
Deciphering the Transcriptomic Complexity of Yak Skin Across Different Ages and Body Sites


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
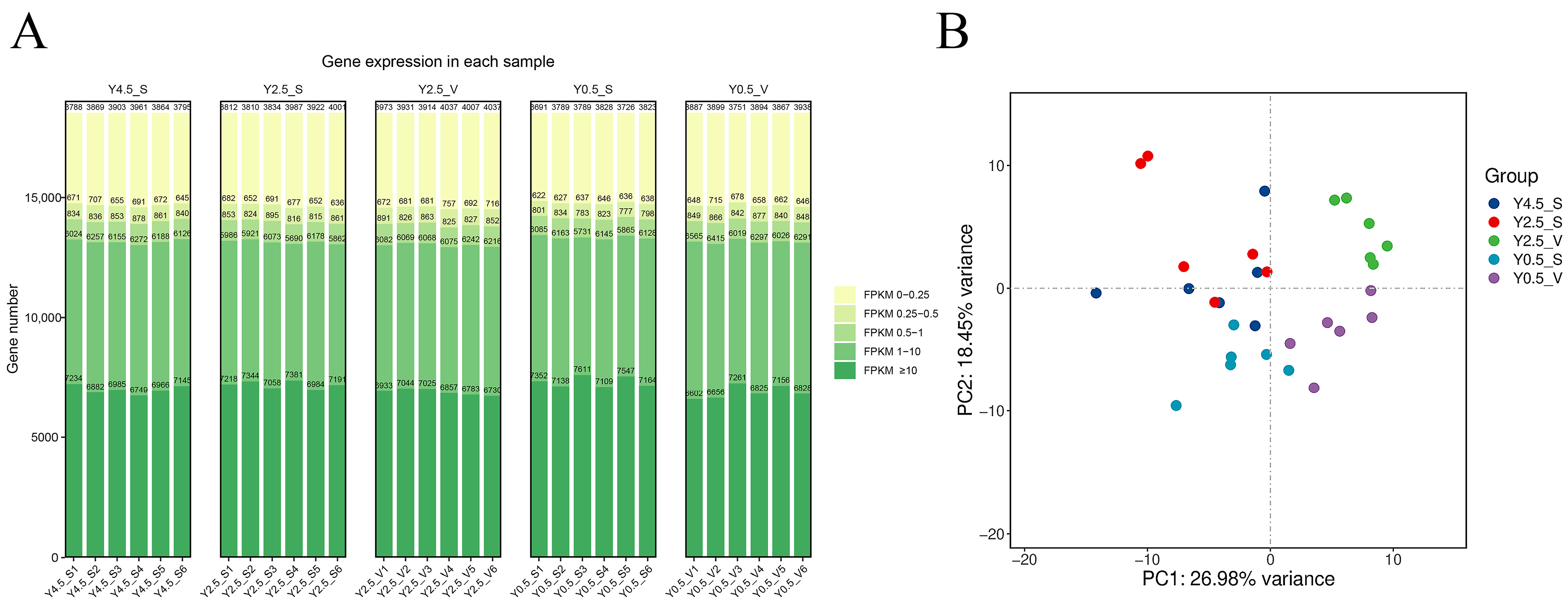
2.1. Overview of RNA-seq
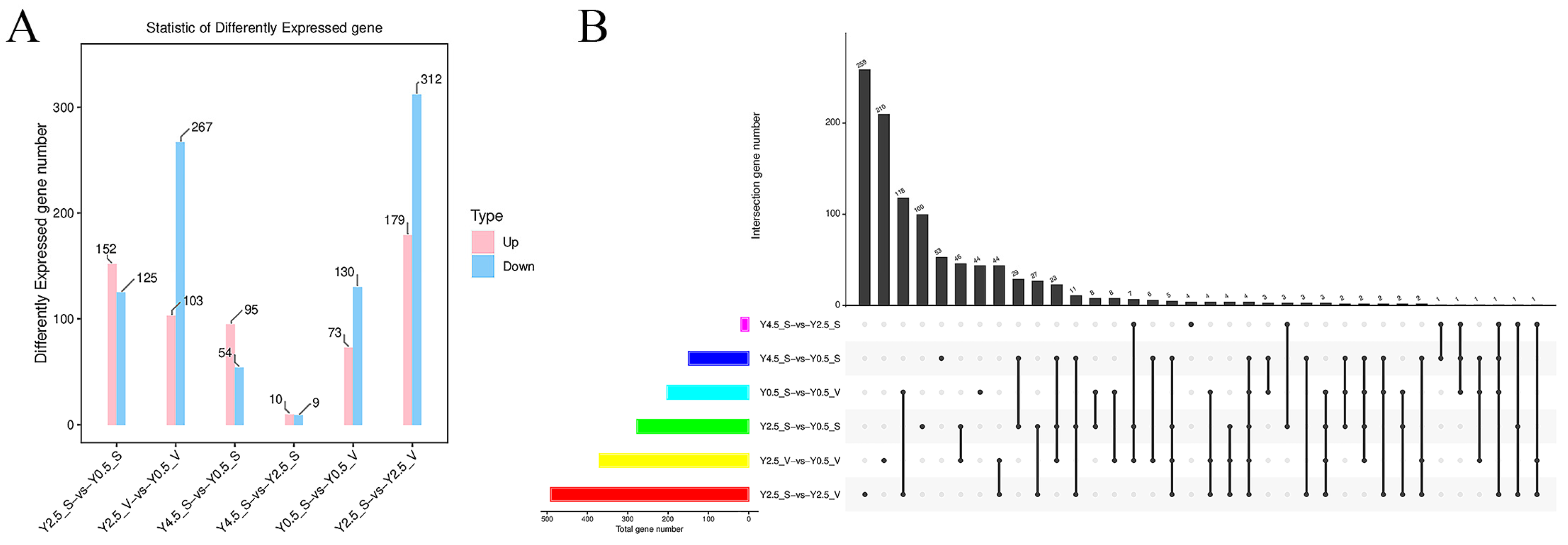
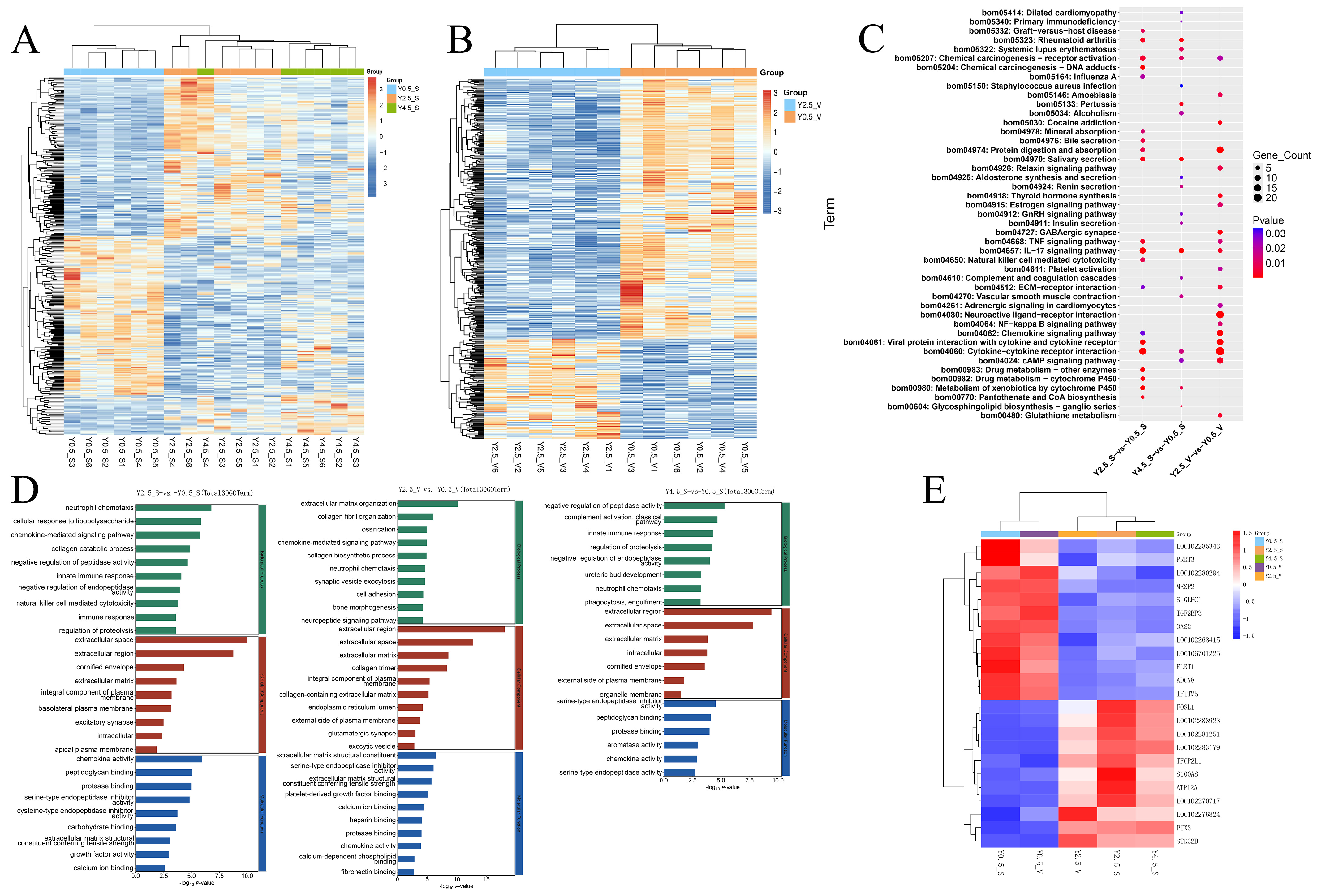
2.2. Expression Patterns and Functional Analysis of DEGs at Different Ages
2.3. Functional Analysis of DEGs at Different Skin Regions
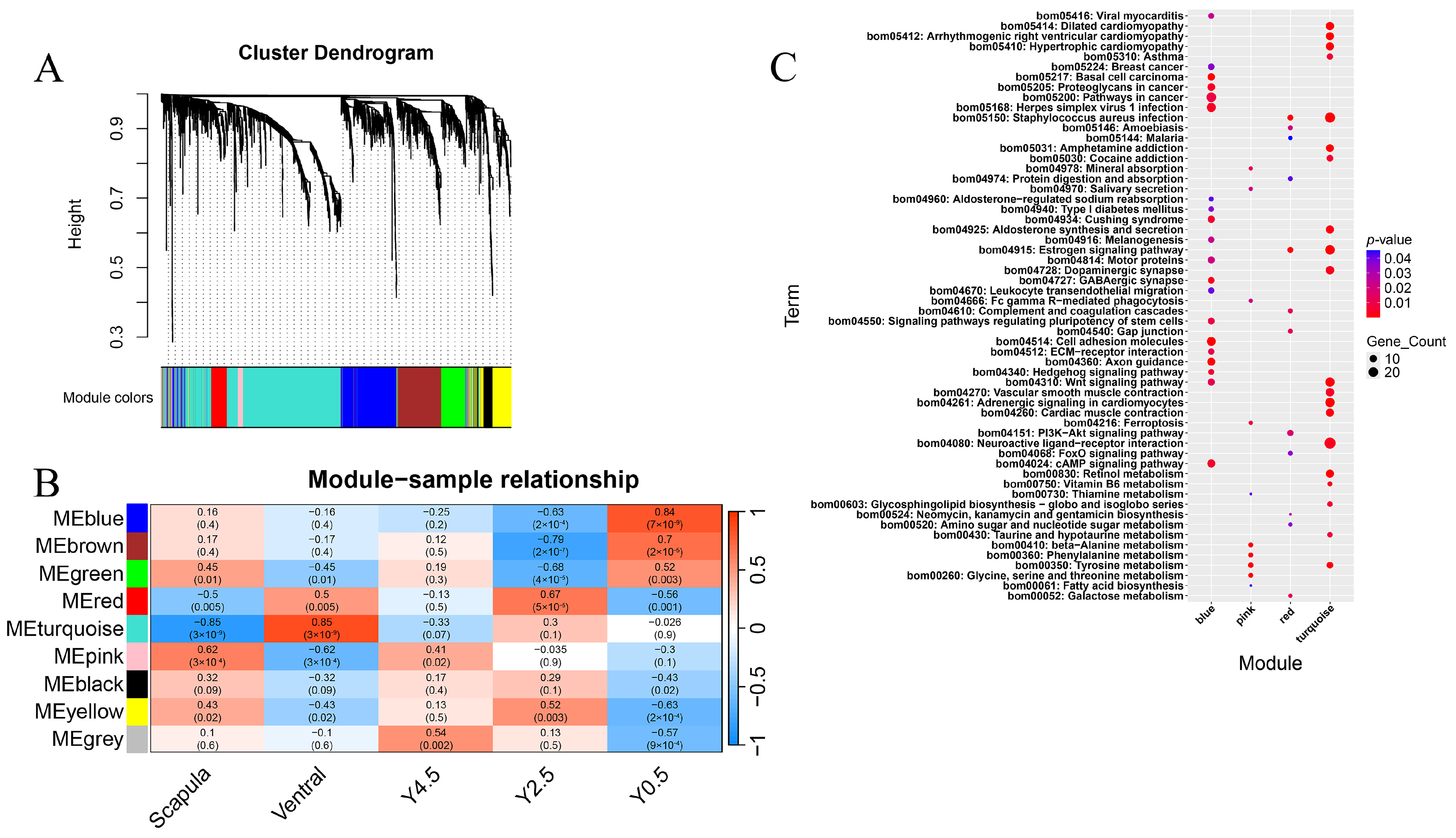
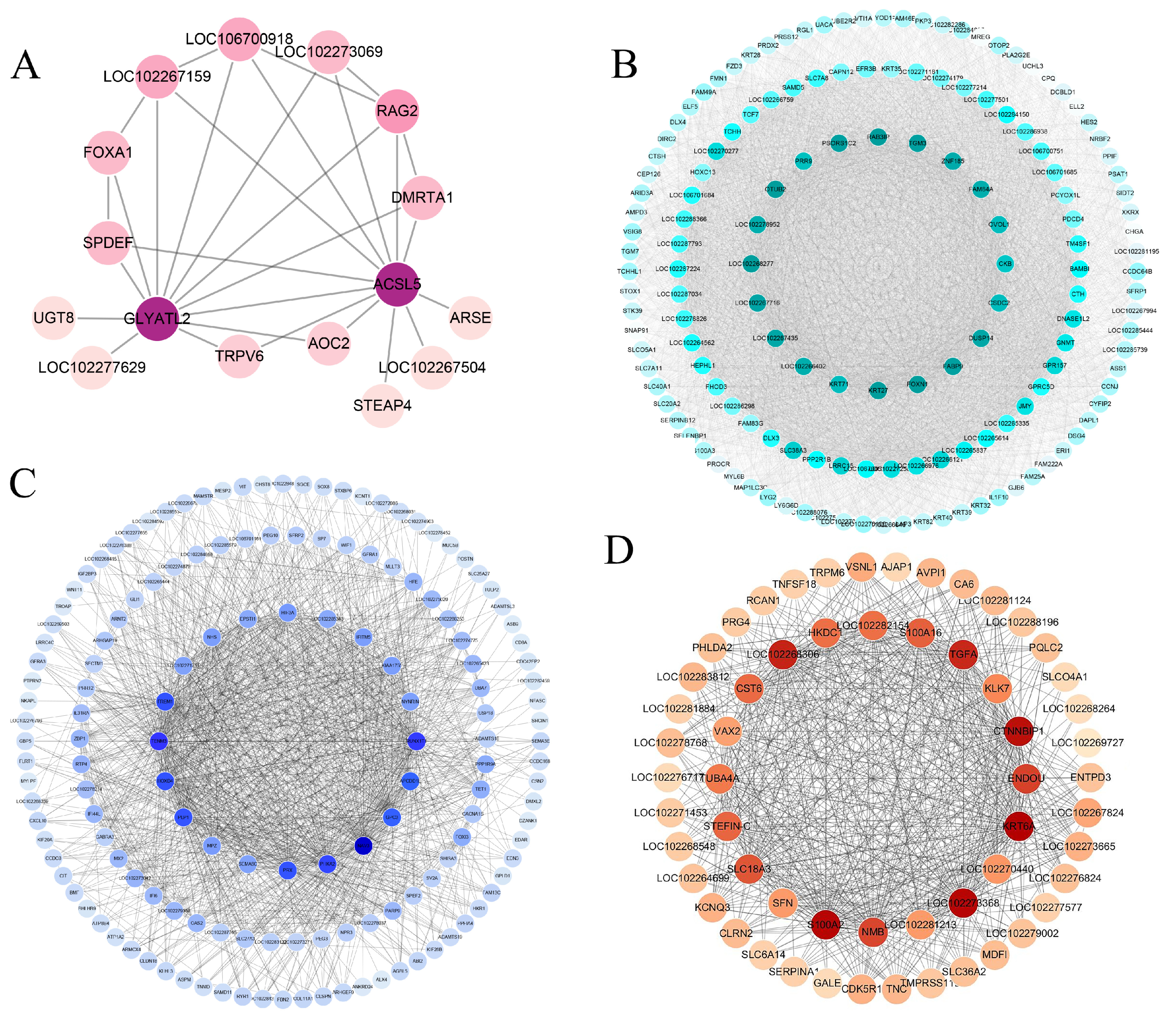
2.4. Weighted Gene Co-Expression Network Analysis
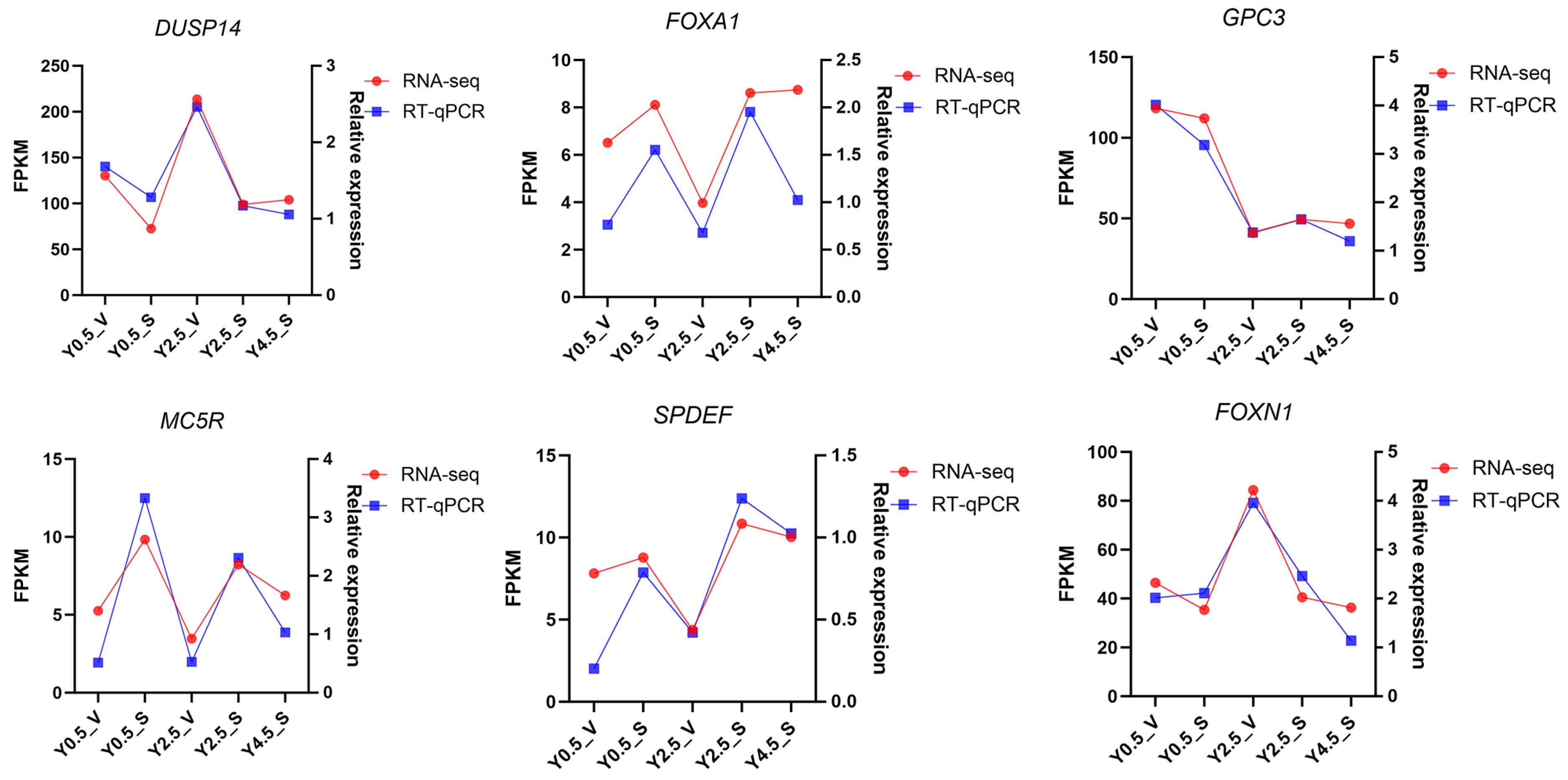
2.5. RT-qPCR Validation of DEGs
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Sample Collection
4.2. RNA Extraction, Library Preparation, and Sequencing
4.3. Differentially Expressed Genes and Functional Enrichment Analysis
4.4. WGCNA
4.5. RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wei, L.; Xiao, Y.; Bi, H.; Yang, H.; Du, Y. Physicochemical and functional properties of gelatin extracted from Yak skin. Int. J. Biol. Macromol. 2017, 95, 1246–1253. [Google Scholar] [CrossRef]
- Fu, C.; Shi, S.; Tian, J.; Gu, H.; Yao, L.; Xiao, J. Non-denatured yak type I collagen accelerates sunburned skin healing by stimulating and replenishing dermal collagen. Biotechnol. Rep. 2023, 37, e00778. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Ginhoux, F.; Collin, M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat. Rev. Immunol. 2008, 8, 935–947. [Google Scholar] [CrossRef]
- Brombin, A.; Patton, E.E. Melanocyte lineage dynamics in development, growth and disease. Development 2024, 151, dev201266. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Long, X.; Zhao, Q.; Zheng, Y.; Song, M.; Ma, S.; Jing, Y.; Wang, S.; He, Y.; Esteban, C.R.; et al. A Single-Cell Transcriptomic Atlas of Human Skin Aging. Dev. Cell 2021, 56, 383–397.e8. [Google Scholar] [CrossRef]
- Sauermann, K.; Clemann, S.; Jaspers, S.; Gambichler, T.; Altmeyer, P.; Hoffmann, K.; Ennen, J. Age related changes of human skin investigated with histometric measurements by confocal laser scanning microscopy in vivo. Ski. Res. Technol. 2002, 8, 52–56. [Google Scholar] [CrossRef]
- He, X.; Gao, X.; Xie, W. Research Progress in Skin Aging and Immunity. Int. J. Mol. Sci. 2024, 25, 4101. [Google Scholar] [CrossRef]
- Streilein, J.W. Skin-associated lymphoid tissues (SALT): Origins and functions. J. Investig. Dermatol. 1983, 80, 12s–16s. [Google Scholar] [CrossRef]
- Russell-Goldman, E.; Murphy, G.F. The Pathobiology of Skin Aging: New Insights into an Old Dilemma. Am. J. Pathol. 2020, 190, 1356–1369. [Google Scholar] [CrossRef]
- Yang, X.; Cui, Y.; Yue, J.; He, H.; Yu, C.; Liu, P.; Liu, J.; Ren, X.; Meng, Y. The histological characteristics, age-related thickness change of skin, and expression of the HSPs in the skin during hair cycle in yak (Bos grunniens). PLoS ONE 2017, 12, e0176451. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ouyang, Y.; Hu, Y.; Jiang, L.; Fu, C.; Lei, L.; Zhang, Y.; Guo, H.; Huang, J.; Chen, J.; et al. Macrophage migration inhibitory factor mediates skin aging via CD74: Insights from single-cell and bulk RNA sequencing data. Clin. Immunol. 2024, 263, 110199. [Google Scholar] [CrossRef]
- Shen, Z.; Chang, C.; Yeh, C.; Lu, C.; Hung, H.; Wang, T.; Wu, K.; Tung, C.; Tsai, T. Hesperetin activates CISD2 to attenuate senescence in human keratinocytes from an older person and rejuvenates naturally aged skin in mice. J. Biomed. Sci. 2024, 31, 15. [Google Scholar] [CrossRef]
- Kimball, A.B.; Alora-Palli, M.B.; Tamura, M.; Mullins, L.A.; Soh, C.; Binder, R.L.; Houston, N.A.; Conley, E.D.; Tung, J.Y.; Annunziata, N.E.; et al. Age-induced and photoinduced changes in gene expression profiles in facial skin of Caucasian females across 6 decades of age. J. Am. Acad. Dermatol. 2018, 78, 29–39.e7. [Google Scholar] [CrossRef]
- Liu, F.; Hamer, M.A.; Deelen, J.; Lall, J.S.; Jacobs, L.; Heemst, D.; Murray, P.G.; Wollstein, A.; Craen, A.J.; Uh, H.W.; et al. The MC1R Gene and Youthful Looks. Curr. Biol. 2016, 26, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Bao, P.; Luo, J.; Liu, Y.; Chu, M.; Ren, Q.; Guo, X.; Tang, B.; Ding, X.; Qiu, Q.; Pan, H.; et al. The seasonal development dynamics of the yak hair cycle transcriptome. BMC Genom. 2020, 21, 355. [Google Scholar] [CrossRef]
- Nocelli, C.; Cappelli, K.; Capomaccio, S.; Pascucci, L.; Mercati, F.; Pazzaglia, I.; Mecocci, S.; Antonini, M.; Renieri, C. Shedding light on cashmere goat hair follicle biology: From morphology analyses to transcriptomic landascape. BMC Genom. 2020, 21, 458. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ding, H.; Wang, Y.; Wang, X.; Zhao, H. Integration Analysis of Hair Follicle Transcriptome and Proteome Reveals the Mechanisms Regulating Wool Fiber Diameter in Angora Rabbits. Int. J. Mol. Sci. 2024, 25, 3260. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, F.; Jin, H.; Dalrymple, B.P.; Cao, Y.; Wei, T.; Vuocolo, T.; Zhang, M.; Piao, Q.; Ingham, A.B. A comparison of transcriptomic patterns measured in the skin of Chinese fine and coarse wool sheep breeds. Sci. Rep. 2017, 7, 14301. [Google Scholar] [CrossRef]
- Wu, C.; Ma, S.; Zhao, B.; Qin, C.; Wu, Y.; Di, J.; Suo, L.; Fu, X. Drivers of plateau adaptability in cashmere goats revealed by genomic and transcriptomic analyses. BMC Genom. 2023, 24, 428. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Zouboulis, C.C. The skin as a mirror of the aging process in the human organism--state of the art and results of the aging research in the German National Genome Research Network 2 (NGFN-2). Exp. Gerontol. 2007, 42, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Usansky, I.; Jaworska, P.; Asti, L.; Kenny, F.N.; Hobbs, C.; Sofra, V.; Song, H.; Logan, M.; Graham, A.; Shaw, T.J. A developmental basis for the anatomical diversity of dermis in homeostasis and wound repair. J. Pathol. 2021, 253, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Boismal, F.; Serror, K.; Dobos, G.; Zuelgaray, E.; Bensussan, A.; Michel, L. Skin aging: Pathophysiology and innovative therapies. Med. Sci. 2020, 36, 1163–1172. [Google Scholar]
- Solá, P.; Mereu, E.; Bonjoch, J.; Casado-Peláez, M.; Prats, N.; Aguilera, M.; Reina, O.; Blanco, E.; Esteller, M.; Di Croce, L.; et al. Targeting lymphoid-derived IL-17 signaling to delay skin aging. Nat. Aging 2023, 3, 688–704. [Google Scholar] [CrossRef]
- Sassi, F.; Tamone, C.; D’Amelio, P. Vitamin D: Nutrient, Hormone, and Immunomodulator. Nutrients 2018, 10, 1656. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D and the skin: Physiology and pathophysiology. Rev. Endocr. Metab. Disord. 2012, 13, 3–19. [Google Scholar] [CrossRef]
- Salzer, M.C.; Lafzi, A.; Berenguer-Llergo, A.; Youssif, C.; Castellanos, A.; Solanas, G.; Peixoto, F.O.; Stephan-Otto, A.C.; Prats, N.; Aguilera, M.; et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018, 175, 1575–1590.e22. [Google Scholar] [CrossRef]
- Stelnicki, E.J.; Kömüves, L.G.; Kwong, A.O.; Holmes, D.; Klein, P.; Rozenfeld, S.; Lawrence, H.J.; Adzick, N.S.; Harrison, M.; Largman, C. HOX homeobox genes exhibit spatial and temporal changes in expression during human skin development. J. Investig. Dermatol. 1998, 110, 110–115. [Google Scholar] [CrossRef]
- Scott, G.A.; Goldsmith, L.A. Homeobox genes and skin development: A review. J. Investig. Dermatol. 1993, 101, 3–8. [Google Scholar] [CrossRef]
- Makpol, S.; Jam, F.A.; Rahim, N.A.; Khor, S.C.; Ismail, Z.; Yusof, Y.A.; Ngah, W.Z.W. Comparable down-regulation of TYR, TYRP1 and TYRP2 genes and inhibition of melanogenesis by tyrostat, tocotrienol-rich fraction and tocopherol in human skin melanocytes improves skin pigmentation. Clin. Ter. 2014, 165, e39–e45. [Google Scholar]
- Shintani, A.; Sakata-Haga, H.; Moriguchi, K.; Tomosugi, M.; Sakai, D.; Tsukada, T.; Taniguchi, M.; Asano, M.; Shimada, H.; Otani, H.; et al. MC5R Contributes to Sensitivity to UVB Waves and Barrier Function in Mouse Epidermis. JID Innov. 2021, 1, 100024. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.W.; Barrett, J.N.P.; Tong, J.; Lin, M.J.; Marohn, M.; Devlin, J.C.; Herrera, A.; Remark, J.; Levine, J.; Liu, P.K.; et al. Skin immune-mesenchymal interplay within tertiary lymphoid structures promotes autoimmune pathogenesis in hidradenitis suppurativa. Immunity 2024, 57, 2827–2842.e5. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Moran, B.; Petrasca, A.; Smith, C.M. IL-17 in inflammatory skin diseases psoriasis and hidradenitis suppurativa. Clin. Exp. Immunol. 2020, 201, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Dawes, J.M.; Antunes-Martins, A.; Perkins, J.R.; Paterson, K.J.; Sisignano, M.; Schmid, R.; Rust, W.; Hildebrandt, T.; Geisslinger, G.; Orengo, C.; et al. Genome-wide transcriptional profiling of skin and dorsal root ganglia after ultraviolet-B-induced inflammation. PLoS ONE 2014, 9, e93338. [Google Scholar] [CrossRef]
- McKittrick, J.; Chen, P.Y.; Bodde, S.G.; Yang, W.; Novitskaya, E.E.; Meyers, M.A. The Structure, Functions, and Mechanical Properties of Keratin. JOM 2012, 64, 449–468. [Google Scholar] [CrossRef]
- Mauro, T.; Bikle, D. Skin and bone crosstalk during aging. Nat. Aging 2022, 2, 874–875. [Google Scholar] [CrossRef]
- Waluk, D.P.; Schultz, N.; Hunt, M.C. Identification of glycine N-acyltransferase-like 2 (GLYATL2) as a transferase that produces N-acyl glycines in humans. FASEB J. 2010, 24, 2795–2803. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Gao, Y.; Lin, J.; Liu, F.; Yang, L.; Zhou, J.; Xue, Y.; Li, Y.; Chang, Z.; Li, J.; et al. Dietary elaidic acid boosts tumoral antigen presentation and cancer immunity via ACSL5. Cell Metab. 2024, 36, 822–838.e8. [Google Scholar] [CrossRef]
- Dubaissi, E.; Hilton, E.N.; Lilley, S.; Collins, R.; Holt, C.; March, P.; Danahay, H.; Gosling, M.; Grencis, R.K.; Roberts, I.S.; et al. The Tmem16a chloride channel is required for mucin maturation after secretion from goblet-like cells in the Xenopus tropicalis tadpole skin. Sci. Rep. 2024, 14, 25555. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, Q.; Jiang, D.; Yu, K.; Yu, W.; Chi, Z.; Chen, S.; Li, M.; Yang, D.; Wang, Z.; et al. Epithelial Gasdermin D shapes the host-microbial interface by driving mucus layer formation. Sci. Immunol. 2022, 7, eabk2092. [Google Scholar] [CrossRef]
- Potter, C.S.; Pruett, N.D.; Kern, M.J.; Baybo, M.A.; Godwin, A.R.; Potter, K.A.; Peterson, R.L.; Sundberg, J.P.; Awgulewitsch, A. The nude mutant gene Foxn1 is a HOXC13 regulatory target during hair follicle and nail differentiation. J. Investig. Dermatol. 2011, 131, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bao, P.; Ye, N.; Zhou, X.; Zhang, Y.; Liang, C.; Guo, X.; Chu, M.; Pei, J.; Yan, P. Identification of the Key Genes Associated with the Yak Hair Follicle Cycle. Genes 2021, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.J.; Benketah, A.; Olivero, C.; Rees, E.; Ziaj, S.; Mukhtar, A.; Lanfredini, S.; Patel, G.K. Hair follicle differentiation-specific keratin expression in human basal cell carcinoma. Clin. Exp. Dermatol. 2020, 45, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gudjonsson, J.E.; Woods, T.L.; Zhang, T.; Johnston, A.; Stoll, S.W.; Elder, J.T. Transgenic expression of S100A2 in hairless mouse skin enhances Cxcl13 mRNA in response to solar-simulated radiation. Arch. Dermatol. Res. 2009, 301, 205–217. [Google Scholar] [CrossRef]
- Cohen, E.; Johnson, C.N.; Wasikowski, R.; Billi, A.C.; Tsoi, L.C.; Kahlenberg, J.M.; Gudjonsson, J.E. Coulombe PA Significance of stress keratin expression in normal and diseased epithelia. iScience 2024, 27, 108805. [Google Scholar] [CrossRef]
- Singh, B.; Coffey, R.J. From wavy hair to naked proteins: The role of transforming growth factor alpha in health and disease. Semin. Cell Dev. Biol. 2014, 28, 12–21. [Google Scholar] [CrossRef]
- Guo, C.J.; Mack, M.R.; Oetjen, L.K.; Trier, A.M.; Council, M.L.; Pavel, A.B.; Guttman-Yassky, E.; Kim, B.S.; Liu, Q. Kallikrein 7 Promotes Atopic Dermatitis-Associated Itch Independently of Skin Inflammation. J. Investig. Dermatol. 2020, 140, 1244–1252.e4. [Google Scholar] [CrossRef]
- Zou, Q.; Yuan, R.; Zhang, Y.; Wang, Y.; Zheng, T.; Shi, R.; Zhang, M.; Li, Y.; Fei, K.; Feng, R.; et al. A single-cell transcriptome atlas of pig skin characterizes anatomical positional heterogeneity. eLife 2023, 12, e86504. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Shi, B.; Zhao, Z.; Deng, Y.; Zhou, X.; Hu, J. Deciphering the Transcriptomic Complexity of Yak Skin Across Different Ages and Body Sites. Int. J. Mol. Sci. 2025, 26, 4601. https://doi.org/10.3390/ijms26104601
Zhang X, Shi B, Zhao Z, Deng Y, Zhou X, Hu J. Deciphering the Transcriptomic Complexity of Yak Skin Across Different Ages and Body Sites. International Journal of Molecular Sciences. 2025; 26(10):4601. https://doi.org/10.3390/ijms26104601
Chicago/Turabian StyleZhang, Xiaolan, Bingang Shi, Zhidong Zhao, Yunqi Deng, Xuelan Zhou, and Jiang Hu. 2025. "Deciphering the Transcriptomic Complexity of Yak Skin Across Different Ages and Body Sites" International Journal of Molecular Sciences 26, no. 10: 4601. https://doi.org/10.3390/ijms26104601
APA StyleZhang, X., Shi, B., Zhao, Z., Deng, Y., Zhou, X., & Hu, J. (2025). Deciphering the Transcriptomic Complexity of Yak Skin Across Different Ages and Body Sites. International Journal of Molecular Sciences, 26(10), 4601. https://doi.org/10.3390/ijms26104601