
Generating Concentration Gradients across Membranes for Molecular Dynamics Simulations of Periodic Systems

Abstract
1. Introduction
1.1. Existing Computational Methods for Characterizing Membrane Permeability
1.2. Need for Generating Concentration Gradients in MD Simulations
2. Results and Discussion
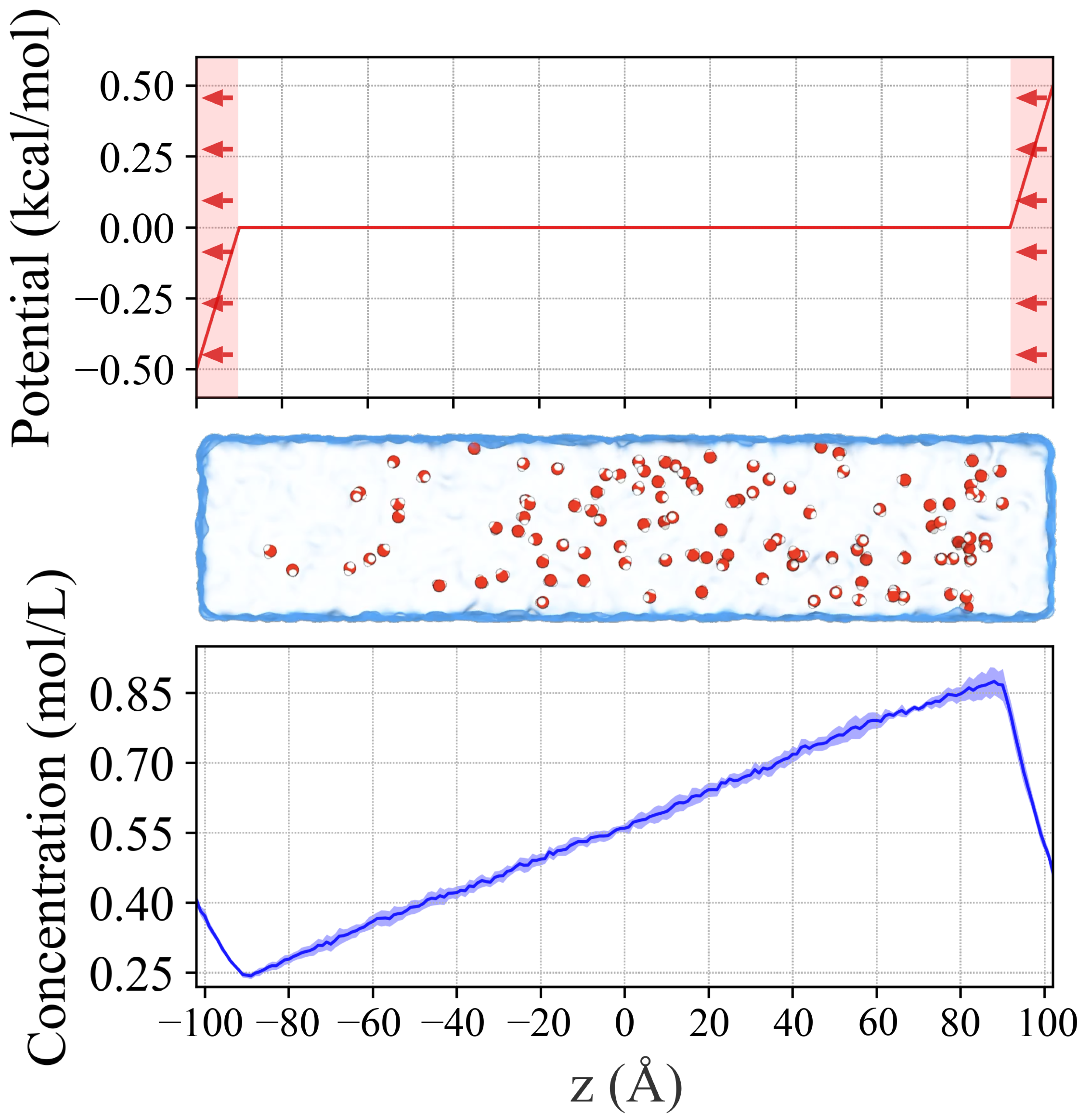
2.1. Generating Concentration Gradient in a Water Box
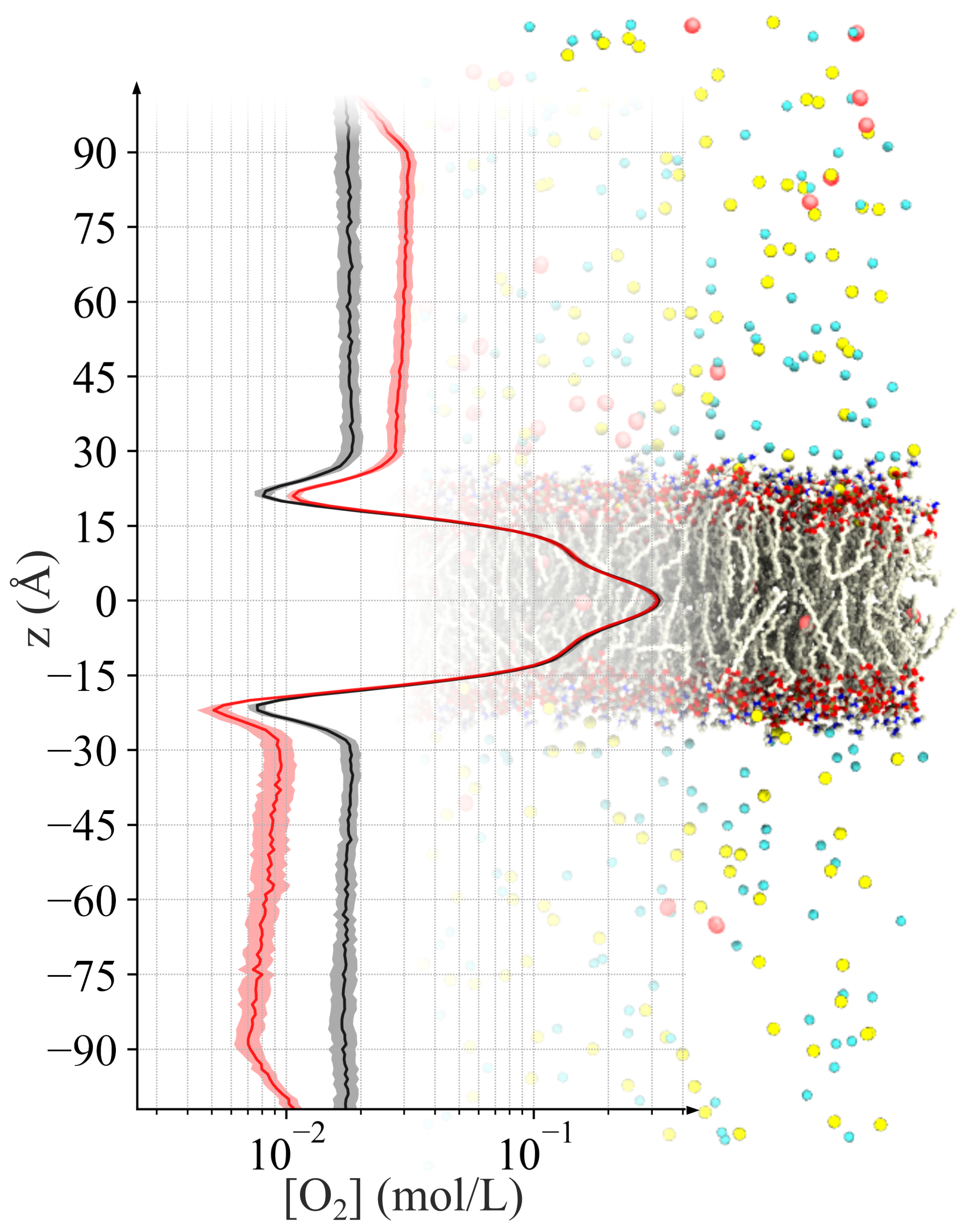
2.2. Generating Oxygen Gradient across a Lipid Bilayer
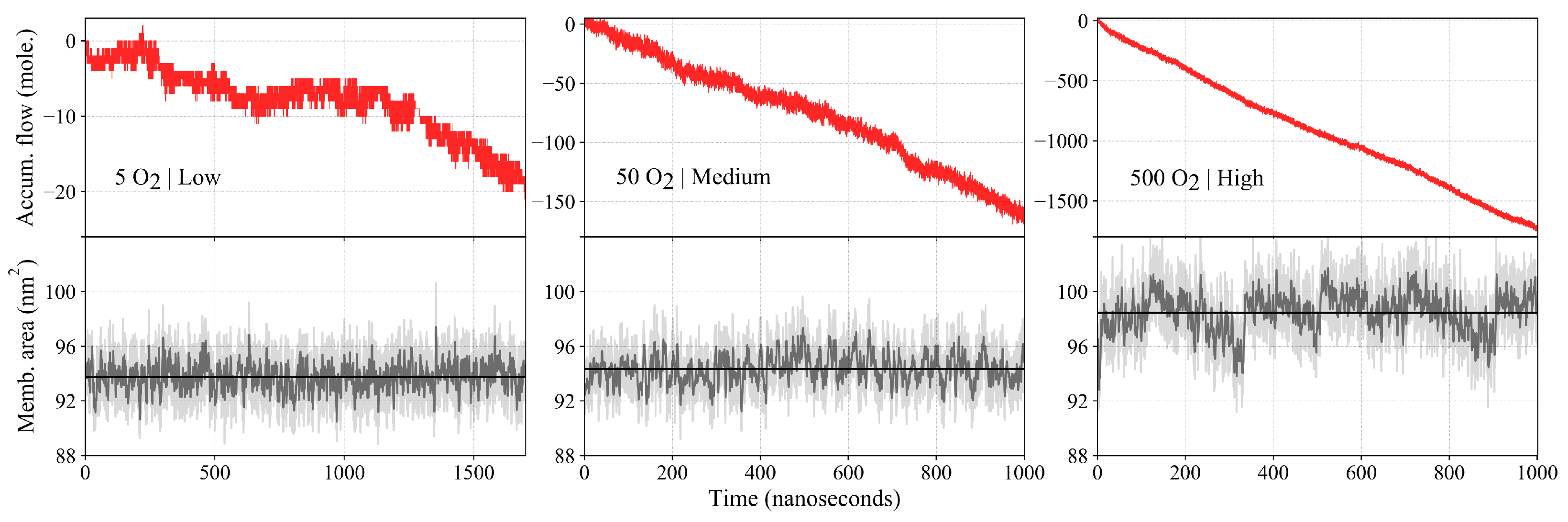
2.3. Oxygen Flux across the Membrane
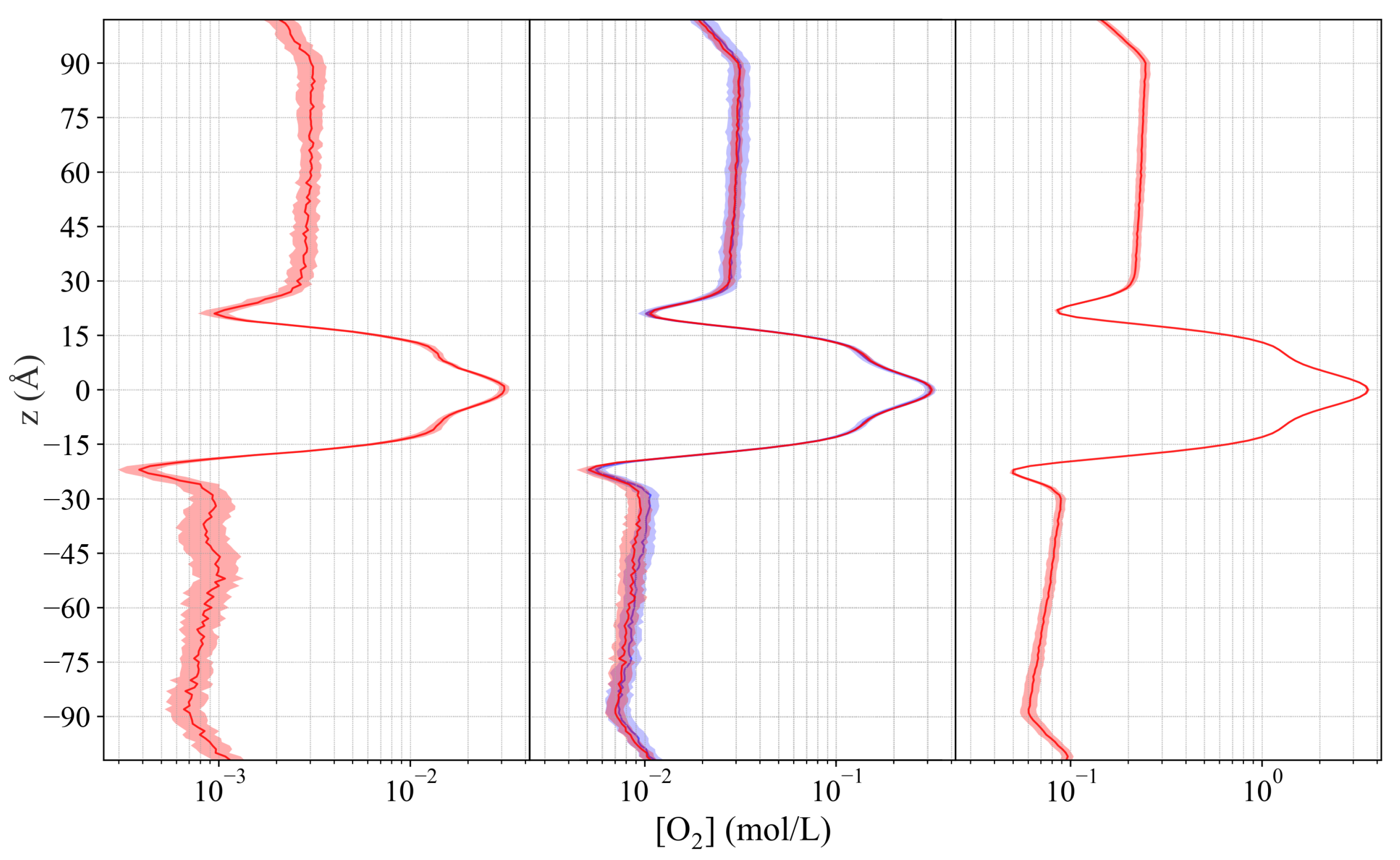
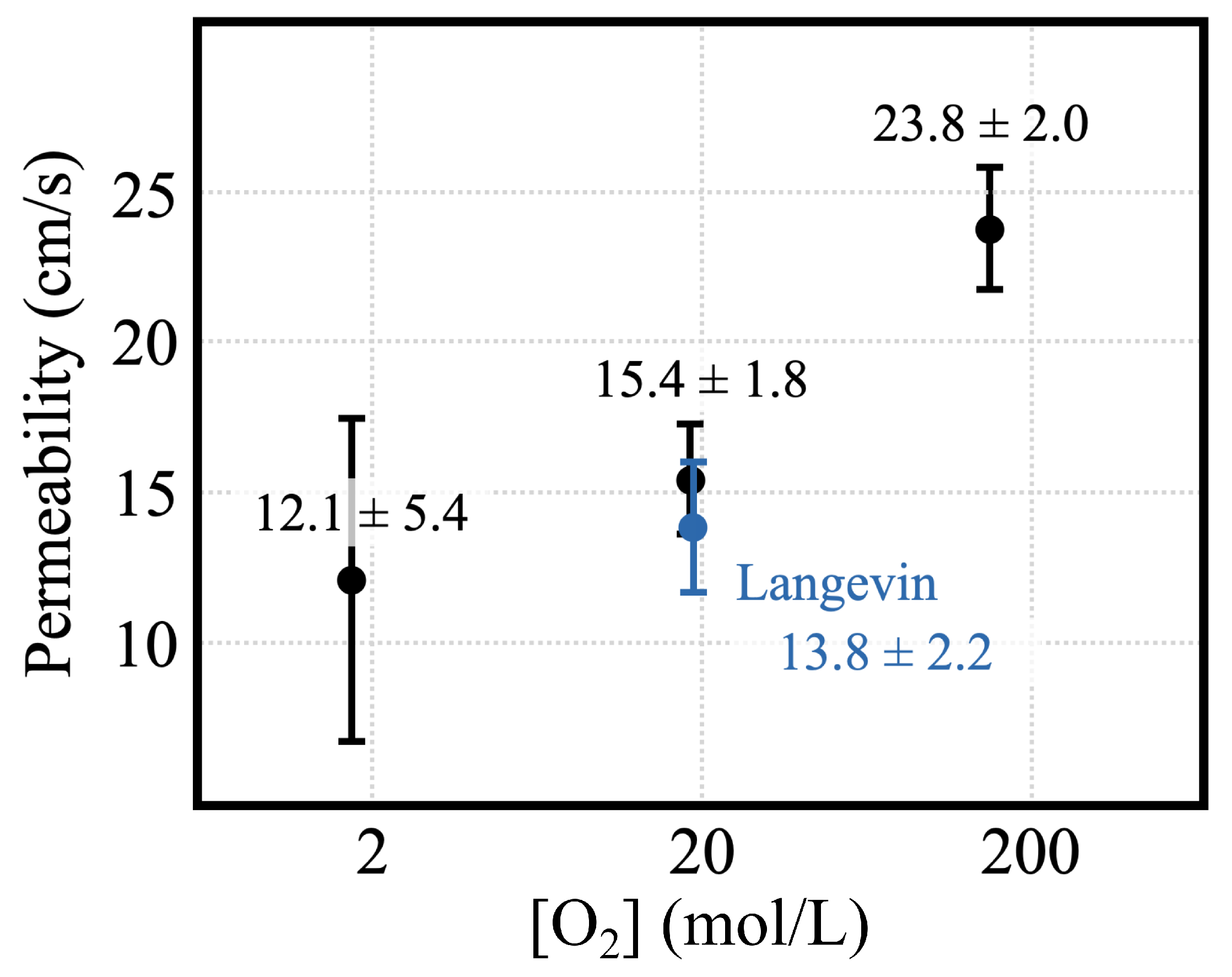
2.4. Oxygen Permeability of POPC Bilayers
2.4.1. Correlation of Permeability and Permeant Concentration
2.4.2. Effect of Thermostat
2.5. Dissipation of the Chemical Potential
2.6. Discrepancy between Computation and Experiments
3. Materials and Methods
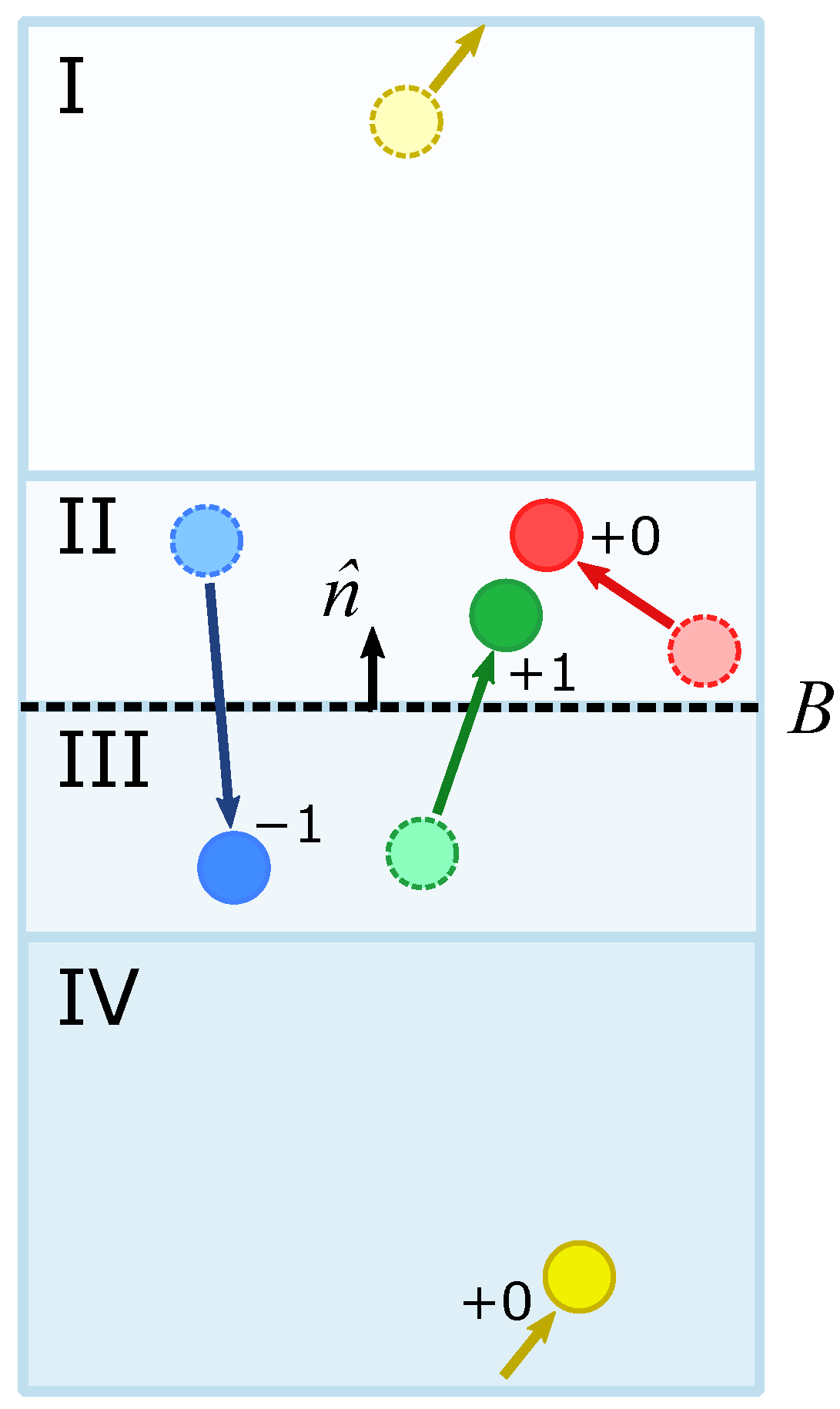
3.1. Generating Concentration Gradients via a Unidirectional Bias at the Boundary
3.2. POPC Bilayers for Concentration Gradient Simulations
3.3. Molecular Dynamics Technical Specifications
3.4. Characterization of Concentration Gradients
3.5. Flux Calculation
3.6. Calculating Membrane Permeability
3.7. Error Analysis via Block Averaging
4. Conclusions
5. Future Work
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CSVR | Canonical Sampling through Velocity Rescaling |
| G-SMD | Grid-Steered Molecular Dynamics |
| ISD | Inhomogeneous Solubility Diffusion |
| MD | Molecular Dynamics |
| PBC | Periodic Boundary Conditions |
| PMF | Potential of Mean Force |
References
- Gorter, E.; Grendel, F. On Bimolecular Layers of Lipoids on the Chromocytes of the Blood. J. Exp. Med. 1924, 41, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Edidin, M. Lipids on the frontier: A century of cell–membrane bilayers. Nat. Rev. Mol. Cell Biol. 2003, 4, 414–418. [Google Scholar] [CrossRef]
- Wu, L.G.; Hamid, E.; Shin, W.; Chiang, H.C. Exocytosis and Endocytosis: Modes, Functions, and Coupling Mechanisms. Annu. Rev. Physiol. 2014, 76, 301–331. [Google Scholar] [CrossRef]
- Jiang, T.; Wen, P.C.; Trebesch, N.; Zhao, Z.; Pant, S.; Kapoor, K.; Shekhar, M.; Tajkhorshid, E. Computational Dissection of Membrane Transport at a Microscopic Level. Trends Biochem. Sci. 2020, 45, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Vermaas, J.V.; Trebesch, N.; Mayne, C.; Thangapandian, S.; Shekhar, M.; Mahinthichaichan, P.; Baylon, J.L.; Jiang, T.; Wang, Y.; Muller, M.P.; et al. Microscopic Characterization of Membrane Transporter Function by in Silico Modeling and Simulation. In Computational Approaches for Studying Enzyme Mechanism Part B; Voth, G.A., Ed.; Academic Press: Cambridge, MA, USA, 2016; Chapter 16; Volume 578, pp. 373–428. [Google Scholar] [CrossRef]
- Pedersen, P.L. Transport ATPases: Structure, motors, mechanism and medicine: A brief overview. J. Bioenerg. Biomembr. 2005, 37, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Carroll, T.P.; Guggino, W.B.; Agre, P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science 1992, 256, 385–387. [Google Scholar] [CrossRef]
- Zeidel, M.L.; Ambudkar, S.V.; Smith, B.L.; Agre, P. Reconstitution of Functional Water Channels in Liposomes Containing Purified Red Cell CHIP28 Protein. Biochemistry 1992, 31, 7436–7440. [Google Scholar] [CrossRef]
- Missner, A.; Pohl, P. 100 Years of the Meyer-Overton Rule: Predicting Membrane Permeability of Gases and Other Small Compounds. Comput. Phys. Commun. 2009, 10, 1405–1414. [Google Scholar]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef]
- Awoonor-Williams, E.; Rowley, C.N. Molecular simulation of nonfacilitate membrane permeation. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 1672–1687. [Google Scholar] [CrossRef]
- Venable, R.M.; Krämer, A.; Pastor, R.W. Molecular dynamics simulations of membrane permeability. Chem. Rev. 2019, 119, 5954–5997. [Google Scholar] [CrossRef]
- Hannesschlaeger, C.; Homer, A.; Pohl, P. Intrinsic membrane permeability to small molecules. Chem. Rev. 2019, 119, 5922–5953. [Google Scholar] [CrossRef]
- Paula, S.; Deamer, D.W. Membrane permeability barriers to ionic and polar solutes. Curr. Top. Membr. 1999, 48, 77–95. [Google Scholar]
- Fick, A. Ueber diffusion. Ann. Der Phys. 1855, 170, 59–86. [Google Scholar] [CrossRef]
- Fick, A. On liquid diffusion. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1855, 10, 30–39. [Google Scholar] [CrossRef]
- Overton, C.E. Studien über die Narkose Zugleich ein Beitrag zur Allgemeinen Pharmakologie; G. Fischer: Jena, Germany, 1901. [Google Scholar]
- Lipnick, R.L. Charles Ernest Overton: Narcosis studies and a contribution to general pharmacology. In Studies of Narcosis; Springer: Dordrecht, The Netherlands, 1991. [Google Scholar]
- Parisio, G.; Stocchero, M.; Ferrarini, A. Passive membrane permeability: Beyond the standard solubility-diffusion model. J. Chem. Theory Comput. 2013, 9, 5236–5246. [Google Scholar] [CrossRef]
- Mansy, S.S. Membrane transport in Primitive Cells. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kleinzeller, A. Charles Ernest Overton’s Concept of a Cell Membrane. Curr. Top. Membr. 1999, 48, 1–22. [Google Scholar]
- Berendsen, H.J.C.; Marrink, S.J. Molecular dynamics of water transport through membranes: Water from solvent to solute. Pure Appl. Chem. 1993, 65, 2513–2520. [Google Scholar] [CrossRef]
- Marrink, S.J.; Berendsen, H.J.C. Simulation of water transport through a lipid membrane. J. Phys. Chem. 1994, 98, 4155–4168. [Google Scholar] [CrossRef]
- Diamond, J.M.; Katz, Y. Interpretation of nonelectrolyte partition coefficients between dimyristoyl lecithin and water. J. Membr. Biol. 1974, 17, 121–154. [Google Scholar] [CrossRef]
- Grossfield, A.; Woolf, T.B. Interaction of tryptophan analogs with POPC lipid bilayers investigated by molecular dynamics calculations. Langmuir 2002, 18, 198–210. [Google Scholar] [CrossRef]
- Bemporad, D.; Luttmann, C.; Essex, J.W. Permeation of Small Molecules through a Lipid Bilayer: A Computer Simulation Study. J. Phys. Chem. B 2004, 108, 4875–4884. [Google Scholar] [CrossRef]
- Wei, C.; Pohorille, A. Permeation of nucleosides through lipid bilayers. J. Phys. Chem. B 2011, 115, 3681–3688. [Google Scholar] [CrossRef]
- Lee, C.T.; Comer, J.; Herndon, C.; Leung, N.; Pavlova, A.; Swift, R.V.; Tung, C.; Rowley, C.N.; Amaro, R.E.; Chipot, C.; et al. Simulation-based approaches for determining membrane permeability of small compounds. J. Chem. Inf. Model. 2016, 56, 721–733. [Google Scholar] [CrossRef]
- Dotson, R.J.; Smith, C.R.; Bueche, K.; Angles, G.; Pias, S.C. Influences of Cholesterol on the Oxygen Permeability of Membranes: Insight from Atomistic Simulations. Biophys. J. 2017, 112, 2336–2347. [Google Scholar] [CrossRef]
- Ghorbani, M.; Wang, E.; Krämer, A.; Klauda, J.B. Molecular dynamics simulations of ethanol permeation through single and double-lipid bilayers. J. Chem. Phys. 2020, 153, 125101. [Google Scholar] [CrossRef]
- Krämer, A.; Ghysels, A.; Wang, E.; Venable, R.M.; Klauda, J.B.; Brooks, B.R.; Pastor, R.W. Membrane permeability of small molecules from unbiased molecular dynamics simulations. J. Chem. Phys. 2020, 153, 124107. [Google Scholar] [CrossRef]
- Paulikat, M.; Piccini, G.M.; Ippoliti, E.; Rossetti, G.; Arnesano, F.; Carloni, P. Physical chemistry of chloroquine permeation through the cell membrane with atomistic detail. J. Chem. Inf. Model. 2023, 63, 7124–7132. [Google Scholar] [CrossRef]
- Khalili-Araghi, F.; Ziervogel, B.; Gumbart, J.C.; Roux, B. Molecular dynamics simulations of membrane proteins under asymmetric ionic concentrations. J. Gen. Physiol. 2013, 142, 465–475. [Google Scholar] [CrossRef]
- Widomska, J.; Raguz, M.; Subczynski, W.K. Oxygen permeability of the lipid bilayer membrane made of calf lens. Biochim. Biophys. Acta 2007, 1768, 2635–2645. [Google Scholar] [CrossRef]
- Al-Abdul-Wahid, M.S.; Yu, C.H.; Batruch, I.; Evanics, F.; Pomes, R.; Prosser, R.S. A combined NMR and molecular dynamics study of the transmembrane solubility and diffusion rate profile of dioxygen in lipid bilayers. Biochemistry 2006, 45, 10719–10728. [Google Scholar] [CrossRef]
- Möller, M.N.; Li, Q.; Chinnaraj, M.; Cheung, H.C.; Lancaster, J.R., Jr.; Denicola, A. Solubility and diffusion of oxygen in phospholipid membranes. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 2923–2930. [Google Scholar] [CrossRef]
- Rettich, T.R.; Handa, P.; Battino, R.; Wilhelm, E. Solubility of gases in liquids. 13. High-precision determination of Henry’s constants for methane and ethane in liquid water at 275 to 328 K. J. Phys. Chem. 1981, 85, 3230–3237. [Google Scholar] [CrossRef]
- Sander, R. Compliation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 2015, 15, 4399–4981. [Google Scholar] [CrossRef]
- Dotson, R.J.; McClenahan, E.; Pias, S.C. Updated evaluation of cholesterol’s influence on membrane oxygen permeability. Adv. Exp. Med. Biol. 2021, 1269, 23–30. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Bussi, G.; Parrinello, M. Stochastic thermostats: Comparison of local and global schemes. Comput. Phys. Commun. 2008, 179, 26–29. [Google Scholar] [CrossRef]
- Yang, L.; Kindt, J.T. Line Tension Assists Membrane Permeation at the Transition Temperature in Mixed-Phase Lipid Bilayers. J. Phys. Chem. B 2016, 120, 11740–11750. [Google Scholar] [CrossRef]
- Stinner, J.N.; Newlon, D.L.; Heisler, N. Extracellular and intracellular carbon dioxide concentration as a function of temperature in the toad Bufo marinus. J. Exp. Biol. 1994, 195, 345–360. [Google Scholar] [CrossRef]
- Prasad, G.V.T.; Coury, L.; Finn, F.; Zeidel, M. Reconstituted aquaporin 1 water channels transport CO2 across membranes. J. Biol. Chem. 1998, 273, 33123–33126. [Google Scholar] [CrossRef]
- Hill, W.G.; Rivers, R.L.; Zeidel, M.L. Role of leaflet asymmetry in the permeability of model biological membranes to protons, solutes, and gases. J. Gen. Physiol. 1999, 114, 405–414. [Google Scholar] [CrossRef]
- Möller, M.N.; Lancaster, J.R.; Denicola, A. The interaction of reactive oxygen and nitrogen species with membranes. Curr. Top. Membr. 2008, 61, 23–42. [Google Scholar]
- Mao, Y.; Zhang, Y. Thermal conductivity, shear viscosity and specific heat of rigid water models. Chem. Phys. Lett. 2012, 542, 37–41. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298K. J. Chem. Phys. 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- van der Spoel, D.; van Maaren, P.J.; Berendsen, H.J.C. A systematic study of water models for molecular siulation: Derivation of water models optimized for use with a reaction field. J. Chem. Phys. 1998, 108, 10220–10230. [Google Scholar] [CrossRef]
- Javanainen, M.; Vattulainen, I.; Monticelli, L. On atomistic models for molecular oxygen. J. Phys. Chem. B 2017, 121, 518–528, Correction in J. Phys. Chem. B 2020, 124, 6943–6946. [Google Scholar] [CrossRef]
- Wells, D.B.; Abramkina, V.; Aksimentiev, A. Exploring Transmembrane Transport Through α-Hemolysin with Grid-Steered Molecular Dynamics. J. Chem. Phys. 2007, 127, 125101. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable Molecular Dynamics With NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Cohen, J.; Arkhipov, A.; Braun, R.; Schulten, K. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys. J. 2006, 91, 1844–1857. [Google Scholar] [CrossRef]
- Wang, Y.; Cohen, J.; Boron, W.F.; Schulten, K.; Tajkhorshid, E. Exploring Gas Permeability of Cellular Membranes and Membrane Channels with Molecular Dynamics. J. Struct. Biol. 2007, 157, 534–544. [Google Scholar] [CrossRef]
- Fiorin, G.; Klein, M.L.; Hénin, J. Using Collective Variables to Drive Molecular Dynamics Simulations. Mol. Phys. 2013, 111, 3345–3362. [Google Scholar] [CrossRef]
- Kučerka, N.; Nieh, M.P.; Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim. Biophys. Acta-Biomembr. 2011, 1808, 2761–2771. [Google Scholar] [CrossRef]
- Flyvbjerg, H.; Petersen, H.G. Error estimates on averages of correlated data. J. Chem. Phys. 1989, 91, 461–466. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Count | Length (s) | Thermostat | (mmol/L) | (mmol/L) | Flow Rate (molecules/100 ns) | Area () | Permeability (cm/s) |
|---|---|---|---|---|---|---|---|
| 5 | 1.7 | CSVR | 2.56 ± 0.19 | 0.92 ± 0.12 | 1.12 ± 0.48 | 9375 ± 5 | 12.07 ± 5.41 |
| 50 | 1.0 | CSVR | 27.77 ± 1.07 | 9.38 ± 0.62 | 16.1 ± 1.58 | 9434 ± 10 | 15.41 ± 1.84 |
| 500 | 1.0 | CSVR | 210.35 ± 6.00 | 88.67 ± 3.21 | 171.4 ± 10.88 | 9847 ± 25 | 23.75 ± 2.01 |
| 50 | 1.0 | Langevin | 27.38 ± 1.63 | 10.52 ± 0.61 | 13.2 ± 1.55 | 9399 ± 12 | 13.84 ± 2.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shinn, E.J.; Tajkhorshid, E. Generating Concentration Gradients across Membranes for Molecular Dynamics Simulations of Periodic Systems. Int. J. Mol. Sci. 2024, 25, 3616. https://doi.org/10.3390/ijms25073616
Shinn EJ, Tajkhorshid E. Generating Concentration Gradients across Membranes for Molecular Dynamics Simulations of Periodic Systems. International Journal of Molecular Sciences. 2024; 25(7):3616. https://doi.org/10.3390/ijms25073616
Chicago/Turabian StyleShinn, Eric Joon, and Emad Tajkhorshid. 2024. "Generating Concentration Gradients across Membranes for Molecular Dynamics Simulations of Periodic Systems" International Journal of Molecular Sciences 25, no. 7: 3616. https://doi.org/10.3390/ijms25073616
APA StyleShinn, E. J., & Tajkhorshid, E. (2024). Generating Concentration Gradients across Membranes for Molecular Dynamics Simulations of Periodic Systems. International Journal of Molecular Sciences, 25(7), 3616. https://doi.org/10.3390/ijms25073616