
Exploring the Phenotypic Heterogeneity and Bioenergetic Profile of the m.13513G>A mtDNA Substitution: A Heteroplasmy Perspective

 , , ,
, , ,  , ,
, ,  , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
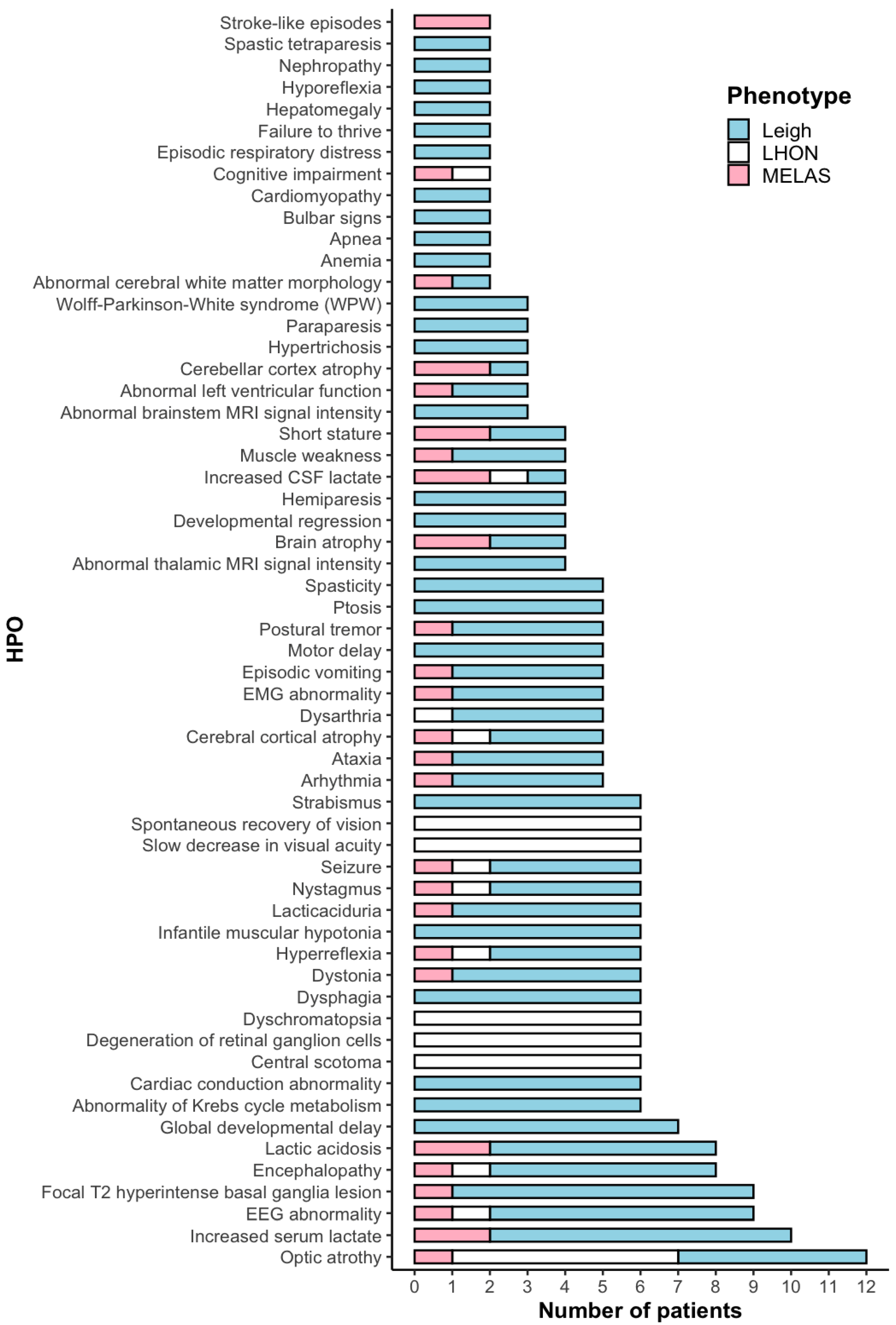
2.1. Clinical Data
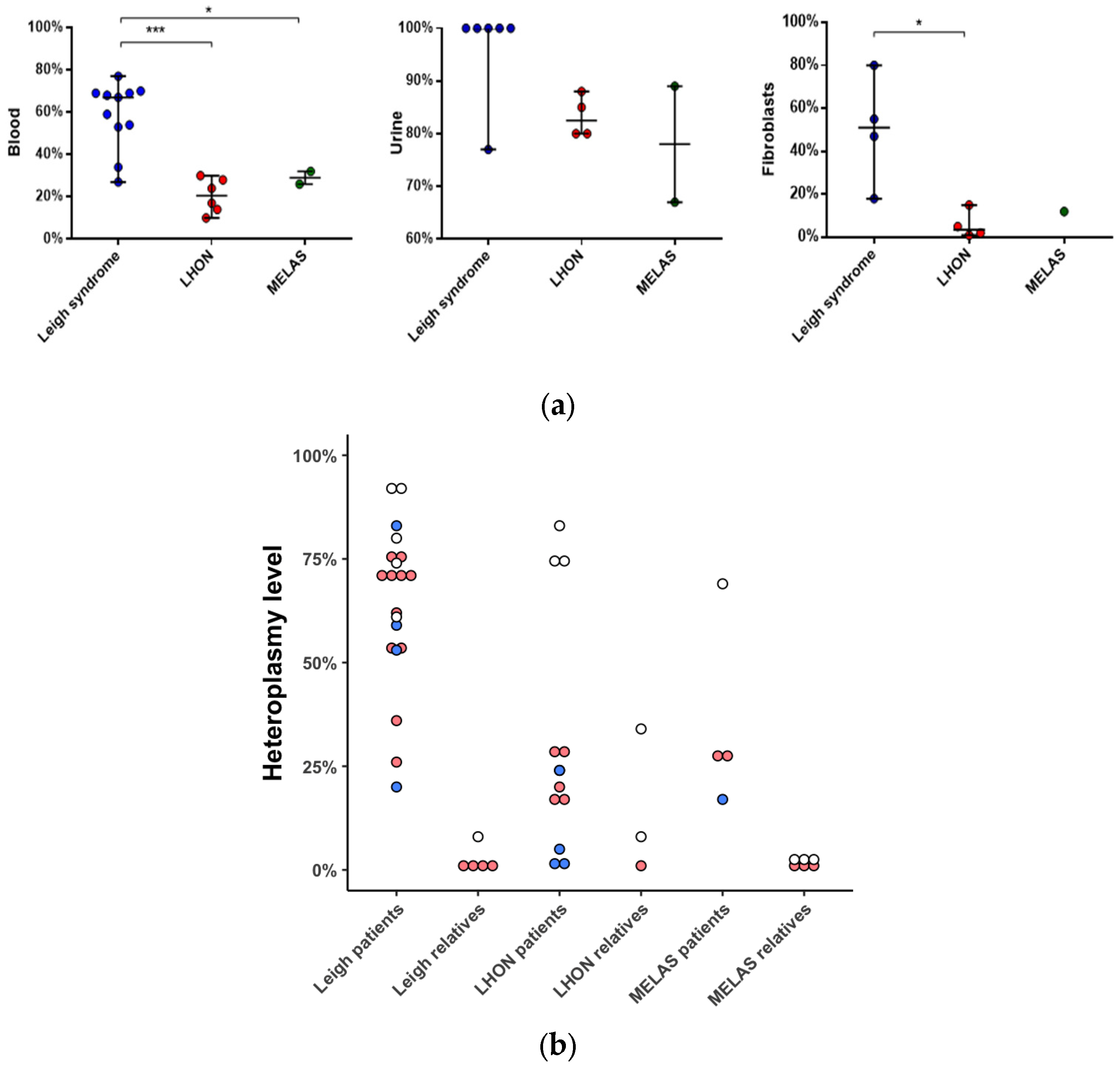
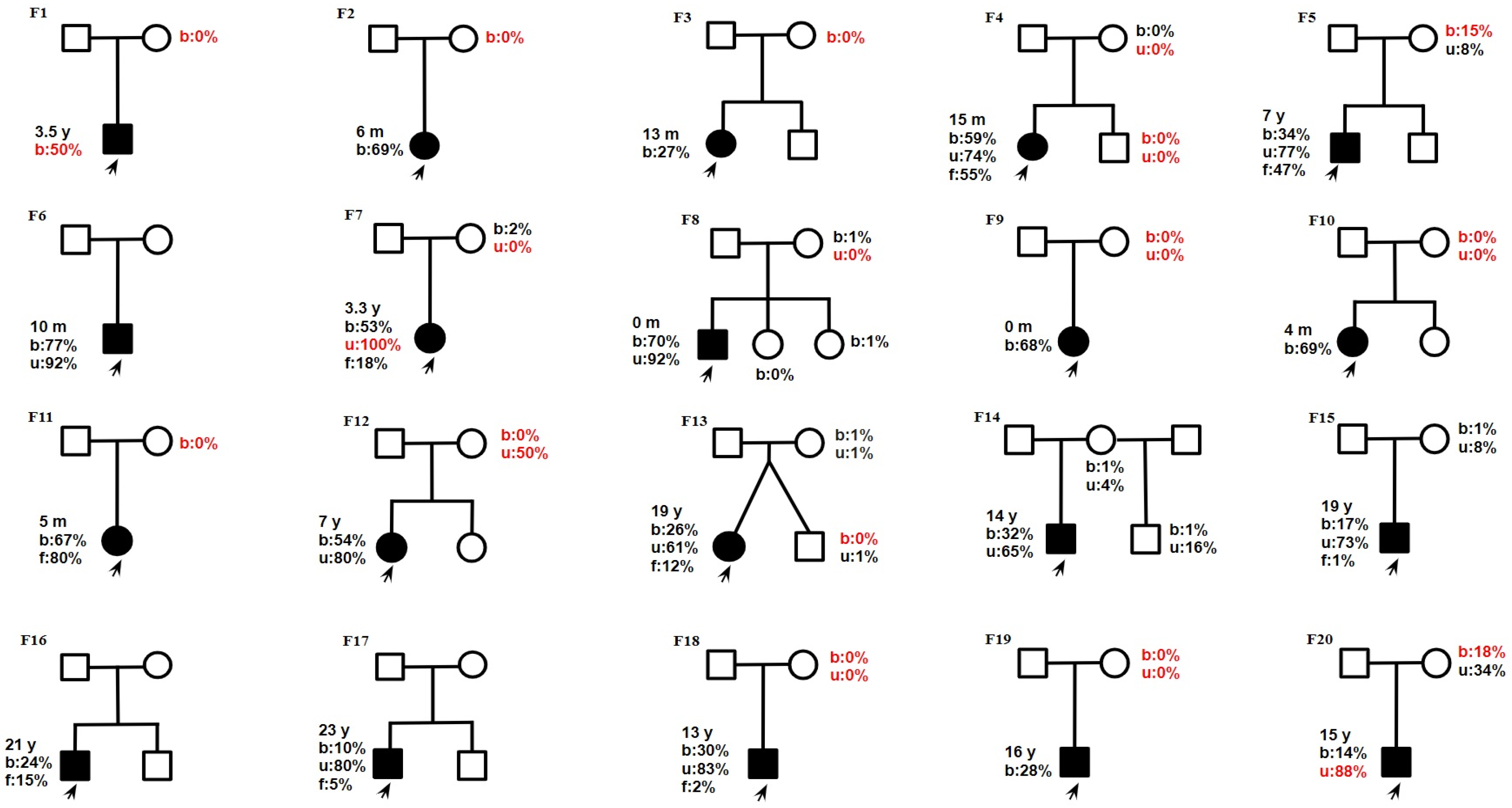
2.2. Molecular Data
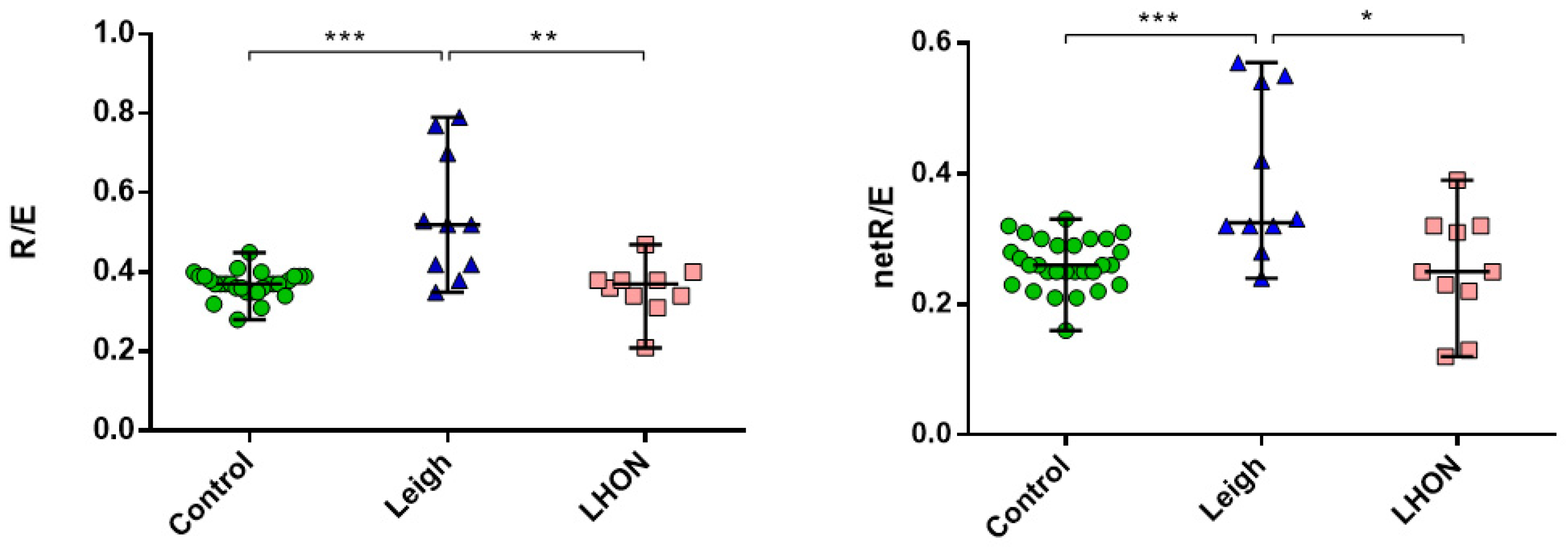
2.3. Mitochondrial Bioenergetics
3. Discussion
4. Materials and Methods
4.1. Editorial Policies and Ethical Considerations
4.2. Patients and Samples
4.3. Heteroplasmy Detection
4.4. Sanger Sequencing
4.5. Massive Parallel Sequencing of the Whole mtDNA
4.6. Amplicon Deep Sequencing
4.7. Cell Lines
4.8. High-Resolution Respirometry
4.9. Mitochondrial Membrane Potential
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PMD | Primary mitochondrial disorder |
| mtDNA | Mitochondrial Deoxyribonucleic acid |
| MELAS | Mitochondrial Encephalomyopathy with Lactate Acidosis and Stroke-like episodes |
| LHON | Leber’s Hereditary Optic Neuropathy |
| OXPHOS | Oxidative phosphorylation |
| DNA | Deoxyribonucleic acid |
References
- Valenti, D.; Vacca, R. Primary and Secondary Mitochondrial Diseases: Etiologies and Therapeutic Strategies. J. Clin. Med. 2022, 11, 4209. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.H.; Willems, P.H.G.M.; Smeitink, J.A.M. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Mitochondrial Complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of Mitochondrial Diseases: Identifying Mutations to Help Diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef] [PubMed]
- Fassone, E.; Rahman, S. Complex I Deficiency: Clinical Features, Biochemistry and Molecular Genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef]
- Scheffler, I.E. Mitochondrial Disease Associated with Complex I (NADH-CoQ Oxidoreductase) Deficiency. J. Inherit. Metab. Dis. 2015, 38, 405–415. [Google Scholar] [CrossRef]
- Rodenburg, R.J. Mitochondrial Complex I-Linked Disease. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1857, 938–945. [Google Scholar] [CrossRef]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr. Protoc. Bioinforma. 2013, 44, 1.23.1-26. [Google Scholar] [CrossRef]
- Ng, Y.S.; Lax, N.Z.; Maddison, P.; Alston, C.L.; Blakely, E.L.; Hepplewhite, P.D.; Riordan, G.; Meldau, S.; Chinnery, P.F.; Pierre, G.; et al. MT-ND5 Mutation Exhibits Highly Variable Neurological Manifestations at Low Mutant Load. EBioMedicine 2018, 30, 86–93. [Google Scholar] [CrossRef]
- Santorelli, F.M.; Tanji, K.; Kulikova, R.; Shanske, S.; Vilarinho, L.; Hays, A.P.; DiMauro, S. Identification of a Novel Mutation in the mtDNA ND5 Gene Associated with MELAS. Biochem. Biophys. Res. Commun. 1997, 238, 326–328. [Google Scholar] [CrossRef]
- Finsterer, J. Phenotypic Heterogeneity of the Mitochondrial DNA Variant m.13513 G > A. J. Pediatr. Genet. 2024, 13, 253–257. [Google Scholar] [CrossRef]
- Na, J.-H.; Lee, Y.-M. Heteroplasmic Mutant Load Differences in Mitochondrial DNA-Associated Leigh Syndrome. Pediatr. Neurol. 2023, 138, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; Boneh, A.; Chow, C.W.; Ohtake, A.; Ryan, M.T.; Thyagarajan, D.; Thorburn, D.R. Low Mutant Load of Mitochondrial DNA G13513A Mutation Can Cause Leigh’s Disease. Ann. Neurol. 2003, 54, 473–478. [Google Scholar] [CrossRef]
- Krylova, T.D.; Sheremet, N.L.; Tabakov, V.Y.; Lyamzaev, K.G.; Itkis, Y.S.; Tsygankova, P.G.; Andreeva, N.A.; Shmelkova, M.S.; Nevinitsyna, T.A.; Kadyshev, V.V.; et al. Three Rare Pathogenic mtDNA Substitutions in LHON Patients with Low Heteroplasmy. Mitochondrion 2020, 50, 139–144. [Google Scholar] [CrossRef]
- Gargano, M.A.; Matentzoglu, N.; Coleman, B.; Addo-Lartey, E.B.; Anagnostopoulos, A.V.; Anderton, J.; Avillach, P.; Bagley, A.M.; Bakštein, E.; Balhoff, J.P.; et al. The Human Phenotype Ontology in 2024: Phenotypes around the World. Nucleic Acids Res. 2024, 52, D1333–D1346. [Google Scholar] [CrossRef]
- Rahman, J.; Noronha, A.; Thiele, I.; Rahman, S. Leigh Map: A Novel Computational Diagnostic Resource for Mitochondrial Disease. Ann. Neurol. 2017, 81, 9–16. [Google Scholar] [CrossRef]
- Eliseeva, D.D.; Kalashnikova, A.K.; Bryukhov, V.V.; Andreeva, N.A.; Zhorzholadze, N.V.; Murakhovskaya, Y.K.; Krilova, T.D.; Tsygankova, P.G.; Zakharova, M.N.; Sheremet, N.L. Hereditary Optic Neuropathy Associated with Demyelinating Diseases of the Central Nervous System. Zhurnal Nevrol. Psikhiatrii Im SS Korsakova 2023, 123, 122. [Google Scholar] [CrossRef] [PubMed]
- Frazier, A.E.; Thorburn, D.R.; Compton, A.G. Mitochondrial Energy Generation Disorders: Genes, Mechanisms, and Clues to Pathology. J. Biol. Chem. 2019, 294, 5386–5395. [Google Scholar] [CrossRef] [PubMed]
- Kistol, D.; Tsygankova, P.; Krylova, T.; Bychkov, I.; Itkis, Y.; Nikolaeva, E.; Mikhailova, S.; Sumina, M.; Pechatnikova, N.; Kurbatov, S.; et al. Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia. Int. J. Mol. Sci. 2023, 24, 1597. [Google Scholar] [CrossRef]
- Ardissone, A.; Ferrera, G.; Lamperti, C.; Tiranti, V.; Ghezzi, D.; Moroni, I.; Lamantea, E. Phenotyping Mitochondrial DNA-related Diseases in Childhood: A Cohort Study of 150 Patients. Eur. J. Neurol. 2023, 30, 2079–2091. [Google Scholar] [CrossRef]
- Pelnena, D.; Burnyte, B.; Jankevics, E.; Lace, B.; Dagyte, E.; Grigalioniene, K.; Utkus, A.; Krumina, Z.; Rozentale, J.; Adomaitiene, I.; et al. Complete mtDNA Sequencing Reveals Mutations m.9185T>C and m.13513G>A in Three Patients with Leigh Syndrome. Mitochondrial DNA Part A 2018, 29, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.-M.; Xin, C.-J.; Wang, G.-L.; Wu, X.-M. Case Report: M.13513 G>A Mutation in a Chinese Patient with Both Leigh Syndrome and Wolff-Parkinson-White Syndrome. Front. Pediatr. 2021, 9, 700898. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, E.; Shimura, M.; Fushimi, T.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; Yamazaki, T.; et al. Clinical Validity of Biochemical and Molecular Analysis in Diagnosing Leigh Syndrome: A Study of 106 Japanese Patients. J. Inherit. Metab. Dis. 2017, 40, 685–693. [Google Scholar] [CrossRef]
- Brautbar, A.; Wang, J.; Abdenur, J.E.; Chang, R.C.; Thomas, J.A.; Grebe, T.A.; Lim, C.; Weng, S.-W.; Graham, B.H.; Wong, L.-J. The Mitochondrial 13513G>A Mutation Is Associated with Leigh Disease Phenotypes Independent of Complex I Deficiency in Muscle. Mol. Genet. Metab. 2008, 94, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, E.M.; Siers, M.H.; Van Den Elzen, C.; Van Engelen, B.G.; Smeitink, J.A.M.; Rodenburg, R.J.; Hol, F.A. The Mitochondrial 13513G>A Mutation Is Most Frequent in Leigh Syndrome Combined with Reduced Complex I Activity, Optic Atrophy and/or Wolff–Parkinson–White. Eur. J. Hum. Genet. 2007, 15, 155–161. [Google Scholar] [CrossRef]
- Sun, C.; Bai, H.; Xu, D.; Xiao, Q.; Liu, Z. Mitochondrial 13513G>A Mutation with Low Mutant Load Presenting as Isolated Leber’s Hereditary Optic Neuropathy Assessed by Next Generation Sequencing. Front. Neurol. 2021, 12, 601307. [Google Scholar] [CrossRef]
- Vázquez-Justes, D.; Carreño-Gago, L.; García-Arumi, E.; Traveset, A.; Montoya, J.; Ruiz-Pesini, E.; López, R.; Brieva, L. Mitochondrial m.13513G>A Point Mutation in ND5 in a 16-Year-Old Man with Leber Hereditary Optic Neuropathy Detected by Next-Generation Sequencing. J. Pediatr. Genet. 2019, 08, 231–234. [Google Scholar] [CrossRef]
- Hsieh, Y.-T.; Yang, M.-T.; Peng, Y.-J.; Hsu, W.-C. Central Retinal Vein Occlusion as the Initial Manifestation of LHON / MELAS Overlap Syndrome with Mitochondrial DNA G13513A Mutation—Case Report and Literature Review. Ophthalmic Genet. 2011, 32, 31–38. [Google Scholar] [CrossRef]
- Barone, V.; La Morgia, C.; Caporali, L.; Fiorini, C.; Carbonelli, M.; Gramegna, L.L.; Bartiromo, F.; Tonon, C.; Morandi, L.; Liguori, R.; et al. Case Report: Optic Atrophy and Nephropathy with m.13513G>A/MT-ND5 mtDNA Pathogenic Variant. Front. Genet. 2022, 13, 887696. [Google Scholar] [CrossRef]
- Bakis, H.; Trimouille, A.; Vermorel, A.; Redonnet, I.; Goizet, C.; Boulestreau, R.; Lacombe, D.; Combe, C.; Martin-Négrier, M.; Rigothier, C. Adult Onset Tubulo-interstitial Nephropathy in MT-ND5-related Phenotypes. Clin. Genet. 2020, 97, 628–633. [Google Scholar] [CrossRef]
- Naganuma, T.; Imasawa, T.; Nukui, I.; Wakasugi, M.; Kitamura, H.; Yatsuka, Y.; Kishita, Y.; Okazaki, Y.; Murayama, K.; Jinguji, Y. Focal Segmental Glomerulosclerosis with a Mutation in the Mitochondrially Encoded NADH Dehydrogenase 5 Gene: A Case Report. Mol. Genet. Metab. Rep. 2023, 35, 100963. [Google Scholar] [CrossRef]
- Piotrowska-Nowak, A.; Krawczyński, M.R.; Kosior-Jarecka, E.; Ambroziak, A.M.; Korwin, M.; Ołdak, M.; Tońska, K.; Bartnik, E. Mitochondrial Genome Variation in Male LHON Patients with the m.11778G > A Mutation. Metab. Brain Dis. 2020, 35, 1317–1327. [Google Scholar] [CrossRef]
- Liutkeviciene, R.; Sidaraite, A.; Kuliaviene, L.; Glebauskiene, B.; Jurkute, N.; Aluzaite-Baranauskiene, L.; Gelzinis, A.; Zemaitiene, R. A Typical Case Presentation with Spontaneous Visual Recovery in Patient Diagnosed with Leber Hereditary Optic Neuropathy Due to Rare Point Mutation in MT-ND4 Gene (m.11253T>C) and Literature Review. Medicina 2021, 57, 202. [Google Scholar] [CrossRef]
- Chen, B.S.; Biousse, V.; Newman, N.J. Mitochondrial DNA 13513G>A Mutation Presenting with Leber’s Hereditary Optic Neuropathy. Clin. Experiment. Ophthalmol. 2019, 47, 1202–1204. [Google Scholar] [CrossRef] [PubMed]
- Legati, A.; Ghezzi, D.; Viscomi, C. Mitochondrial DNA Sequencing and Heteroplasmy Quantification by Next Generation Sequencing. In Mitochondrial DNA; Nicholls, T.J., Uhler, J.P., Falkenberg, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2023; Volume 2615, pp. 381–395. ISBN 978-1-07-162921-5. [Google Scholar]
- Wei, Y.; Huang, Y.; Yang, Y.; Qian, M. MELAS/LS Overlap Syndrome Associated with Mitochondrial DNA Mutations: Clinical, Genetic, and Radiological Studies. Front. Neurol. 2021, 12, 648740. [Google Scholar] [CrossRef] [PubMed]
- Loos, M.A.; Gomez, G.; Mayorga, L.; Caraballo, R.H.; Eiroa, H.D.; Obregon, M.G.; Rugilo, C.; Lubieniecki, F.; Taratuto, A.L.; Saccoliti, M.; et al. Clinical and Molecular Characterization of Mitochondrial DNA Disorders in a Group of Argentinian Pediatric Patients. Mol. Genet. Metab. Rep. 2021, 27, 100733. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA Heteroplasmy in Disease and Targeted Nuclease-based Therapeutic Approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef]
- Tsyba, N.; Feng, G.; Grub, L.K.; Held, J.P.; Strozak, A.M.; Burkewitz, K.; Patel, M.R. Tissue-Specific Heteroplasmy Segregation Is Accompanied by a Sharp mtDNA Decline in Caenorhabditis Elegans Soma. iScience 2023, 26, 106349. [Google Scholar] [CrossRef]
- Balmaceda, V.; Komlódi, T.; Szibor, M.; Gnaiger, E.; Moore, A.L.; Fernandez-Vizarra, E.; Viscomi, C. The Striking Differences in the Bioenergetics of Brain and Liver Mitochondria Are Enhanced in Mitochondrial Disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2024, 1870, 167033. [Google Scholar] [CrossRef]
- Baker, Z.N.; Forny, P.; Pagliarini, D.J. Mitochondrial Proteome Research: The Road Ahead. Nat. Rev. Mol. Cell Biol. 2024, 25, 65–82. [Google Scholar] [CrossRef]
- Folmes, C.D.L.; Martinez-Fernandez, A.; Perales-Clemente, E.; Li, X.; Mcdonald, A.; Oglesbee, D.; Hrstka, S.C.; Perez-Terzic, C.; Terzic, A.; Nelson, T.J. Disease-Causing Mitochondrial Heteroplasmy Segregated Within Induced Pluripotent Stem Cell Clones Derived from a Patient with MELAS. Stem Cells 2013, 31, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Kidere, D.; Zayakin, P.; Livcane, D.; Makrecka-Kuka, M.; Stavusis, J.; Lace, B.; Lin, T.-K.; Liou, C.-W.; Inashkina, I. Impact of the m.13513G>A Variant on the Functions of the OXPHOS System and Cell Retrograde Signaling. Curr. Issues Mol. Biol. 2023, 45, 1794–1809. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Ji, Y.; Zhang, L.; Wang, X.; Hu, C.; Zhang, J.; Zhu, Y.; Mo, J.Q.; Guan, M.-X. Leber’s Hereditary Optic Neuropathy-Associated ND6 14484T > C Mutation Caused Pleiotropic Effects on the Complex I, RNA Homeostasis, Apoptosis and Mitophagy. Hum. Mol. Genet. 2022, 31, 3299–3312. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yao, S.; Yang, M.; Guo, Q.; Li, Y.; Li, L.; Lei, B. Superoxide Dismutase 2 Ameliorates Mitochondrial Dysfunction in Skin Fibroblasts of Leber’s Hereditary Optic Neuropathy Patients. Front. Neurosci. 2022, 16, 917348. [Google Scholar] [CrossRef]
- Burr, S.P.; Chinnery, P.F. Origins of Tissue and Cell-Type Specificity in Mitochondrial DNA (mtDNA) Disease. Hum. Mol. Genet. 2024, 33, R3–R11. [Google Scholar] [CrossRef]
- Grba, D.N.; Chung, I.; Bridges, H.R.; Agip, A.-N.A.; Hirst, J. Investigation of Hydrated Channels and Proton Pathways in a High-Resolution Cryo-EM Structure of Mammalian Complex I. Sci. Adv. 2023, 9, eadi1359. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Sazanov, L.A. Mammalian Mitochondrial Complex I Structure and Disease-Causing Mutations. Trends Cell Biol. 2018, 28, 835–867. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-Resolution Respirometry: OXPHOS Protocols for Human Cells and Permeabilized Fibers from Small Biopsies of Human Muscle. In Mitochondrial Bioenergetics; Palmeira, C.M., Moreno, A.J., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 810, pp. 25–58. ISBN 978-1-61779-381-3. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krylova, T.; Itkis, Y.; Tsygankova, P.; Chistol, D.; Lyamzaev, K.; Tabakov, V.; Mikhaylova, S.; Nikitina, N.; Rudenskaya, G.; Murtazina, A.; et al. Exploring the Phenotypic Heterogeneity and Bioenergetic Profile of the m.13513G>A mtDNA Substitution: A Heteroplasmy Perspective. Int. J. Mol. Sci. 2025, 26, 4565. https://doi.org/10.3390/ijms26104565
Krylova T, Itkis Y, Tsygankova P, Chistol D, Lyamzaev K, Tabakov V, Mikhaylova S, Nikitina N, Rudenskaya G, Murtazina A, et al. Exploring the Phenotypic Heterogeneity and Bioenergetic Profile of the m.13513G>A mtDNA Substitution: A Heteroplasmy Perspective. International Journal of Molecular Sciences. 2025; 26(10):4565. https://doi.org/10.3390/ijms26104565
Chicago/Turabian StyleKrylova, Tatiana, Yulia Itkis, Polina Tsygankova, Denis Chistol, Konstantin Lyamzaev, Vyacheslav Tabakov, Svetlana Mikhaylova, Natalia Nikitina, Galina Rudenskaya, Aysylu Murtazina, and et al. 2025. "Exploring the Phenotypic Heterogeneity and Bioenergetic Profile of the m.13513G>A mtDNA Substitution: A Heteroplasmy Perspective" International Journal of Molecular Sciences 26, no. 10: 4565. https://doi.org/10.3390/ijms26104565
APA StyleKrylova, T., Itkis, Y., Tsygankova, P., Chistol, D., Lyamzaev, K., Tabakov, V., Mikhaylova, S., Nikitina, N., Rudenskaya, G., Murtazina, A., Markova, T., Semenova, N., Buchinskaya, N., Saifullina, E., Aksyanova, H., Sparber, P., Andreeva, N., Venediktova, N., Ivanushkina, A., ... Zakharova, E. (2025). Exploring the Phenotypic Heterogeneity and Bioenergetic Profile of the m.13513G>A mtDNA Substitution: A Heteroplasmy Perspective. International Journal of Molecular Sciences, 26(10), 4565. https://doi.org/10.3390/ijms26104565