
Blood-Based Epigenetic Age Acceleration and Incident Colorectal Cancer Risk: Findings from a Population-Based Case–Control Study
 ,
,  ,
,
Abstract
1. Introduction
2. Results
2.1. General Baseline Characteristics of the Studied CRC and Control Groups
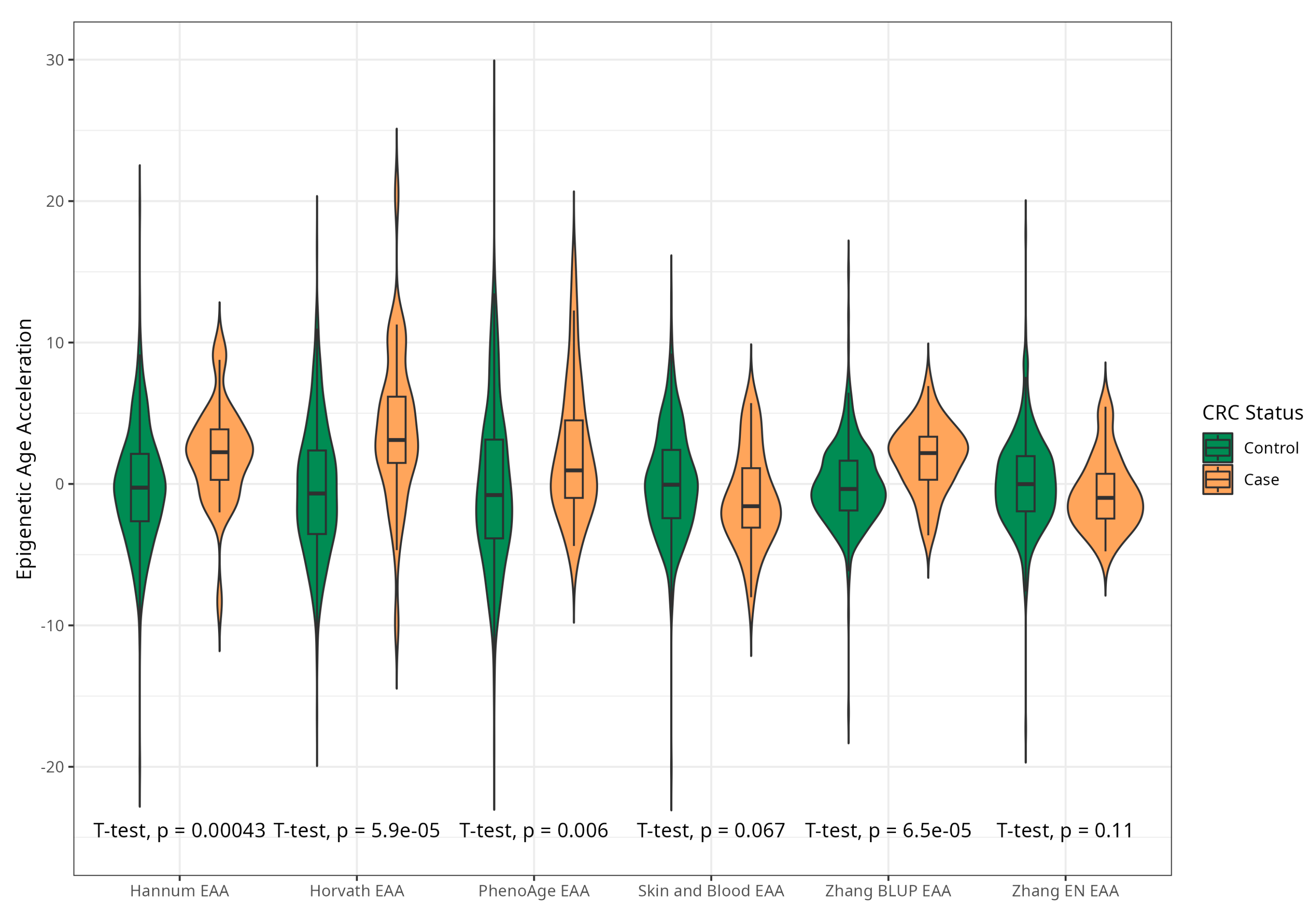
2.2. Association between Baseline Epigenetic Age Acceleration by Six Markers and Risk of CRC
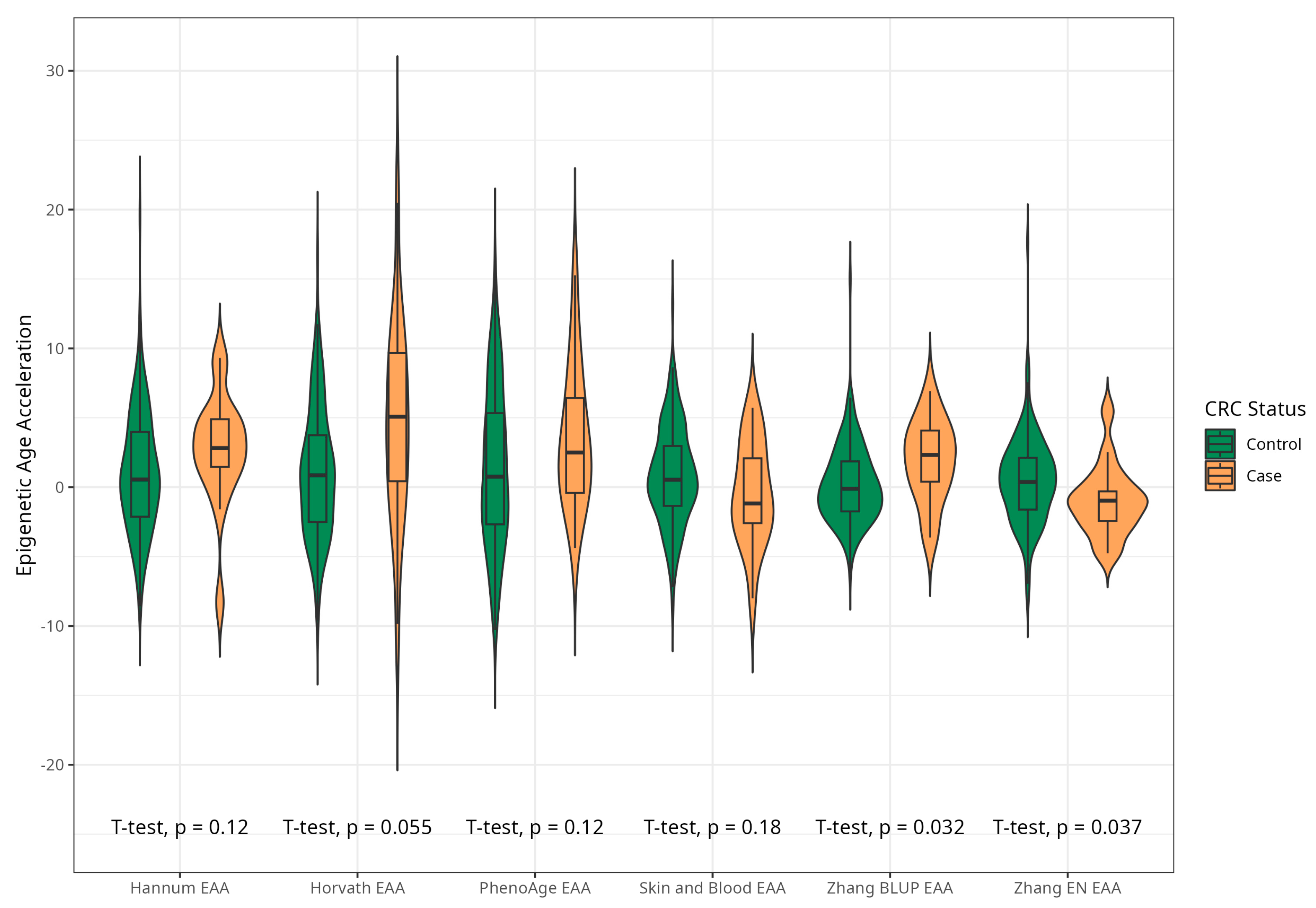
2.3. Sensitivity Analyses: Associations between Baseline Epigenetic Age Acceleration by Six Markers and Risk of CRC Excluding Early Cases and Against Extended Control
3. Discussion
3.1. The Relationship between Measures of Epigenetic Age Acceleration and CRC
3.2. Study Limitations and Strengths
4. Materials and Methods
4.1. Study Population and Design
4.2. Study Sample Selection
4.3. Data Collection
4.4. DNAm Data Profiling
4.5. DNAm Data Preprocessing and Quality Control (QC)
4.6. Epigenetic Age and Epigenetic Age Acceleration
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Prospects: The 2017 Revision, Key Findings and Advance Tables. Working Paper No. ESA/P/WP/248. 2017. Available online: https://population.un.org/wpp/publications/files/wpp2017_keyfindings.pdf (accessed on 20 February 2024).
- Centers for Disease Control and Prevention, National Center for Health Statistics. National Vital Statistics System, Mortality 2018–2021 on CDC WONDER Online Database, Released in 2021. Data Are from the Multiple Cause of Death Files, 2018–2021, as Compiled from Data Provided by the 57 Vital Statistics Jurisdictions through the Vital Statistics Cooperative Program. Available online: http://wonder.cdc.gov/ucd-icd10-expanded.html (accessed on 1 March 2024).
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef]
- Maugeri, A.; Barchitta, M.; Magnano San Lio, R.; Li Destri, G.; Agodi, A.; Basile, G. Epigenetic Aging and Colorectal Cancer: State of the Art and Perspectives for Future Research. Int. J. Mol. Sci. 2021, 22, 200. [Google Scholar] [CrossRef] [PubMed]
- GLOBOCAN Estimated Age-Standardized Cancer Incidence and Mortality Worldwide. 2020. Available online: http://globocan.iarc.fr/gco.iarc.fr/today/online-analysis-table?v=2020 (accessed on 20 February 2024).
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Sinicrope, F.A.; Brenner, D.E.; Carethers, J.M. Colorectal cancer prevention and treatment. Gastroenterology 2000, 118 (Suppl. S1), S115–S128. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Jonson, P.; Fadlalla, K.; Moore, A.; Huang, C.-H.; Berry, K.; Maxuitenko, Y.Y.; Chen, X.; Keeton, A.B.; Zhou, G.; et al. Novel sulindac derivatives for colorectal cancer chemoprevention that target cGMP phosphodiesterases to suppress Wnt/β-catenin transcriptional activity. Cancer Insight 2023, 3, 28. [Google Scholar] [CrossRef]
- Johnson, C.M.; Wei, C.; Ensor, J.E.; Smolenski, D.J.; Amos, C.I.; Levin, B.; Berry, D.A. Meta-analyses of Colorectal Cancer Risk Factors. Cancer Causes Control 2013, 24, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Botteri, E.; Borroni, E.; Sloan, E.K.; Bagnardi, V.; Bosetti, C.; Peveri, G.; Santucci, C.; Specchia, C.; van den Brandt, P.; Gallus, S.; et al. Smoking and Colorectal Cancer Risk, Overall and by Molecular Subtypes: A Meta-Analysis. Am. J. Gastroenterol. 2020, 115, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- McNabb, S.; Harrison, T.A.; Albanes, D.; Berndt, S.; Brenner, H.; Caan, B.J.; Campbell, P.T.; Cao, Y.; Chang-Claude, J.; Chan, A.; et al. Meta-analysis of 16 studies of the association of alcohol with colorectal cancer. Int. J. Cancer 2020, 146, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Nikitenko, T.M.; Shcherbakova, L.V.; Malyutina, S.K.; Mustafina, S.V.; Verevkin, E.G.; Ragino, Y.I.; Voytsitsky, V.E.; Pyatibratova, A.B.; Rymar, O.D. The metabolic syndrome as a risk factor for colorectal cancer. Obes. Metab. 2017, 14, 24–32. (In Russian) [Google Scholar] [CrossRef]
- Murphy, C.C.; Zaki, T.A. Changing epidemiology of colorectal cancer—birth cohort effects and emerging risk factors. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Jylhävä, J.; Pedersen, N.L.; Hägg, S. Biological age predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Berstein, F.M.; McCartney, D.L.; Lu, A.T.; Tsilidis, K.K.; Bouras, E.; Haycock, P.; Burrows, K.; Phipps, A.I.; Buchanan, D.D.; Cheng, I.; et al. Assessing the causal role of epigenetic clocks in the development of multiple cancers: A Mendelian randomization study. eLife 2022, 11, e75374. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [PubMed]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jockel, K.H.; Erbel, R.; Muhleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, W.; Lin, F.; Huang, Q.; Zhong, J.; Gao, H.; Song, Y.; Liang, H. DNA methylation profile is a quantitative measure of biological aging in children. Aging 2019, 11, 10031–10051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, O.; Vallerga, C.L.; Walker, R.M.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.; Fowdar, J.; Kennedy, M.; et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019, 11, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, A.; Kuh, D.; Paul, D.S.; Rakyan, V.K.; Leslie, R.D.; Zheng, S.C.; Widschwendter, M.; Beck, S.; Teschendorff, A.E. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016, 17, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Youn, A.; Wang, S. The MiAge Calculator: A DNA methylation-based mitotic age calculator of human tissue types. Epigenetics 2018, 13, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E. A comparison of epigenetic mitotic-like clocks for cancer risk prediction. Genome Med. 2020, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Oblak, L.; van der Zaag, J.; Higgins-Chen, A.T.; Levine, M.E.; Boks, M.P. A systematic review of biological, social and environmental factors associated with epigenetic clock acceleration. Ageing Res. Rev. 2021, 69, 101348. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef] [PubMed]
- Fransquet, P.D.; Wrigglesworth, J.; Woods, R.L.; Ernst, M.E.; Ryan, J. The epigenetic clock as a predictor of disease and mortality risk: A systematic review and meta-analysis. Clin. Epigenetics 2019, 11, 62. [Google Scholar] [CrossRef]
- Wang, C.; Ni, W.; Yao, Y.; Just, A.; Heiss, J.; Wei, Y.; GaoBrent, X.; Coull, A.; Kosheleva, A.; Baccarelli, A.A.; et al. DNA methylation-based biomarkers of age acceleration and all-cause death, myocardial infarction, stroke, and cancer in two cohorts: The NAS, and KORA F4. EBioMedicine 2021, 63, 103151. [Google Scholar] [CrossRef] [PubMed]
- Durso, D.F.; Bacalini, M.G.; Sala, C.; Pirazzini, C.; Marasco, E.; Bonafé, M.; do Valle, Í.; Gentilini, D.; Castellani, G.; Faria, A.M.C.; et al. Acceleration of leukocytes’ epigenetic age as an early tumor and sex-specific marker of breast and colorectal cancer. Oncotarget 2017, 8, 23237–23245. [Google Scholar] [CrossRef]
- Dugué, P.A.; Bassett, J.K.; Joo, J.E.; Jung, C.H.; Ming Wong, E.; Moreno-Betancur, M.; Schmidt, D.; Makalic, E.; Li, S.; Severi, G.; et al. DNA methylation-based biological aging and cancer risk and survival: Pooled analysis of seven prospective studies. Int. J. Cancer 2018, 142, 1611–1619. [Google Scholar] [CrossRef]
- Dugue, P.A.; Bassett, J.K.; Wong, E.M.; Joo, J.H.E.; Li, S.; Yu, C.; Schmidt, D.F.; Makalic, E.; Wong Doo, N.; Buchanan, D.D.; et al. Biological Aging Measures Based on Blood DNA Methylation and Risk of Cancer: A Prospective Study. JNCI Cancer Spectr. 2021, 5, pkaa109. [Google Scholar] [CrossRef]
- Widayati, T.A.; Schneider, J.; Panteleeva, K.; Chernysheva, E.; Hrbkova, N.; Beck, S.; Voloshin, V.; Chervova, O. Open access-enabled evaluation of epigenetic age acceleration in colorectal cancer and development of a classifier with diagnostic potential. Front. Genet. 2023, 14, 1258648. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Maden, S.K.; Luebeck, G.E.; Li, C.I.; Newcomb, P.A.; Ulrich, C.M.; Joo, J.E.; Buchanan, D.D.; Milne, R.L.; Southey, M.C.; et al. Dysfunctional epigenetic aging of the normal colon and colorectal cancer risk. Clin. Epigenetics 2020, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, Y.; Boakye, D.; Li, X.; Chang-Claude, J.; Hoffmeister, M.; Brenner, H. Whole blood DNA methylation aging markers predict colorectal cancer survival: A prospective cohort study. Clin. Epigenetics 2020, 12, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Hillary, R.F.; Stevenson, A.J.; McCartney, D.L.; Campbell, A.; Walker, R.M.; Howard, D.M.; Ritchie, C.W.; Horvath, S.; Hayward, C.; McIntosh, A.M.; et al. Epigenetic measures of ageing predict the prevalence and incidence of leading causes of death and disease burden. Clin. Epigenetics 2020, 12, 115. [Google Scholar] [CrossRef] [PubMed]
- Stefler, D.; Malyutina, S.; Maximov, V.; Orlov, P.; Ivanoschuk, D.; Nikitin, Y.; Gafarov, V.; Ryabikov, A.; Voevoda, M.; Bobak, M.; et al. Leukocyte telomere length and risk of coronary heart disease and stroke mortality: Prospective evidence from a Russian cohort. Sci. Rep. 2018, 8, 16627. [Google Scholar] [CrossRef]
- Maximov, V.; Malyutina, S.; Orlov, P.; Ivanoschuk, D.; Mikhailova, S.V.; Shapkina, M.Y.; Hubacek, J.; Holmes, M.; Bobak, M.; Voevoda, M. Copy Number of the Mitochondrial DNA of Leucocytes as an Aging Marker and Risk Factors for the Development of Age-Related Diseases in Humans. Adv. Gerontol. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Malyutina, S.; Chervova, O.; Tillmann, T.; Maximov, V.; Ryabikov, A.; Gafarov, V.; Hubacek, J.A.; Pikhart, H.; Beck, S.; Bobak, M. The Relationship between Epigenetic Age and Myocardial Infarction/Acute Coronary Syndrome in a Population-Based Nested Case-Control Study. J. Pers. Med. 2022, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Malyutina, S.; Maximov, V.; Chervova, O.; Orlov, P.; Voloshin, V.; Ryabikov, A.; Voevoda, M.; Nikitenko, T. Leukocyte telomere length and mitochondrial DNA copy number association with colorectal cancer risk in an aging population. Global Transl. Med. 2023, 2, 184. [Google Scholar] [CrossRef]
- Chervova, O.; Chernysheva, E.; Panteleeva, K.; Widayati, T.A.; Hrbkova, N.; Schneider, J.; Maximov, V.; Ryabikov, A.; Tillmann, T.; Pikhart, H.; et al. Evaluation of Epigenetic Age Acceleration Scores and Their Associations with CVD-Related Phenotypes in a Population Cohort. Biology 2023, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Malyutina, S.; Maximov, V.; Chervova, O.; Orlov, P.; Ivanova, A.; Mazdorova, E.; Ryabikov, A.; Simonova, G.; Voevoda, M. The relationship between all-cause natural mortality and copy number of mitochondrial DNA in a 15-year follow-up study. Int. J. Mol. Sci. 2023, 24, 10469. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Li, L.; Xu, R. Association of Epigenetic Clock with Consensus Molecular Subtypes and Overall Survival of Colorectal Cancer. Cancer Epidemiol. Biomarkers Prev. 2019, 28, 1720–1724. [Google Scholar] [CrossRef] [PubMed]
- McCartney, D.L.; Min, J.L.; Richmond, R.C.; Lu, A.T.; Sobczyk, M.K.; Davies, G.; Broer, L.; Guo, X.; Jeong, A.; Jung, J.; et al. Genome-wide association studies identify 137 genetic loci for DNA methylation biomarkers of aging. Genome Biol. 2022, 22, 194. [Google Scholar] [CrossRef]
- Hartman, R.J.G.; Huisman, S.E.; den Ruijter, H.M. Sex differences in cardiovascular epigenetics-a systematic review. Biol. Sex Differ. 2018, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Peasey, A.; Bobak, M.; Kubinova, R.; Malyutina, S.; Pajak, A.; Tamosiunas, A.; Pikhart, H.; Nicholson, A.; Marmot, M. Determinants of cardiovascular disease and other non-communicable diseases in Central and Eastern Europe: Rationale and design of the HAPIEE study. BMC Public Health 2006, 6, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Ryden, L.; Standl, E.; Bartnik, M.; Van den Berghe, G.; Betteridge, J.; de Boer, M.-J.; Cosentino, F.; Jonsson, B.; Laakso, M.; Malmberg, K.; et al. Guidelines on diabetes, pre-diabetes, and cardiovascular diseases: Executive summary. The Task Force on Diabetes and Cardiovascular Diseases of the European Society of Cardiology (ESC) and of the European Association for the Study of Diabetes (EASD). Eur. Heart J. 2007, 28, 88–136. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Kalco, S.R.; Cantor, C.R. Pulsed-field gel electrophoresis and the technology of large DNA molecules. In Genome Analysis: A Practical Approach; Davies, K.E., Ed.; IRL Press: Oxford, UK, 1988; pp. 41–72. ISBN 1-85221-110-5. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org (accessed on 20 February 2024).
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.; Minfi, A. A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina Bead Chips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Langie, S.A.; De Boever, P.; Taylor, J.A.; Niu, L. RELIC: A novel dye-bias correction method for Illumina Methylation BeadChip. BMC Genom. 2017, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Chervova, O.; Conde, L.; Guerra-Assuncao, J.A.; Moghul, I.; Webster, A.P.; Berner, A.; Cadieux, E.L.; Tian, Y.; Voloshin, V.; Jesuset, T.F.; et al. The Personal Genome Project-UK, an open access resource of human multi-omics data. Sci. Data 2019, 6, 257. [Google Scholar] [CrossRef]
- Fortin, J.P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina Human Methylation EPIC array with minfi. Bioinformatics 2017, 33, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Pelegí-Sisó, D.; De Prado, P.; Ronkainen, J.; Bustamante, M.; González, J.R. Methylclock: A Bioconductor package to estimate DNA methylation age. Bioinformatics 2021, 37, 1759–1760. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Cases (Incident CRC) | Controls | p-Value |
|---|---|---|---|
| Observed, n | 35 | 354 | |
| Baseline age, years (mean, SD) | 60.2 (7.31) | 58.0 (7.05) | 0.088 |
| Women, no. (%) | 18 (51.4) | 203 (57.3) | 0.500 |
| Systolic blood pressure, mmHg (mean, SD) | 142.6 (24.78) | 141.8 (25.82) | 0.851 |
| Diastolic blood pressure, mmHg (mean, SD) | 90.1 (14.01) | 88.6 (13.70) | 0.543 |
| Body mass index, kg/sqm (mean, SD) | 27.7 (4.52) | 28.2 (5.21) | 0.578 |
| Waist/hip ratio, unit (mean, SD) | 0.89 (0.09) | 0.88 (0.08) | 0.434 |
| Total cholesterol, mmol/L (mean, SD) | 6.32 (1.16) | 6.49 (1.28) | 0.445 |
| LDL cholesterol, mmol/L (mean, SD) | 3.95 (1.00) | 4.22 (1.13) | 0.167 |
| HDL cholesterol, mmol/L (mean, SD) | 1.73 (0.50) | 1.55 (0.33) | 0.005 |
| TG, mmol/L (mean, SD) | 1.42 (0.71) | 1.58 (0.81) | 0.244 |
| Glucose, plasma, mmol/L (mean, SD) | 5.98 (1.40) | 5.98 (1.47) | 0.988 |
| Hypertension (%) | 22 (62.9) | 208 (58.8) | 0.638 |
| HT treatment (among HT), no. (%) | 11 (50.0) | 91 (43.8) | 0.575 |
| Diabetes mellitus type 2, no. (%) | 3 (9.1) | 34 (9.9) | 0.884 |
| DMT2 treatment (among DMT2), no. (%) | 1 (33.3) | 11 (32.4) | 0.972 |
| Smoking, no. (%) | 0.404 | ||
| Present smoker | 5 (14.3) | 86 (24.3) | |
| Former smoking | 5 (14.3) | 48 (13.6) | |
| Never smoking | 25 (71.4) | 220 (62.4) | |
| Frequency of drinking (%) | 0.869 | ||
| 5+/week | 5 (14.3) | 47 (13.3) | |
| 1–4/week | 12 (34.3) | 155 (43.8) | |
| 1–3/month | 8 (22.9) | 68 (19.2) | |
| <1/month | 9 (25.7) | 77 (21.8) | |
| Non-drinkers | 1 (2.9) | 7 (2.0) | |
| Education (%) | 0.815 | ||
| Primary | 4 (11.4) | 33 (9.3) | |
| Vocational | 7 (20.0) | 85 (24.0) | |
| Middle | 16 (45.7) | 139 (39.3) | |
| High | 8 (22.9) | 97 (27.4) | |
| Marital status (%) | 0.664 | ||
| Single | 11 (31.4) | 99 (28.0) | |
| Married | 24 (68.6) | 255 (72.0) | |
| Epigenetic Age, years, mean (SD) | |||
| Horvath | 58.9 (7.82) | 53.0 (6.51) | <0.001 |
| Hannum | 40.3 (6.19) | 36.3 (6.41) | 0.001 |
| PhenoAge | 53.3 (8.29) | 49.0 (8.02) | 0.003 |
| Skin and Blood | 58.6 (7.29) | 58.1 (6.47) | 0.650 |
| Zhang BLUP | 65.8 (7.06) | 62.1 (6.39) | 0.001 |
| Zhang EN | 59.6 (7.43) | 58.4 (7.13) | 0.347 |
| Epigenetic Age Acceleration, years, mean (SD) | |||
| Horvath | 3.91 (5.45) | −0.43 (4.53) | <0.001 |
| Hannum | 2.15 (3.41) | −0.24 (4.10) | <0.001 |
| PhenoAge | 2.29 (4.73) | −0.22 (5.69) | 0.005 |
| Skin and Blood | −1.03 (3.45) | 0.08 (3.73) | 0.091 |
| Zhang BLUP | 1.83 (2.41) | −0.11 (2.90) | <0.001 |
| Zhang EN | −0.68 (2.58) | 0.08 (3.27) | 0.185 |
| EAA Measure | No., Case/Control | Model 1 | Model 2 | Model 3 | Model 4 |
|---|---|---|---|---|---|
| OR (95% CI) | OR (95% CI) | OR (95% CI) | OR (95% CI) | ||
| Horvath, per decile | 35/354 | 1.43 (1.22–1.67) | 1.43 (1.22–1.68) | 1.44 (1.23—1.69) | 1.44 (1.21–1.76) |
| p-value for trend | <0.001 | <0.001 | <0.001 | <0.001 | |
| Hannum, per decile | 35/354 | 1.29 (1.11–1.48) | 1.29 (1.11–1.48) | 1.29 (1.11–1.49) | 1.23 (1.06–1.42) |
| p-value for trend | 0.001 | 0.001 | 0.001 | 0.007 | |
| PhenoAge, per decile | 35/354 | 1.20 (1.05–1.37) | 1.23 (1.07–1.41) | 1.25 (1.08–1.44) | 1.20 (1.04–1.39) |
| p-value for trend | 0.009 | 0.004 | 0.003 | 0.014 | |
| SkinBlood, per decile | 35/354 | 0.88 (0.77–0.99) | 0.88 (0.77–0.99) | 0.87 (0.76–0.99) | 0.86 (0.76–0.99) |
| p-value for trend | 0.044 | 0.040 | 0.029 | 0.033 | |
| BLUP, per decile | 35/354 | 1.34 (1.16–1.55) | 1.33 (1.15–1.54) | 1.32 (1.14–1.54) | 1.35 (1.16–1.57) |
| p-value for trend | <0.001 | <0.001 | <0.001 | <0.001 | |
| EN, per decile | 35/354 | 0.90 (0.78–1.02) | 0.90 (0.79–1.02) | 0.89 (0.78–1.01) | 0.87 (0.76–0.99) |
| p-value for trend | 0.104 | 0.091 | 0.078 | 0.040 |
| EAA Measure | No., Case/Control | Tertiles | Absolute Difference T1–T2 T2–T3 | Model 1 | Model 2 | Model 3 | Model 4 |
|---|---|---|---|---|---|---|---|
| OR (95% CI) | OR (95% CI) | OR (95% CI) | OR (95% CI) | ||||
| Horvath | 35/354 | T1(ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 3.96 | 1.60 (0.37–6.99) | 2.12 (0.52–8.71) | 2.15 (0.52—8.88) | 3.73 (0.72–19.38) | ||
| T3 | 18.88 | 12.04 (3.21–45.07) | 10.85 (3.15–37.39) | 11.38 (3.28–39.56) | 16.09 (3.59–72.12) | ||
| p-value for trend | <0.001 | <0.001 | <0.001 | <0.001 | |||
| Hannum | 35/354 | T1(ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 3.24 | 5.95 (1.29–27.47) | 6.39 (1.38–29.62) | 6.33 (1.36–29.55) | 5.35 (1.13–25.26) | ||
| T3 | 18.07 | 12.73 (2.90–55.95) | 13.57 (3.07–60.00) | 13.52 (3.04–60.11) | 9.79 (2.15–44.48) | ||
| p-value for trend | <0.001 | <0.001 | <0.001 | 0.001 | |||
| PhenoAge | 35/354 | T1(ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 4.46 | 6.59 (1.87–23.22) | 6.96 (1.97–24.58) | 6.97 (1.97–24.68) | 6.66 (1.84–24.06) | ||
| T3 | 23.79 | 5.42 (1.50–19.61) | 6.20 (1.71–22.52) | 6.65 (1.79–24.65) | 5.12 (1.35–19.45) | ||
| p-value for trend | 0.015 | 0.007 | 0.005 | 0.022 | |||
| SkinBlood | 35/354 | T1(ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 2.87 | 0.57 (0.25–1.35) | 0.58 (0.24–1.36) | 0.55 (0.23–1.32) | 0.55 (0.21–1.40) | ||
| T3 | 11.98 | 0.46 (0.19–1.12) | 0.45 (0.18–1.09) | 0.41 (0.16–1.00) | 0.42 (0.16–1.07) | ||
| p-value for trend | 0.081 | 0.071 | 0.048 | 0.067 | |||
| BLUP | 35/354 | T1(ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 2.29 | 1.41 (0.43–4.57) | 1.44 (0.44–4.68) | 1.43 (0.44–4.69) | 1.64 (0.48–5.63) | ||
| T3 | 13.95 | 5.25 (1.92–14.34) | 5.09 (1.86–13.92) | 5.27 (1.91–14.52) | 5.79 (1.97–17.03) | ||
| p-value for trend | <0.001 | <0.001 | <0.001 | <0.001 | |||
| EN | 35/354 | T1(ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 2.54 | 0.83 (0.37–1.82) | 0.76 (0.34–1.70) | 0.69 (0.30–1.60) | 0.63 (0.26–1.53) | ||
| T3 | 16.49 | 0.40 (0.16–1.04) | 0.38 (0.15–0.98) | 0.36 (0.14–0.94) | 0.28 (0.10–0.79) | ||
| p-value for trend | 0.062 | 0.046 | 0.035 | 0.015 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malyutina, S.; Chervova, O.; Maximov, V.; Nikitenko, T.; Ryabikov, A.; Voevoda, M. Blood-Based Epigenetic Age Acceleration and Incident Colorectal Cancer Risk: Findings from a Population-Based Case–Control Study. Int. J. Mol. Sci. 2024, 25, 4850. https://doi.org/10.3390/ijms25094850
Malyutina S, Chervova O, Maximov V, Nikitenko T, Ryabikov A, Voevoda M. Blood-Based Epigenetic Age Acceleration and Incident Colorectal Cancer Risk: Findings from a Population-Based Case–Control Study. International Journal of Molecular Sciences. 2024; 25(9):4850. https://doi.org/10.3390/ijms25094850
Chicago/Turabian StyleMalyutina, Sofia, Olga Chervova, Vladimir Maximov, Tatiana Nikitenko, Andrew Ryabikov, and Mikhail Voevoda. 2024. "Blood-Based Epigenetic Age Acceleration and Incident Colorectal Cancer Risk: Findings from a Population-Based Case–Control Study" International Journal of Molecular Sciences 25, no. 9: 4850. https://doi.org/10.3390/ijms25094850
APA StyleMalyutina, S., Chervova, O., Maximov, V., Nikitenko, T., Ryabikov, A., & Voevoda, M. (2024). Blood-Based Epigenetic Age Acceleration and Incident Colorectal Cancer Risk: Findings from a Population-Based Case–Control Study. International Journal of Molecular Sciences, 25(9), 4850. https://doi.org/10.3390/ijms25094850