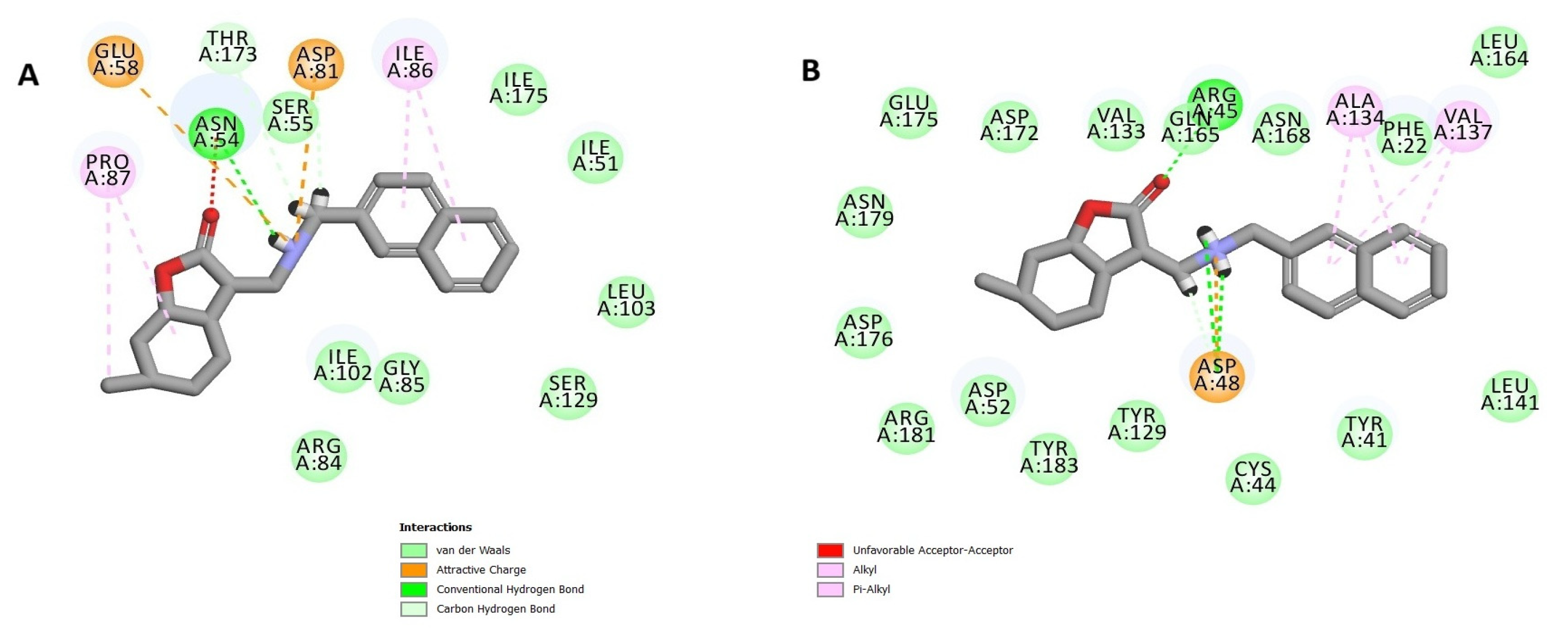
3.10.2. Docking Study (CDOCKER)
The CDOCKER method enables the generation of all possible conformations of the compound within the protein’s active site. These conformations were then assessed according to both the CDOCKER energy and the interactions observed between the ligand and the active site. This approach requires preparing the crystal structure (as previously described) and the examined compound, which involves using the Accelrys Discovery Studio protocol and applying a force field. Before initiating this study, it is crucial to validate the method by comparing the conformation of the reference compound to the conformations generated by the docking approach, ensuring that the RMSD (Root Mean Square Deviation) remains within the acceptable range.
3.10.3. In Silico ADMET prediction
The ADMET properties of compounds
28 and
30 were predicted using ADMET descriptors in the Accelrys Discovery Studio 3.5 software. Multiple mathematical models were employed for quantitative prediction. These models included the following: aqueous solubility (predict solubility in water at 25 °C), blood–brain barrier (BBB) penetration, cytochrome P450 (CYP2D6) inhibition, human intestinal absorption (HIA), and plasma protein binding (PPB) [
47]. An ADMET model was also generated to predict the human intestinal absorption (HIA) and blood–brain barrier (BBB) penetration of the tested compounds. The model shows the ADMET_PSA_2D and ADMET_ALogP98 plot with 95 and 99% confidence limit ellipses (
Figure S87 in Supplementary Materials).




 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}