
Free Energy Barriers for Passive Drug Transport through the Mycobacterium tuberculosis Outer Membrane: A Molecular Dynamics Study

 , , , , and
, , , , and
Abstract
1. Introduction
- Passive diffusion: This is the movement of substances directly across the lipid bilayer of a membrane from an area of high concentration to an area of low concentration that requires no energy input.
- Facilitated diffusion: This is the movement of substances across a membrane through the special porin proteins. Porins form large, water-filled channels that allow small hydrophilic molecules to enter the cell without expending energy. This absorption pathway is suitable for a limited range of compounds because the diameter of the channel at the narrowest point determines the exclusion limit, and parameters such as the channel length and the number of open pores determine the transport rate.
- Active transport: This is the transfer of molecules and ions against a concentration gradient, coupled to energy consumption. This process is performed by the special transporter proteins.
2. Results and Discussion
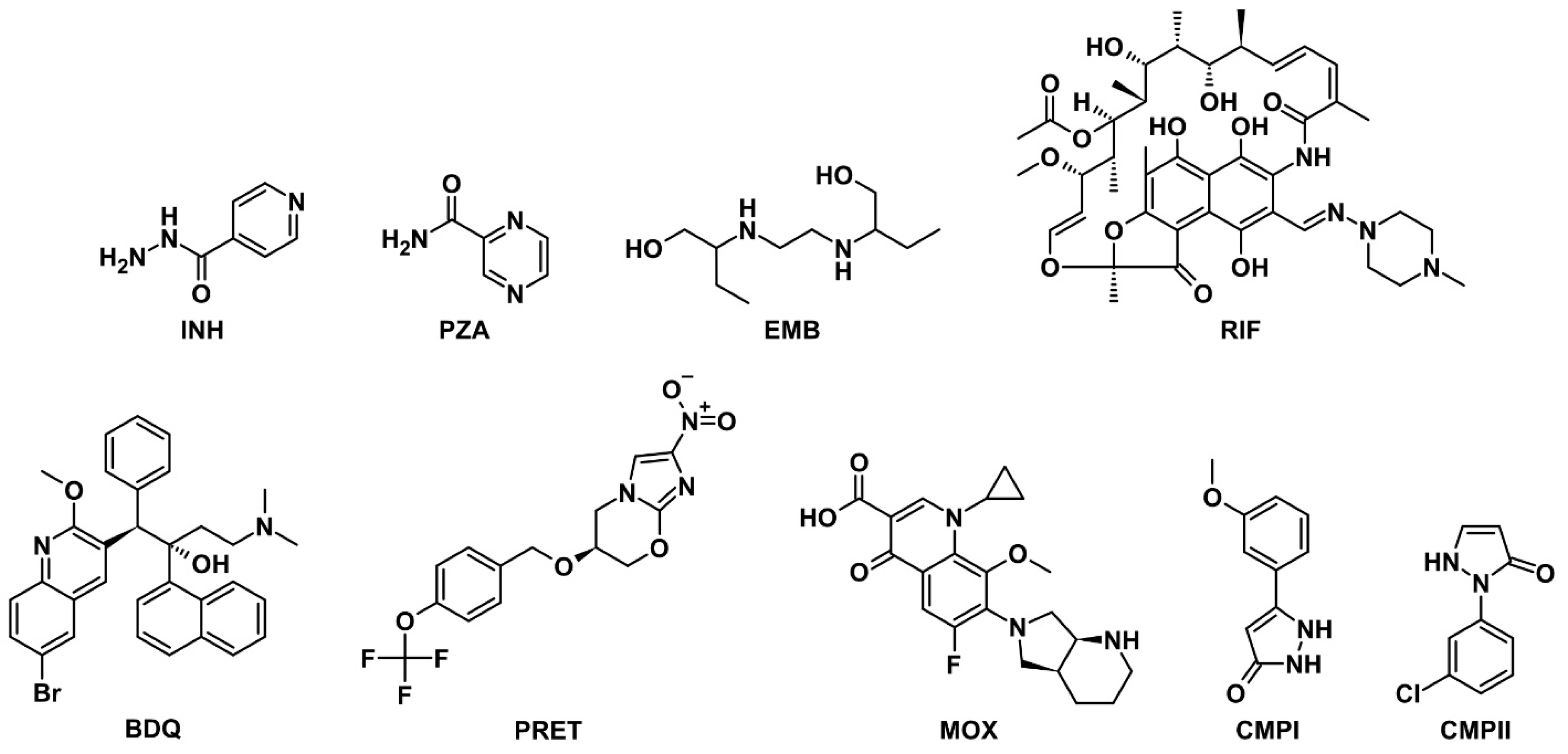
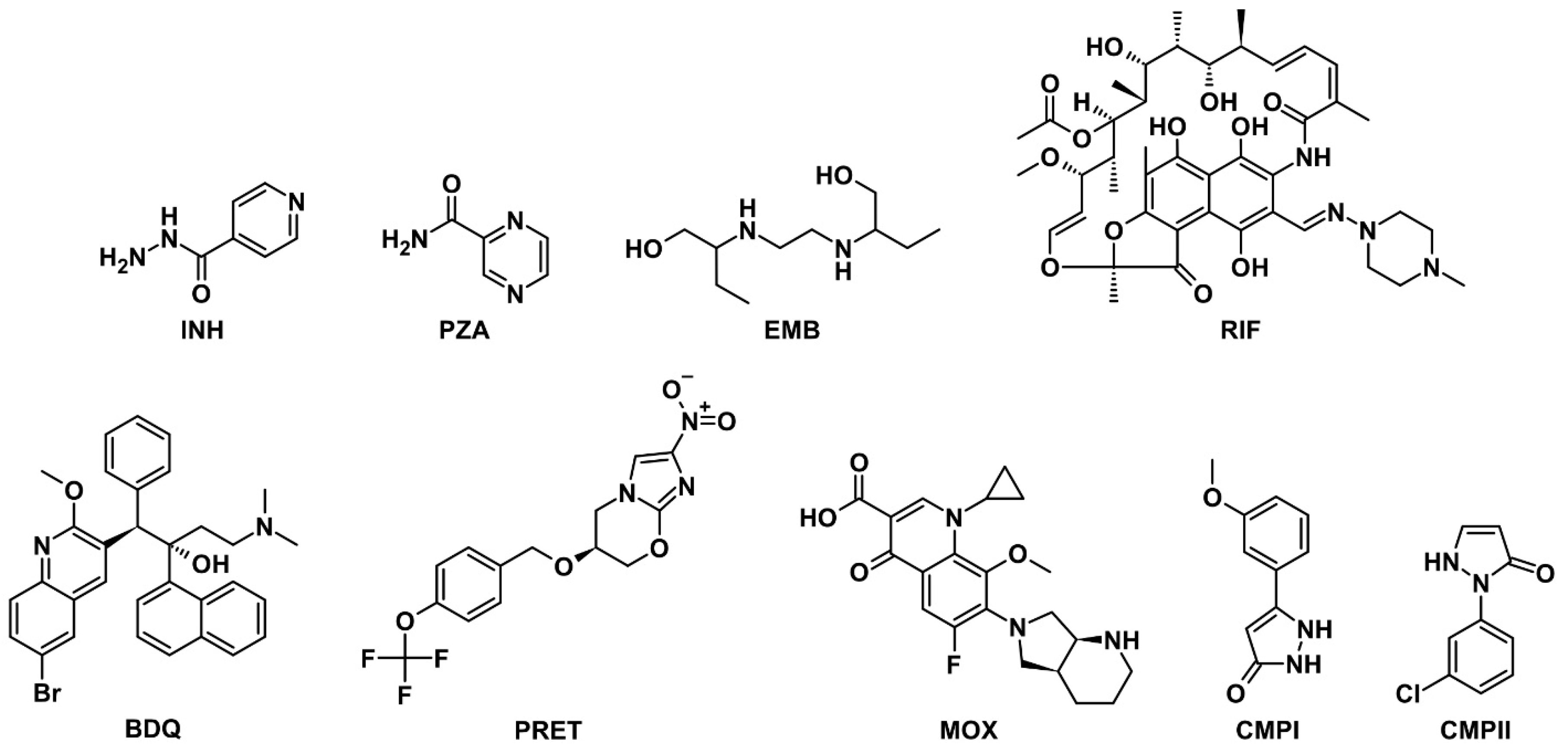
2.1. Selection and Appraisal of Model Drug Molecules
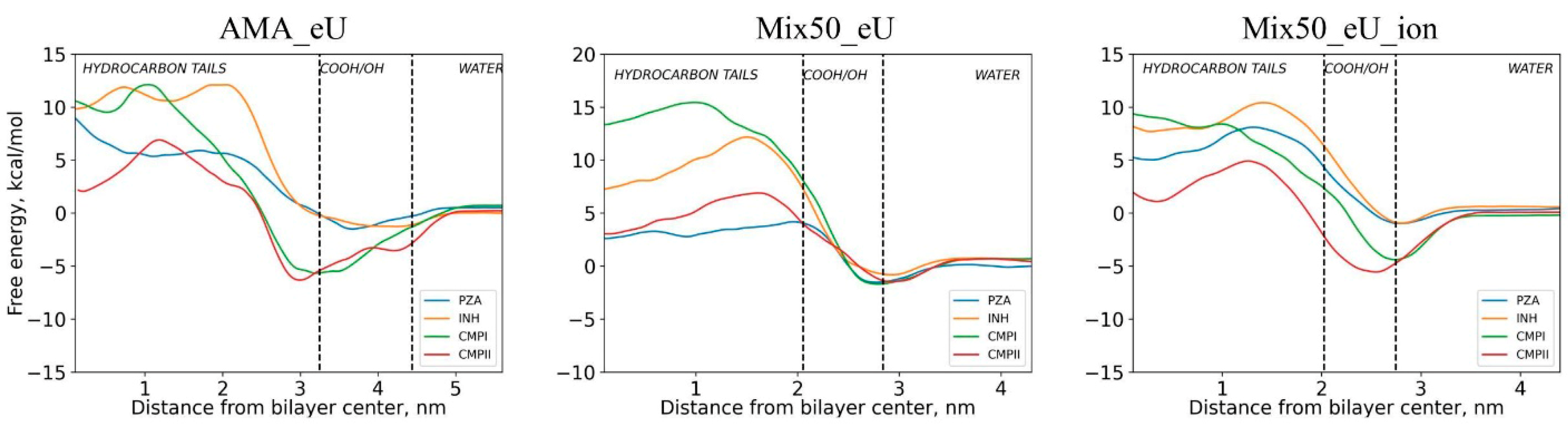
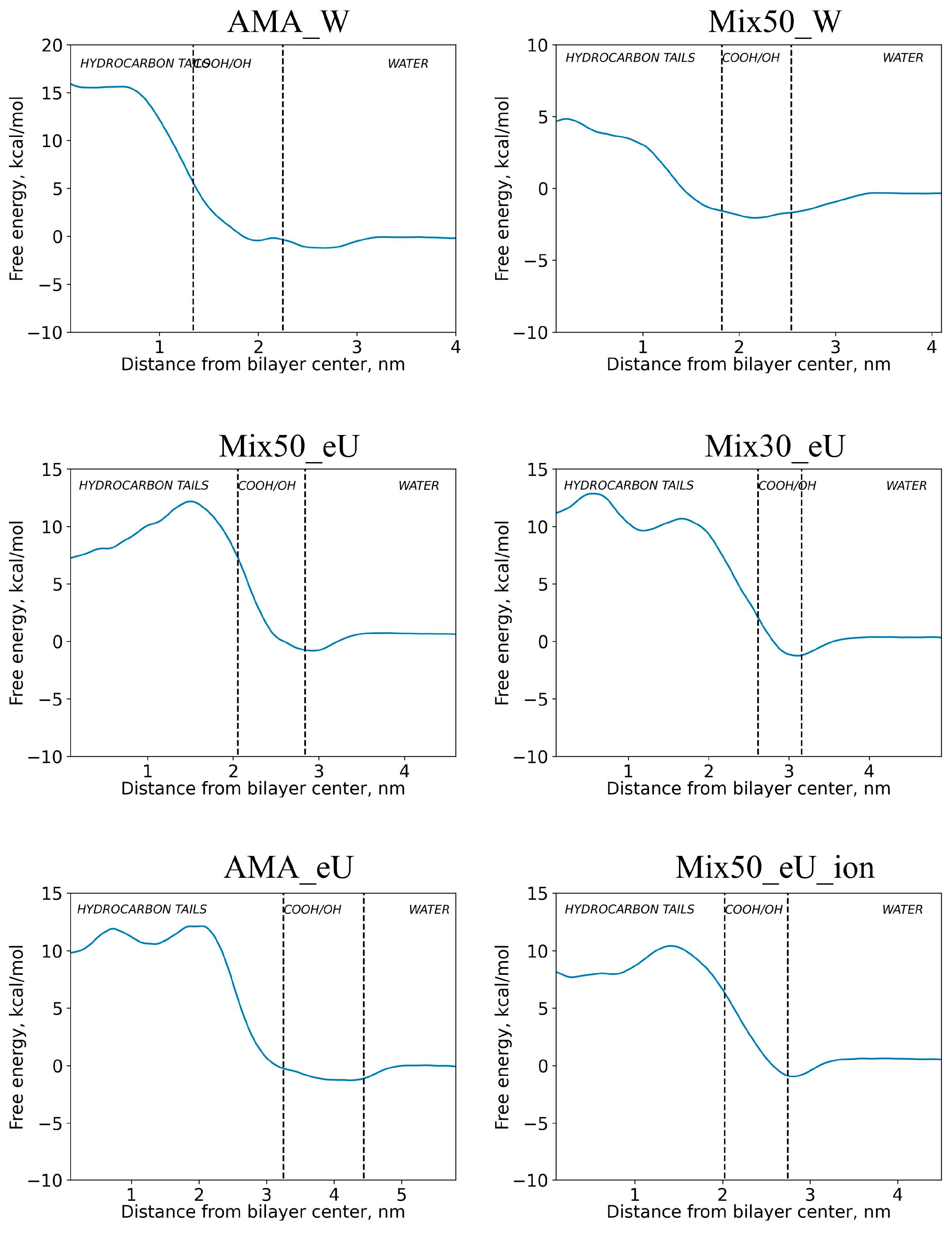
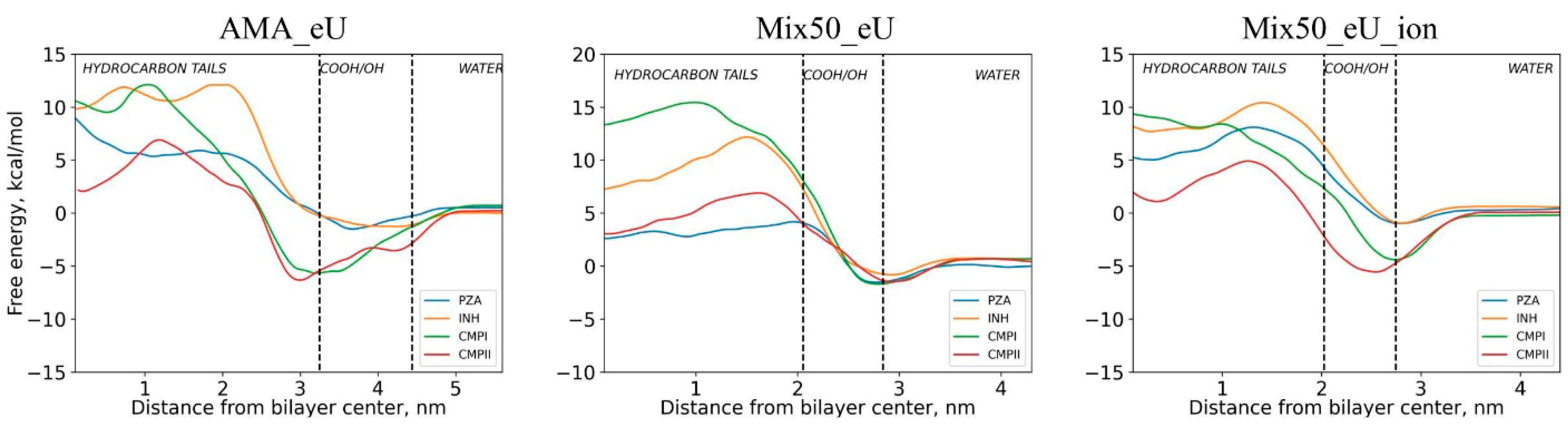
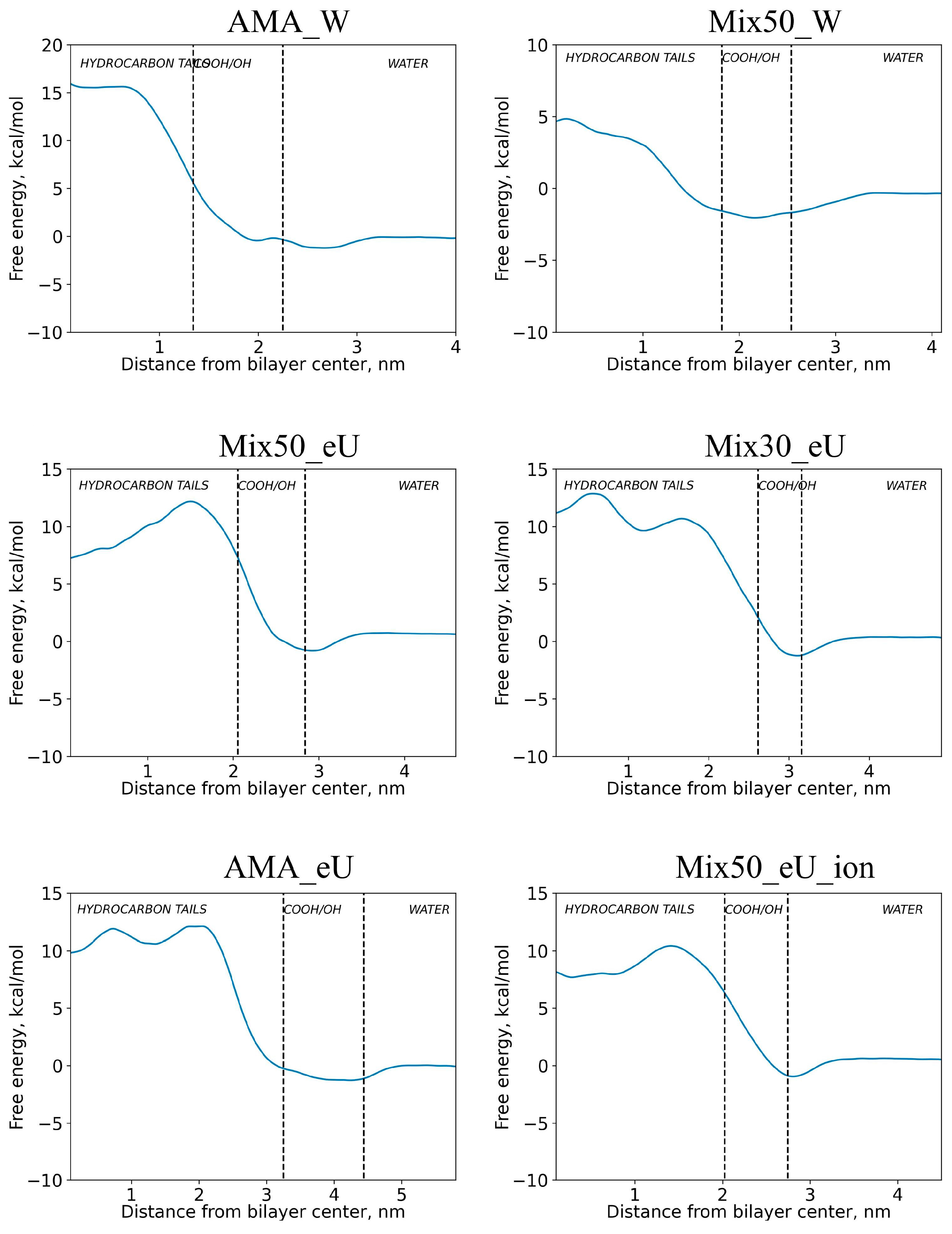
2.2. Free Energy Profiles of Transmembrane Diffusion
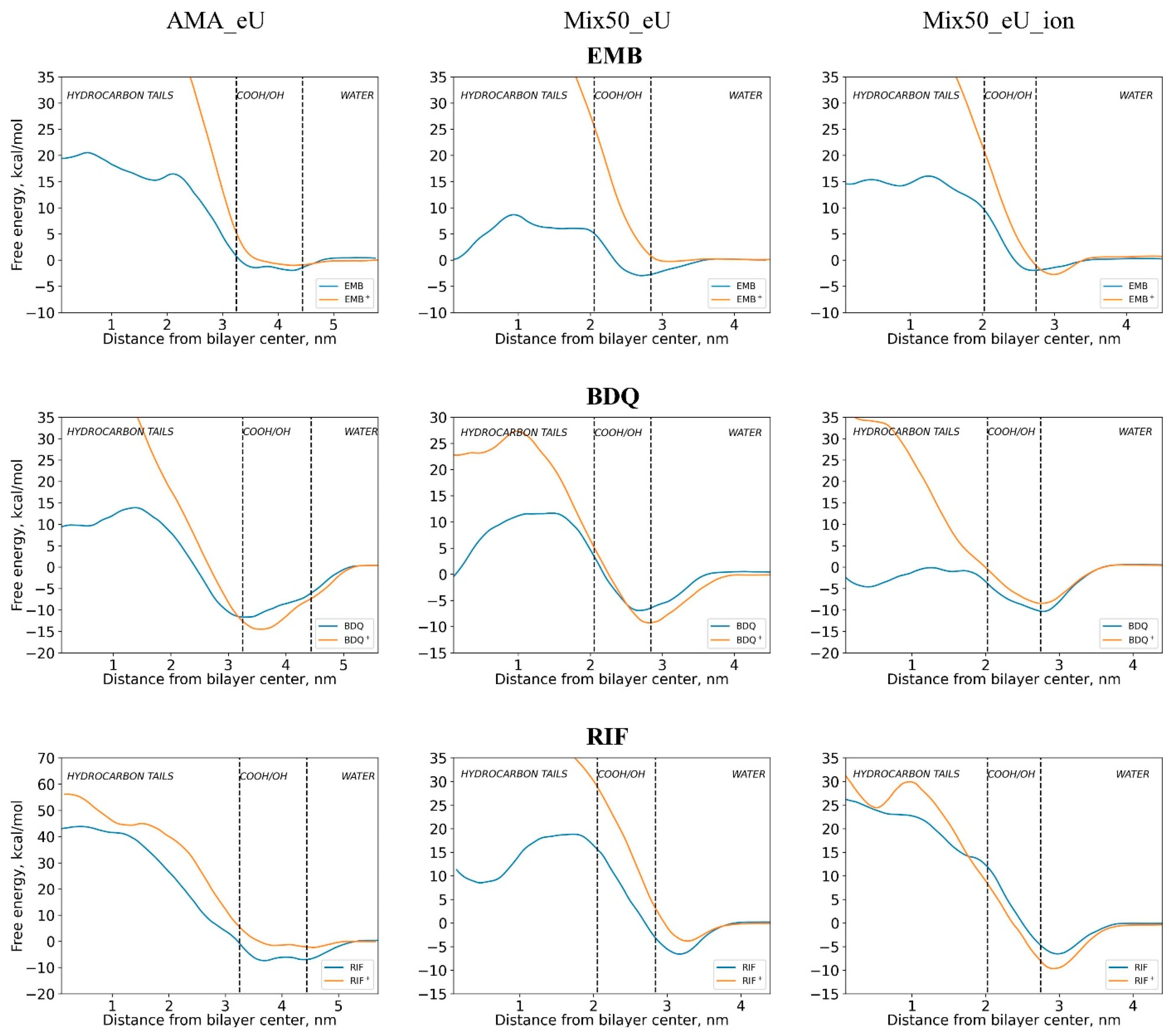
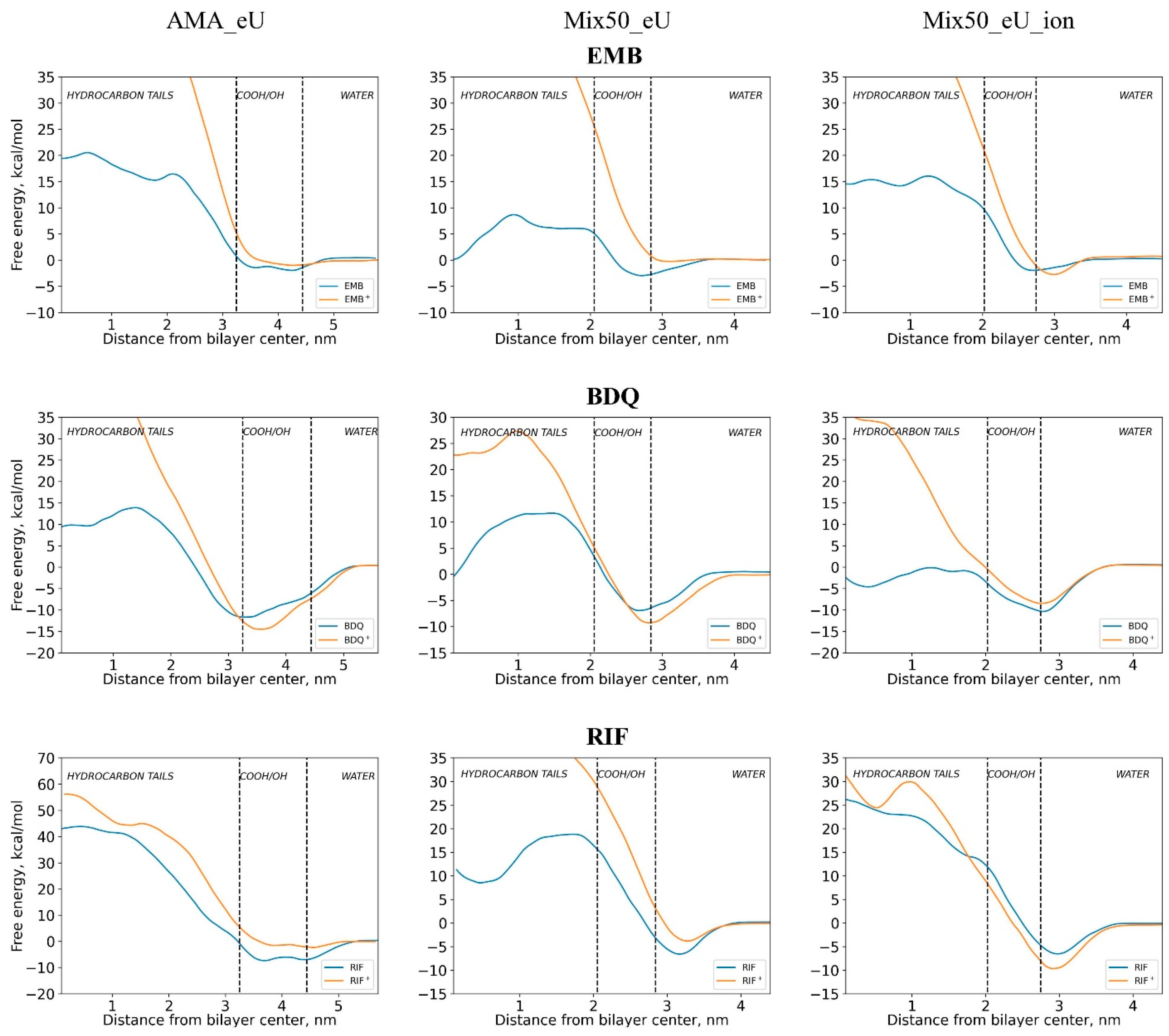
2.3. Influence of Molecular Charge on Diffusion Barriers
2.4. Influence of Membrane Type on Diffusion Barriers
2.5. Rotational Mobility of Diffusing Molecules in Different Regions of the Membrane
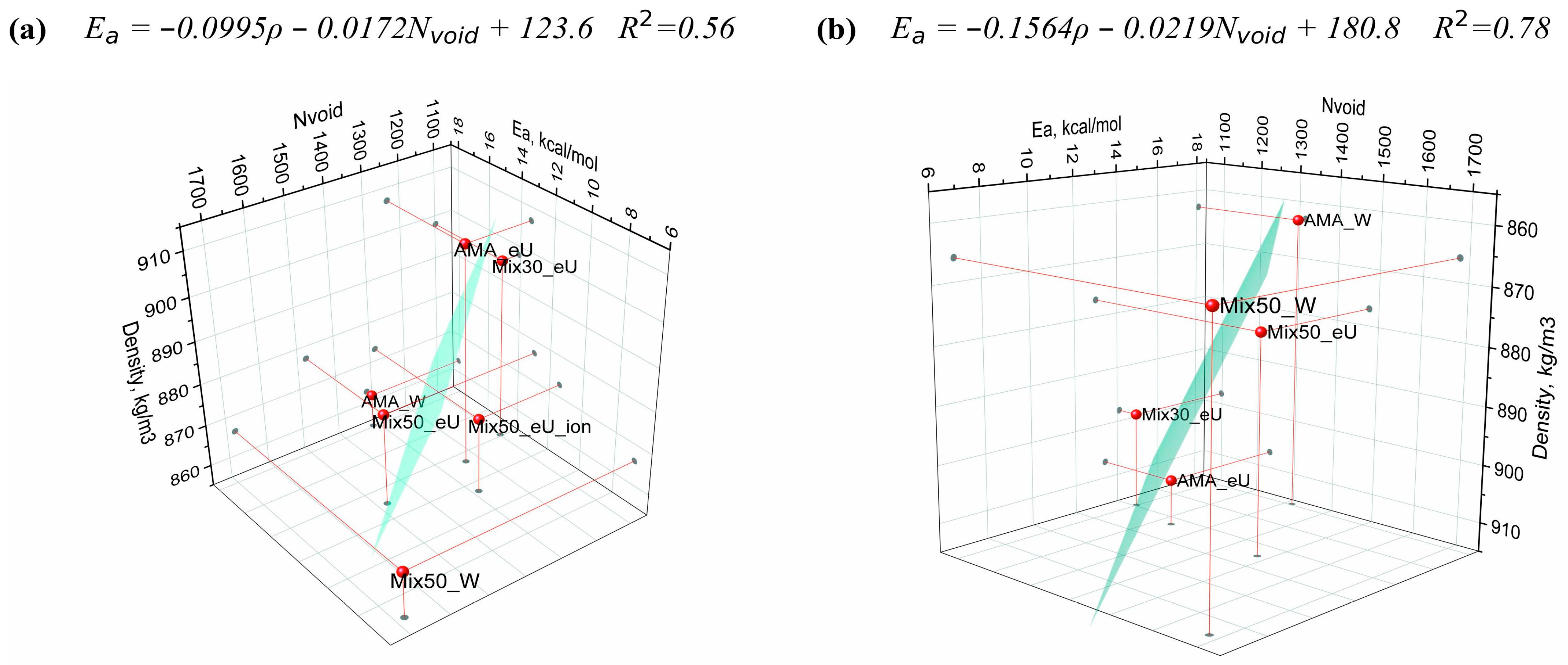
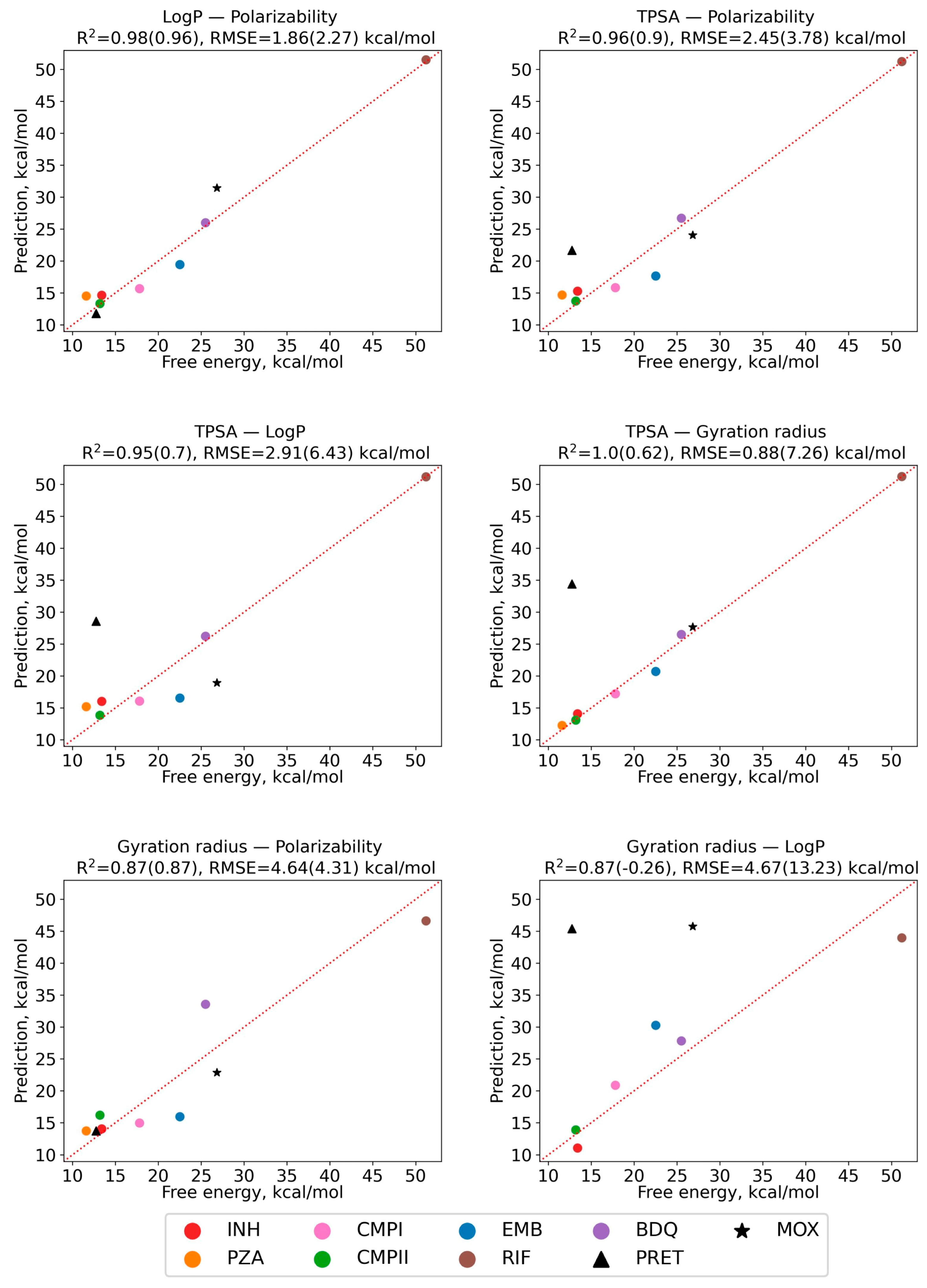
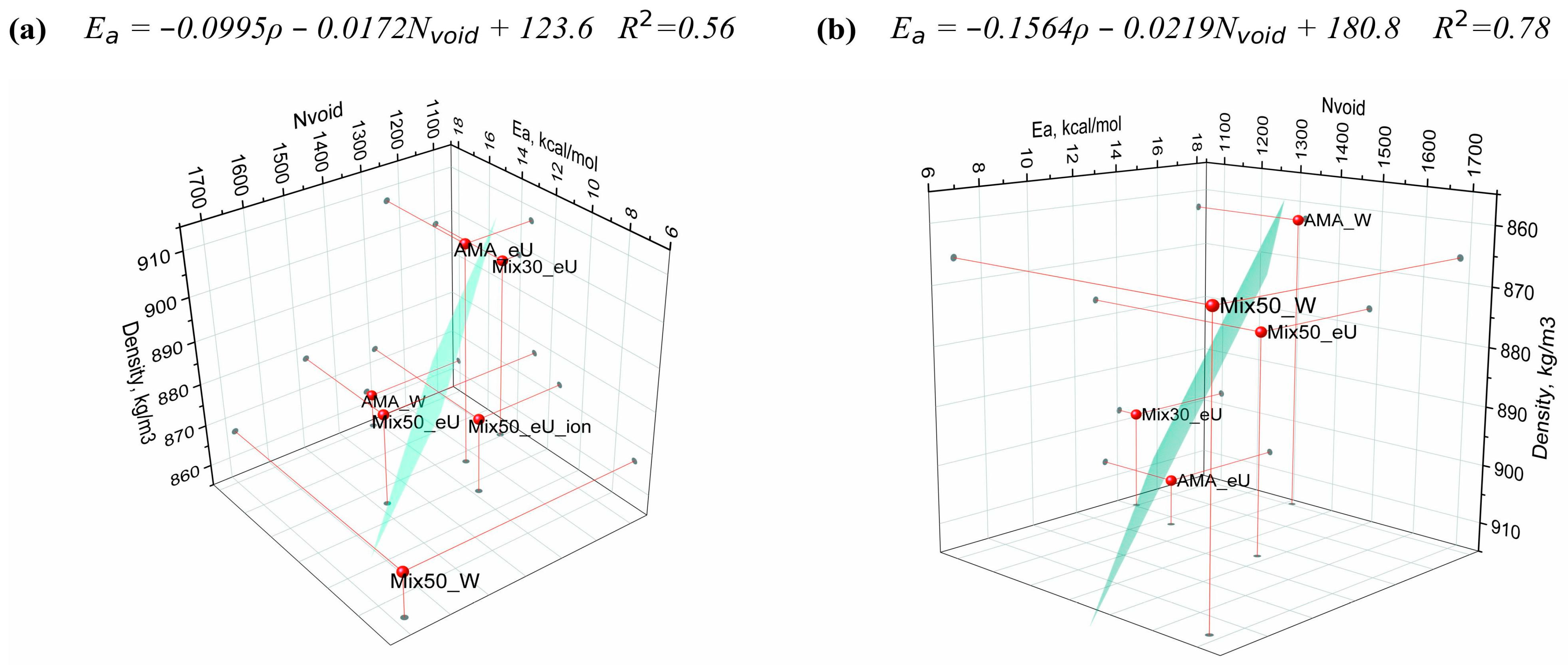
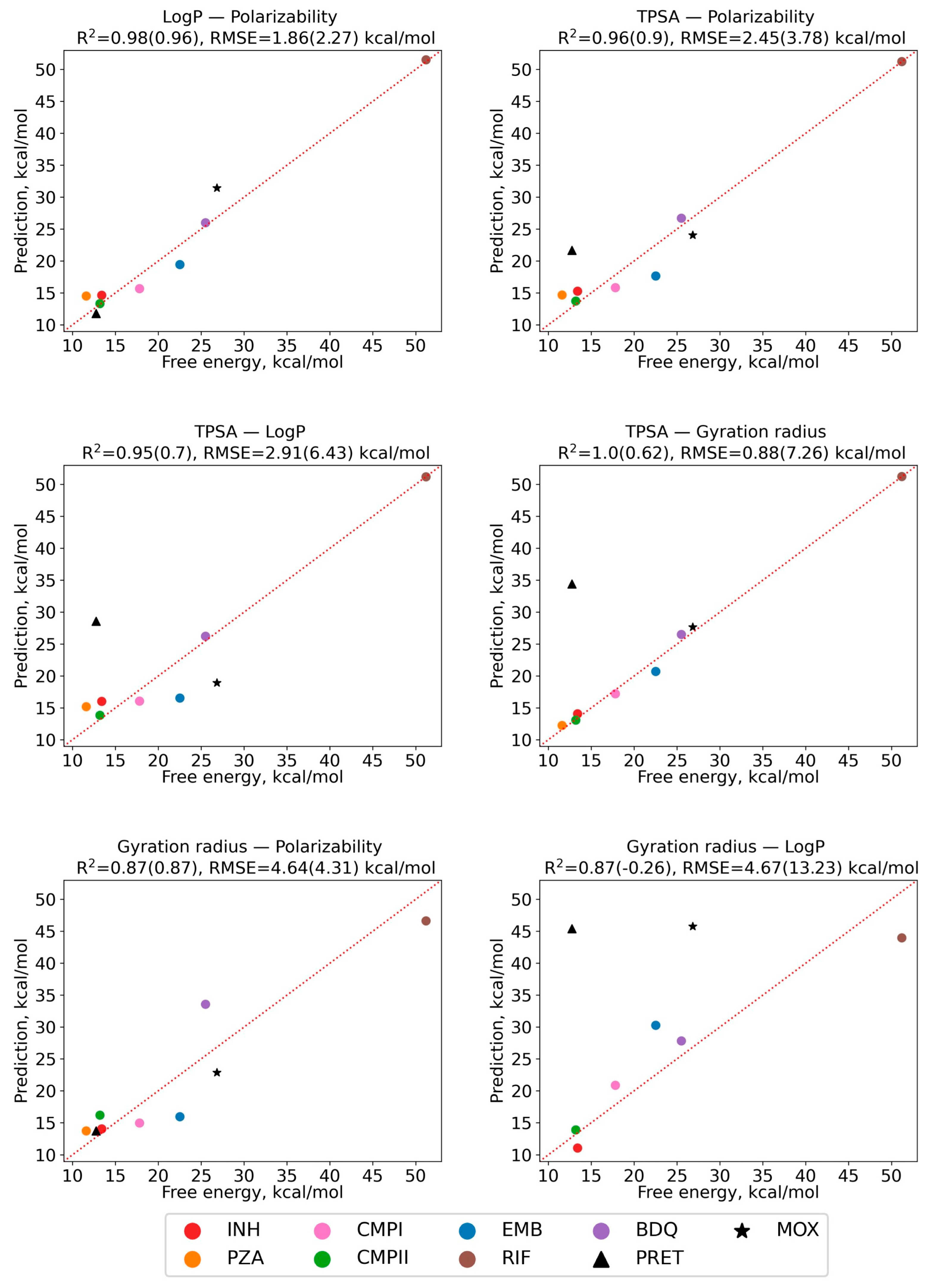
2.6. Regression Model of Diffusion Activation Barriers Based on Solute Molecular Properties
3. Materials and Methods
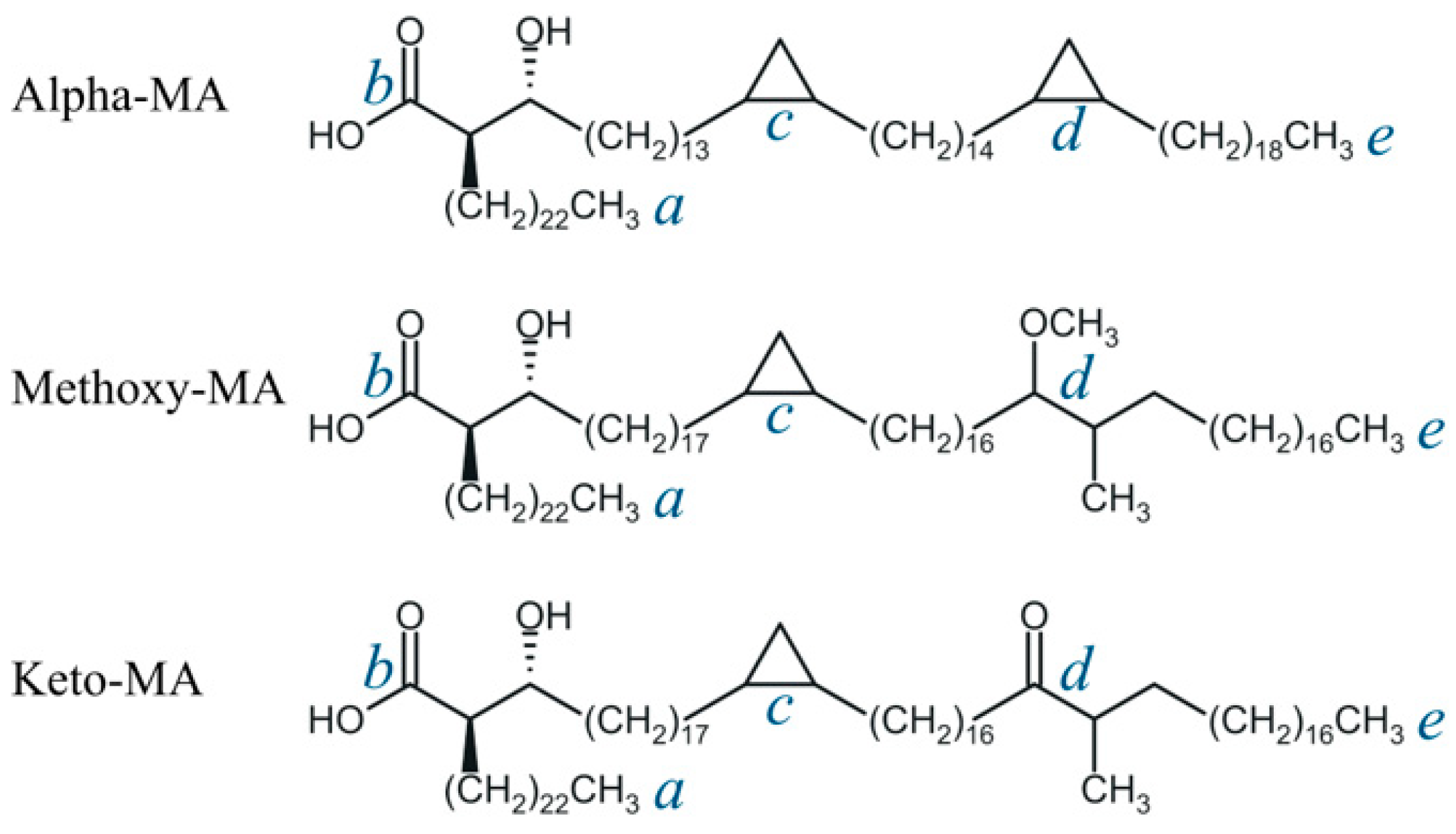
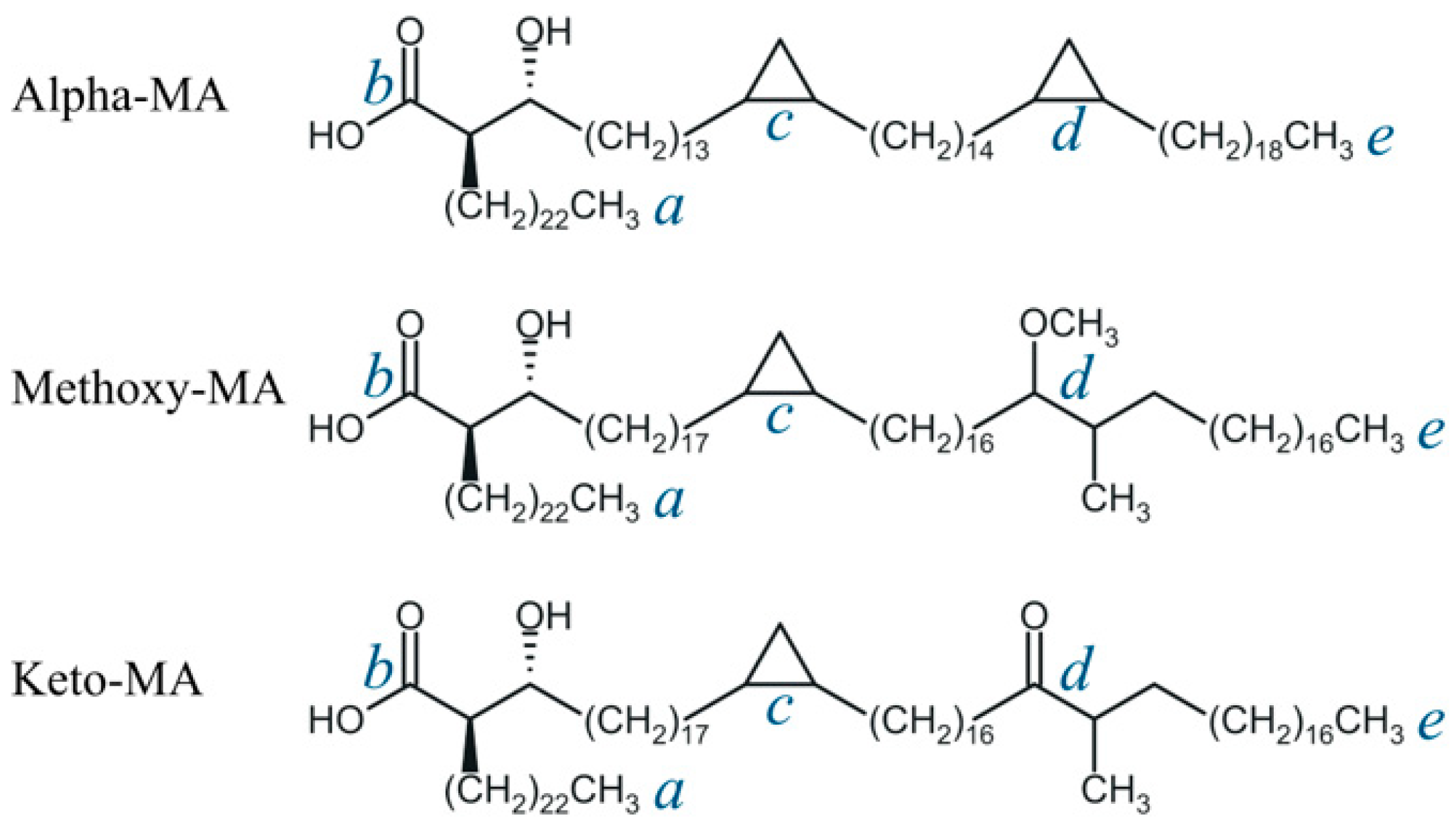
3.1. Molecular Properties of Drugs and Mycolic Acids
3.2. Model Membranes
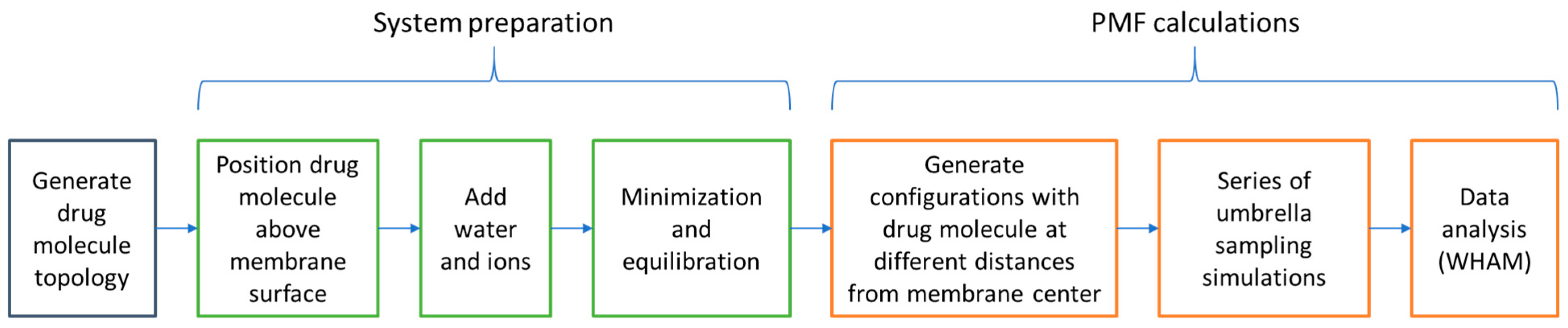
3.3. Molecular Dynamics Simulations
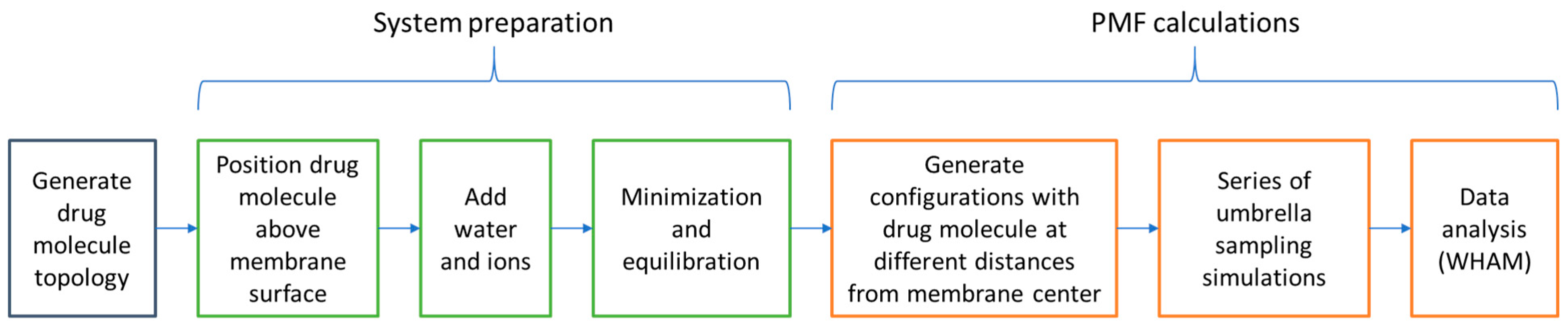
3.4. Umbrella Sampling and PMF Analysis
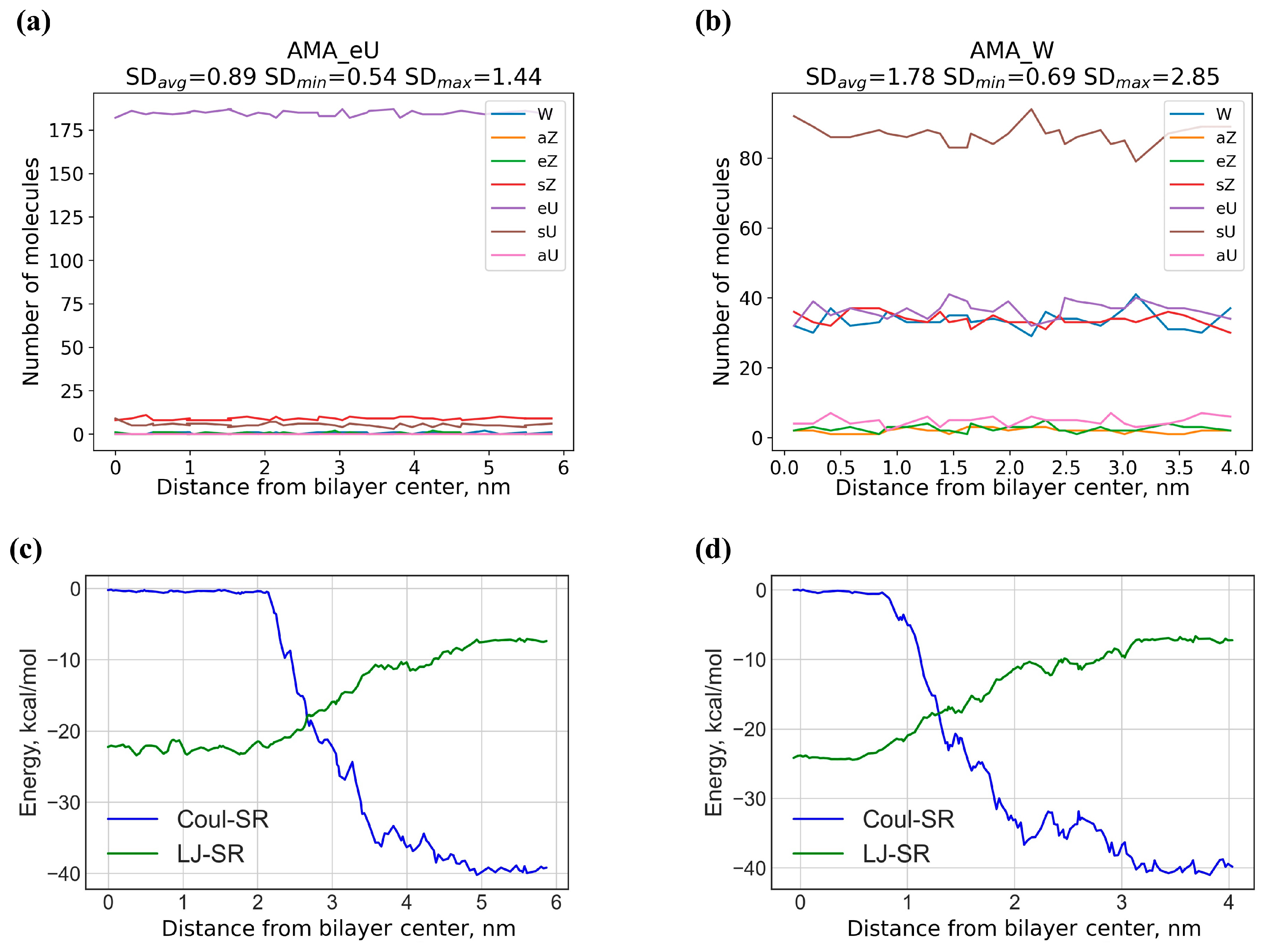
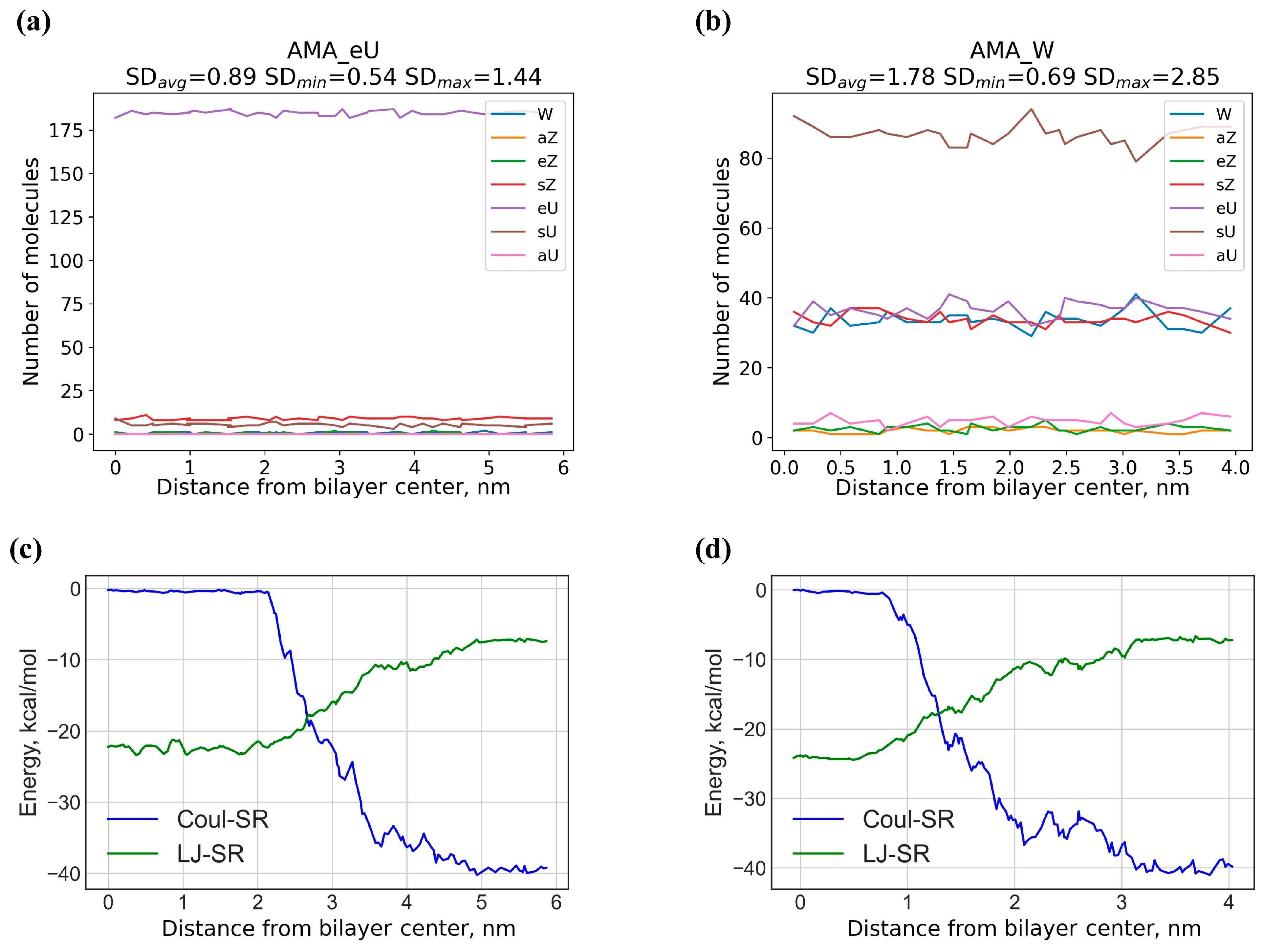
3.5. Analysis of MD Trajectories
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022; ISBN 978-92-4-006172-9. [Google Scholar]
- The Path That Ends AIDS: UNAIDS Global AIDS Update 2023; Joint United Nations Programme on HIV/AIDS: Geneva, Switzerland, 2023.
- Rao, M.; Ippolito, G.; Mfinanga, S.; Ntoumi, F.; Yeboah-Manu, D.; Vilaplana, C.; Zumla, A.; Maeurer, M. Latent TB Infection (LTBI)—Mycobacterium tuberculosis pathogenesis and the dynamics of the granuloma battleground. Int. J. Infect. Dis. 2019, 80, S58–S61. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.M.G.J.; Dodd, P.J. The global burden of latent tuberculosis infection: A re-estimation using mathematical modelling. PLoS Med. 2016, 13, e1002152. [Google Scholar] [CrossRef]
- Dulberger, C.L.; Rubin, E.J.; Boutte, C.C. The mycobacterial cell envelope—A moving target. Nat. Rev. Microbiol. 2020, 18, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Craggs, P.D.; De Carvalho, L.P.S. Bottlenecks and opportunities in antibiotic discovery against Mycobacterium tuberculosis. Curr. Opin. Microbiol. 2022, 69, 102191. [Google Scholar] [CrossRef] [PubMed]
- Bhat, Z.S.; Rather, M.A.; Maqbool, M.; Lah, H.U.; Yousuf, S.K.; Ahmad, Z. Cell wall: A versatile fountain of drug targets in Mycobacterium tuberculosis. Biomed. Pharmacother. 2017, 95, 1520–1534. [Google Scholar] [CrossRef]
- Batt, S.M.; Minnikin, D.E.; Besra, G.S. The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host’s immune system. Biochem. J. 2020, 477, 1983–2006. [Google Scholar] [CrossRef]
- Marrakchi, H.; Lanéelle, M.-A.; Daffé, M. Mycolic acids: Structures, biosynthesis, and beyond. Chem. Biol. 2014, 21, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Aoyagi, Y.; Ridell, M.; Minnikin, D.E. Separation and characterization of individual mycolic acids in representative mycobacteria. Microbiology 2001, 147, 1825–1837. [Google Scholar] [CrossRef]
- Takayama, K.; Wang, C.; Besra, G.S. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2005, 18, 81–101. [Google Scholar] [CrossRef]
- Villeneuve, M.; Kawai, M.; Watanabe, M.; Aoyagi, Y.; Hitotsuyanagi, Y.; Takeya, K.; Gouda, H.; Hirono, S.; Minnikin, D.E.; Nakahara, H. Differential conformational behaviors of α-mycolic acids in Langmuir monolayers and computer simulations. Chem. Phys. Lipids 2010, 163, 569–579. [Google Scholar] [CrossRef]
- Lambert, P.A. Cellular impermeability and uptake of biocides and antibiotics in Gram-positive bacteria and mycobacteria: Antimicrobial penetration of walls. J. Appl. Microbiol. 2002, 92, 46S–54S. [Google Scholar] [CrossRef]
- Savintseva, L.A.; Steshin, I.S.; Avdoshin, A.A.; Panteleev, S.V.; Rozhkov, A.V.; Shirokova, E.A.; Livshits, G.D.; Vasyankin, A.V.; Radchenko, E.V.; Ignatov, S.K.; et al. Conformational dynamics and stability of bilayers formed by mycolic acids from the Mycobacterium tuberculosis outer membrane. Molecules 2023, 28, 1347. [Google Scholar] [CrossRef]
- Sarathy, J.; Dartois, V.; Lee, E. The role of transport mechanisms in Mycobacterium tuberculosis drug resistance and tolerance. Pharmaceuticals 2012, 5, 1210–1235. [Google Scholar] [CrossRef]
- Jarlier, V.; Nikaido, H. Mycobacterial cell wall: Structure and role in natural resistance to antibiotics. FEMS Microbiol. Lett. 1994, 123, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nikaido, H. A mutant of Mycobacterium smegmatis defective in the biosynthesis of mycolic acids accumulates meromycolates. Proc. Natl. Acad. Sci. USA 1999, 96, 4011–4016. [Google Scholar] [CrossRef] [PubMed]
- Trias, J.; Benz, R. Permeability of the cell wall of Mycobacterium smegmatis. Mol. Microbiol. 1994, 14, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Jarlier, V.; Nikaido, H. Permeability barrier to hydrophilic solutes in Mycobacterium chelonei. J. Bacteriol. 1990, 172, 1418–1423. [Google Scholar] [CrossRef]
- Hong, X.; Hopfinger, A.J. Molecular modeling and simulation of Mycobacterium tuberculosis cell wall permeability. Biomacromolecules 2004, 5, 1066–1077. [Google Scholar] [CrossRef]
- Merget, B.; Zilian, D.; Müller, T.; Sotriffer, C.A. MycPermCheck: The Mycobacterium tuberculosis permeability prediction tool for small molecules. Bioinformatics 2013, 29, 62–68. [Google Scholar] [CrossRef]
- Lee, S.-H.; Choi, M.; Kim, P.; Myung, P.K. 3D-QSAR and cell wall permeability of antitubercular nitroimidazoles against Mycobacterium tuberculosis. Molecules 2013, 18, 13870–13885. [Google Scholar] [CrossRef]
- Janardhan, S.; Ram Vivek, M.; Narahari Sastry, G. Modeling the permeability of drug-like molecules through the cell wall of Mycobacterium tuberculosis: An analogue based approach. Mol. BioSyst. 2016, 12, 3377–3384. [Google Scholar] [CrossRef]
- Nagamani, S.; Sastry, G.N. Mycobacterium tuberculosis cell wall permeability model generation using chemoinformatics and machine learning approaches. ACS Omega 2021, 6, 17472–17482. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Antonyan, G.V.; Ignatov, S.K.; Palyulin, V.A. Machine learning prediction of mycobacterial cell wall permeability of drugs and drug-like compounds. Molecules 2023, 28, 633. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, C.; Lanéelle, M.-A.; Senaratne, R.H.; Draper, P.; Lanéelle, G.; Daffé, M. Mechanisms of pyrazinamide resistance in mycobacteria: Importance of lack of uptake in addition to lack of pyrazinamidase activity. Microbiology 1999, 145, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Bardou, F.; Raynaud, C.; Ramos, C.; Lanéelle, M.A.; Lanŕelle, G. Mechanism of isoniazid uptake in Mycobacterium tuberculosis. Microbiology 1998, 144, 2539–2544. [Google Scholar] [CrossRef] [PubMed]
- Beggs, W.H.; Auran, N.E. Uptake and binding of 14C-ethambutol by tubercle bacilli and the relation of binding to growth inhibition. Antimicrob. Agents Chemother. 1972, 2, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Giraud-Gatineau, A.; Coya, J.M.; Maure, A.; Biton, A.; Thomson, M.; Bernard, E.M.; Marrec, J.; Gutierrez, M.G.; Larrouy-Maumus, G.; Brosch, R.; et al. The antibiotic bedaquiline activates host macrophage innate immune resistance to bacterial infection. eLife 2020, 9, e55692. [Google Scholar] [CrossRef]
- Le-Deygen, I.M.; Safronova, A.S.; Mamaeva, P.V.; Kolmogorov, I.M.; Skuredina, A.A.; Kudryashova, E.V. Drug–membrane interaction as revealed by spectroscopic methods: The role of drug structure in the example of Rifampicin, Levofloxacin and Rapamycin. Biophysica 2022, 2, 353–365. [Google Scholar] [CrossRef]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta BBA—Proteins Proteom. 2009, 1794, 808–816. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Bioassay Record for AID 1626, High Throughput Screen to Identify Inhibitors of Mycobacterium tuberculosis H37Rv. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1626 (accessed on 4 December 2023).
- National Center for Biotechnology Information. PubChem Bioassay Record for AID 449762, High Throughput Screening Assay used to Identify Novel Compounds that Inhibit Mycobacterium tuberculosis in 7H9 Media. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/449762 (accessed on 4 December 2023).
- National Center for Biotechnology Information. PubChem Bioassay Record for AID 488890, Elucidation of physiology of non-replicating, drug-tolerant Mycobacterium tuberculosis. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/488890 (accessed on 4 December 2023).
- Piccaro, G.; Poce, G.; Biava, M.; Giannoni, F.; Fattorini, L. Activity of lipophilic and hydrophilic drugs against dormant and replicating Mycobacterium tuberculosis. J. Antibiot. 2015, 68, 711–714. [Google Scholar] [CrossRef]
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Testa, B.; Fahr, A. Lipophilicity and Its relationship with passive drug permeation. Pharm. Res. 2011, 28, 962–977. [Google Scholar] [CrossRef]
- Tapfumaneyi, P.; Imran, M.; Alavi, S.E.; Mohammed, Y. Science of, and insights into, thermodynamic principles for dermal formulations. Drug Discov. Today 2023, 28, 103521. [Google Scholar] [CrossRef] [PubMed]
- Orsi, M.; Essex, J.W. Permeability of drugs and hormones through a lipid bilayer: Insights from dual-resolution molecular dynamics. Soft. Matter 2010, 6, 3797. [Google Scholar] [CrossRef]
- Orsi, M.; Noro, M.G.; Essex, J.W. Dual-resolution molecular dynamics simulation of antimicrobials in biomembranes. J. R. Soc. Interface 2011, 8, 826–841. [Google Scholar] [CrossRef]
- Paloncýová, M.; Fabre, G.; DeVane, R.H.; Trouillas, P.; Berka, K.; Otyepka, M. Benchmarking of force fields for molecule–membrane interactions. J. Chem. Theory Comput. 2014, 10, 4143–4151. [Google Scholar] [CrossRef]
- Basu, S.; Mandal, S.; Maiti, P.K. Drug-bacterial membrane interactions: A tale of two force fields. Available online: http://biorxiv.org/lookup/doi/10.1101/2023.09.27.559676 (accessed on 26 November 2023).
- Meng, F.; Xu, W. Drug permeability prediction using PMF method. J. Mol. Model. 2013, 19, 991–997. [Google Scholar] [CrossRef]
- Bennion, B.J.; Be, N.A.; McNerney, M.W.; Lao, V.; Carlson, E.M.; Valdez, C.A.; Malfatti, M.A.; Enright, H.A.; Nguyen, T.H.; Lightstone, F.C.; et al. Predicting a drug’s membrane permeability: A computational model validated with in vitro permeability assay data. J. Phys. Chem. B 2017, 121, 5228–5237. [Google Scholar] [CrossRef]
- Brown, C.M.; Corey, R.A.; Gao, Y.; Choi, Y.K.; Gilleron, M.; Destainville, N.; Fullam, E.; Im, W.; Stansfeld, P.J.; Chavent, M. From molecular dynamics to supramolecular organization: The role of PIM lipids in the originality of the mycobacterial plasma membrane. Available online: http://biorxiv.org/lookup/doi/10.1101/2022.06.29.498153 (accessed on 26 November 2023).
- Chemicalize—Instant Cheminformatics Solutions. Available online: https://chemicalize.com/ (accessed on 4 December 2023).
- RDKit: Open-Source Cheminformatics Software. Available online: https://www.rdkit.org/ (accessed on 1 September 2023).
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Dragotakes, Q.; Stouffer, K.M.; Fu, M.S.; Sella, Y.; Youn, C.; Yoon, O.I.; De Leon-Rodriguez, C.M.; Freij, J.B.; Bergman, A.; Casadevall, A. Macrophages use a bet-hedging strategy for antimicrobial activity in phagolysosomal acidification. J. Clin. Investig. 2020, 130, 3805–3819. [Google Scholar] [CrossRef]
- Bansal-Mutalik, R.; Nikaido, H. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc. Natl. Acad. Sci. USA 2014, 111, 4958–4963. [Google Scholar] [CrossRef] [PubMed]
- Innes, E.; Yiu, H.H.P.; McLean, P.; Brown, W.; Boyles, M. Simulated biological fluids—A systematic review of their biological relevance and use in relation to inhalation toxicology of particles and fibres. Crit. Rev. Toxicol. 2021, 51, 217–248. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Corradi, V.; Souza, P.C.T.; Ingólfsson, H.I.; Tieleman, D.P.; Sansom, M.S.P. Computational modeling of realistic cell membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2017, 38, 1114–1124. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Hub, J.S.; De Groot, B.L.; Van Der Spoel, D. g_wham—A free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- The PLUMED consortium Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. [CrossRef]
- Harris, C.R.; Millman, K.J.; Van Der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise. In KDD’96: Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, Portland, Oregon, USA, 2–4 August 1996; AAAI Press: Portland, Oregon, 1996; pp. 226–231. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Designation | Molar Mass, g/mol | Gyration Radius, nm | TPSA, Å2 | LogP [35] (cLogP) | Polarizability α, Å3 |
|---|---|---|---|---|---|---|
| Isoniazid | INH | 137.1 | 0.23 | 68.0 | −0.71 (−0.69) | 13.7 |
| Pyrazinamid | PZA | 123.1 | 0.20 | 68.9 | −0.71 (−1.22) | 11.4 |
| Etambutol | EMB | 204.3 | 0.34 | 64.5 | −0.12 (−0.06) | 23.4 |
| Rifampicin | RIF | 823.0 | 0.48 | 220.2 | 3.85 (3.50) | 85.5 |
| Bedaquilin | BDQ | 555.5 | 0.47 | 45.6 | 6.37 (7.13) | 61.9 |
| Pretomanid | PRET | 359.3 | 0.50 | 88.7 | (4.14) | 28.8 |
| Moxifloxacin | MOX | 401.4 | 0.41 | 82.1 | (−0.49) | 39.6 |
| MOPP a | CMPI | 190.2 | 0.30 | 57.9 | (0.33) | 19.3 |
| CPMP b | CMPII | 208.6 | 0.29 | 32.7 | (1.61) | 20.9 |
| Diffusing Molecule | Energy of Adsorption at the Membrane Surface Layer Eads Relative to the Bulk Solution, kcal/mol | Free Energy Barrier Relative to Minimum Point Ea of Energy Profile (Relative to Bulk Solution E0a), kcal/mol | ||||
|---|---|---|---|---|---|---|
| AMA_eU | Mix50_eU | Mix50_eU_ion | AMA_eU | Mix50_eU | Mix50_eU_ion | |
| Neutral forms | ||||||
| INH | 1.3 | 1.5 | 1.5 | 13.4 (12.1) | 13 (11.5) | 11.4 (9.9) |
| PZA | 1.7 | 1.5 | 1.4 | 11.6 (9.6) | 5.7 (4.2) | 9.1 (7.7) |
| CMPI | 4.2 | 2.4 | 4.2 | 17.8 (11.4) | 17.1 (14.8) | 14.1 (9.9) |
| CMPII | 3.8 | 2.0 | 5.6 | 13.2 (6.7) | 8.4 (6.3) | 10.5 (4.8) |
| EMB | 2.4 | 3.0 | 2.2 | 22.5 (20.1) | 11.6 (8.6) | 18.0 (15.7) |
| RIF | 7.3 | 6.8 | 6.5 | 51.2 (43.6) | 25.4 (18.6) | 33.3 (26.9) |
| BDQ | 9.2 | 7.4 | 10.9 | 25.5 (13.5) | 18.6 (11.2) | 10.2 (−0.7) |
| Ionized forms | ||||||
| EMB+ | 0.9 | 0.4 | 3.5 | 63.3 (62.4) | 62.8 (62.5) | 65.8 (62.3) |
| RIF+ | 2.3 | 3.7 | 9.3 | 58.6 (56.3) | 48.2 (44.4) | 41.4 (32.2) |
| BDQ+ | 13 | 9.1 | 8.9 | 64.3 (49.4) | 36.6 (27.4) | 44.8 (36.0) |
| AMA_W | Mix50_W | Mix50_eU | Mix30_eU | AMA_eU | Mix50_eU_ion | |
|---|---|---|---|---|---|---|
| Thickness d, nm | 4.40 | 4.50 | 5.00 | 5.90 | 7.80 | 5.10 |
| , kg/m3 | 863 | 866 | 877 | 898 | 907 | 873 |
| Number of void spheres, Nvoid | 1315 | 1679 | 1477 | 1098 | 1234 | 1285 |
| Volume of membrane a Vm, nm3 | 407.2 | 427.8 | 420.4 | 411.2 | 404.4 | 427.0 |
| Fraction of free volume, % | 0.59 | 0.70 | 0.64 | 0.49 | 0.54 | 0.54 |
| Diffusion barrier Ea (E0a), kcal/mol | 18.1 (17.0) | 6.9 (5.6) | 13.0 (11.5) | 14.1 (12.5) | 13.4 (12.1) | 11.4 (9.9) |
| Adsorption energy Eads, kcal/mol | 1.1 | 1.2 | 1.5 | 1.6 | 1.3 | 1.5 |
| AMA_eU | Mix50_eU | |
|---|---|---|
| Total number of void spheres | 1234 | 1477 |
| Number of void spheres not assigned to any cluster | 559 | 547 |
| Ratio of the non-clustered void spheres to the total void spheres, % | 45 | 37 |
| Number of clusters | 53 | 50 |
| Maximum cluster size | 58 | 132 |
| Average cluster size | 13 | 19 |
| Membrane | AMA_W | AMA_eU | Mix30_eU | Mix50_eU | Mix50_W | Mix50_eU_ion |
|---|---|---|---|---|---|---|
| Number of MA molecules in a box AMA:KMA:MMA | 200:0:0 | 200:0:0 | 140:30:30 | 100:50:50 | 100:50:50 | 100:50:50 |
| Initial conformations of AMA:KMA:MMA | W:–:– | eU:–:– | eU:W:W | eU:W:W | W:W:W | eU:W:W |
| Time of equilibration, ns | 1200 | 300 | 300 | 300 | 300 | 100 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steshin, I.S.; Vasyankin, A.V.; Shirokova, E.A.; Rozhkov, A.V.; Livshits, G.D.; Panteleev, S.V.; Radchenko, E.V.; Ignatov, S.K.; Palyulin, V.A. Free Energy Barriers for Passive Drug Transport through the Mycobacterium tuberculosis Outer Membrane: A Molecular Dynamics Study. Int. J. Mol. Sci. 2024, 25, 1006. https://doi.org/10.3390/ijms25021006
Steshin IS, Vasyankin AV, Shirokova EA, Rozhkov AV, Livshits GD, Panteleev SV, Radchenko EV, Ignatov SK, Palyulin VA. Free Energy Barriers for Passive Drug Transport through the Mycobacterium tuberculosis Outer Membrane: A Molecular Dynamics Study. International Journal of Molecular Sciences. 2024; 25(2):1006. https://doi.org/10.3390/ijms25021006
Chicago/Turabian StyleSteshin, Ilya S., Alexander V. Vasyankin, Ekaterina A. Shirokova, Alexey V. Rozhkov, Grigory D. Livshits, Sergey V. Panteleev, Eugene V. Radchenko, Stanislav K. Ignatov, and Vladimir A. Palyulin. 2024. "Free Energy Barriers for Passive Drug Transport through the Mycobacterium tuberculosis Outer Membrane: A Molecular Dynamics Study" International Journal of Molecular Sciences 25, no. 2: 1006. https://doi.org/10.3390/ijms25021006
APA StyleSteshin, I. S., Vasyankin, A. V., Shirokova, E. A., Rozhkov, A. V., Livshits, G. D., Panteleev, S. V., Radchenko, E. V., Ignatov, S. K., & Palyulin, V. A. (2024). Free Energy Barriers for Passive Drug Transport through the Mycobacterium tuberculosis Outer Membrane: A Molecular Dynamics Study. International Journal of Molecular Sciences, 25(2), 1006. https://doi.org/10.3390/ijms25021006