
Neurogenic Effects of Inorganic Arsenic and Cdk5 Knockdown in Zebrafish Embryos: A Perspective on Modeling Autism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Risk Genes of Autism and Zebrafish
3. Autism and Overproduction and Reduction of Specific Neurons
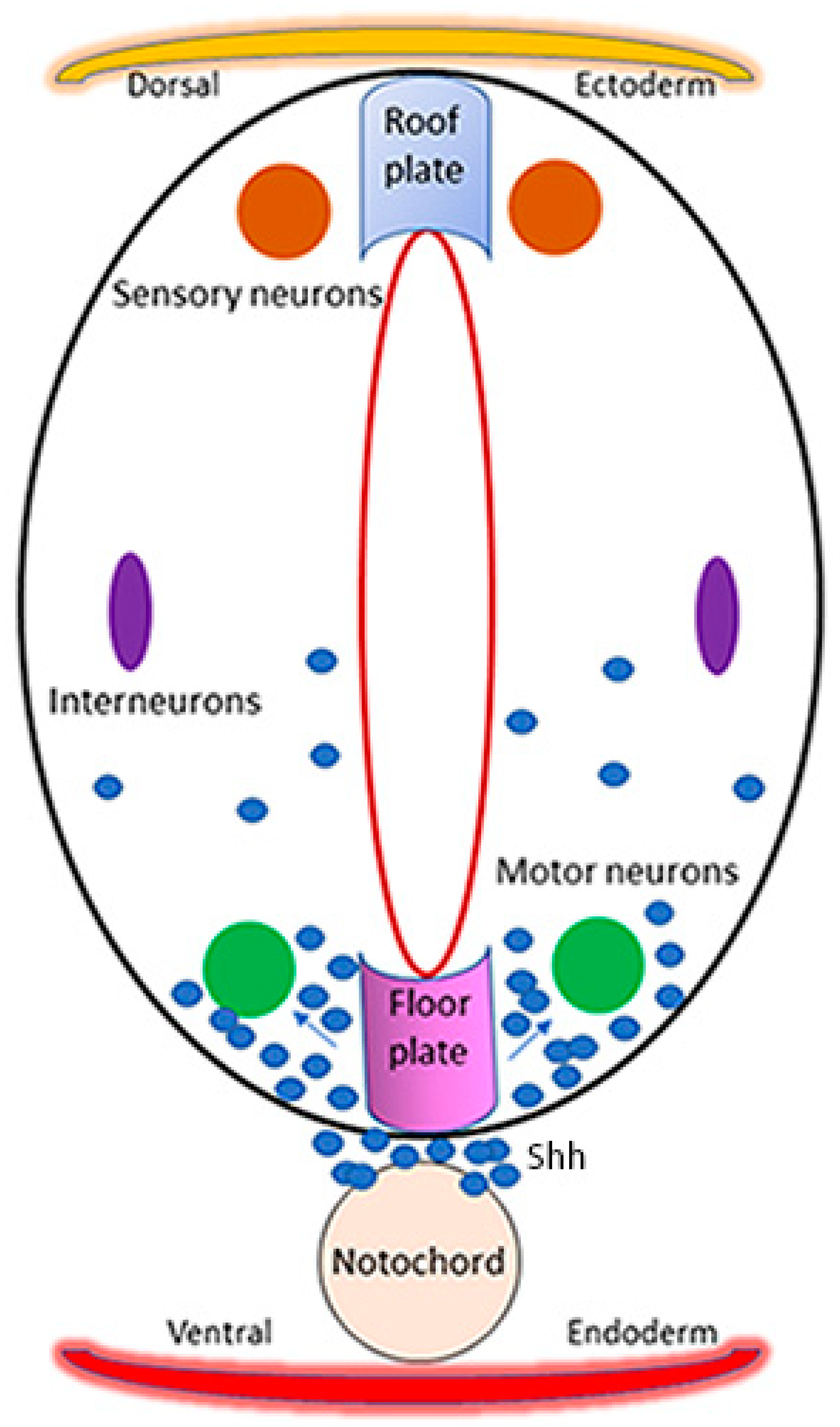
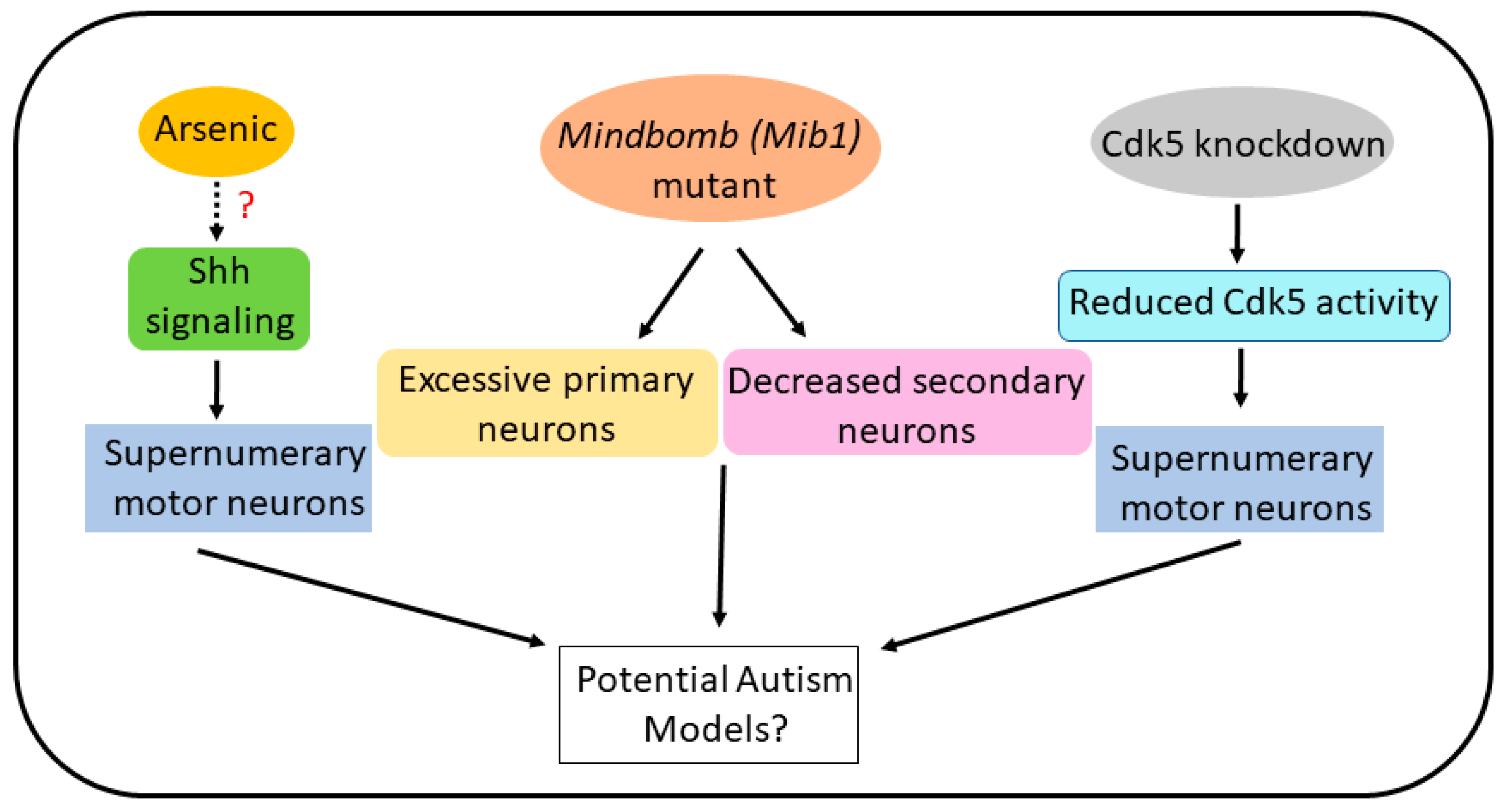
4. Arsenic and Zebrafish Motor Neurons: Relevance to Autism
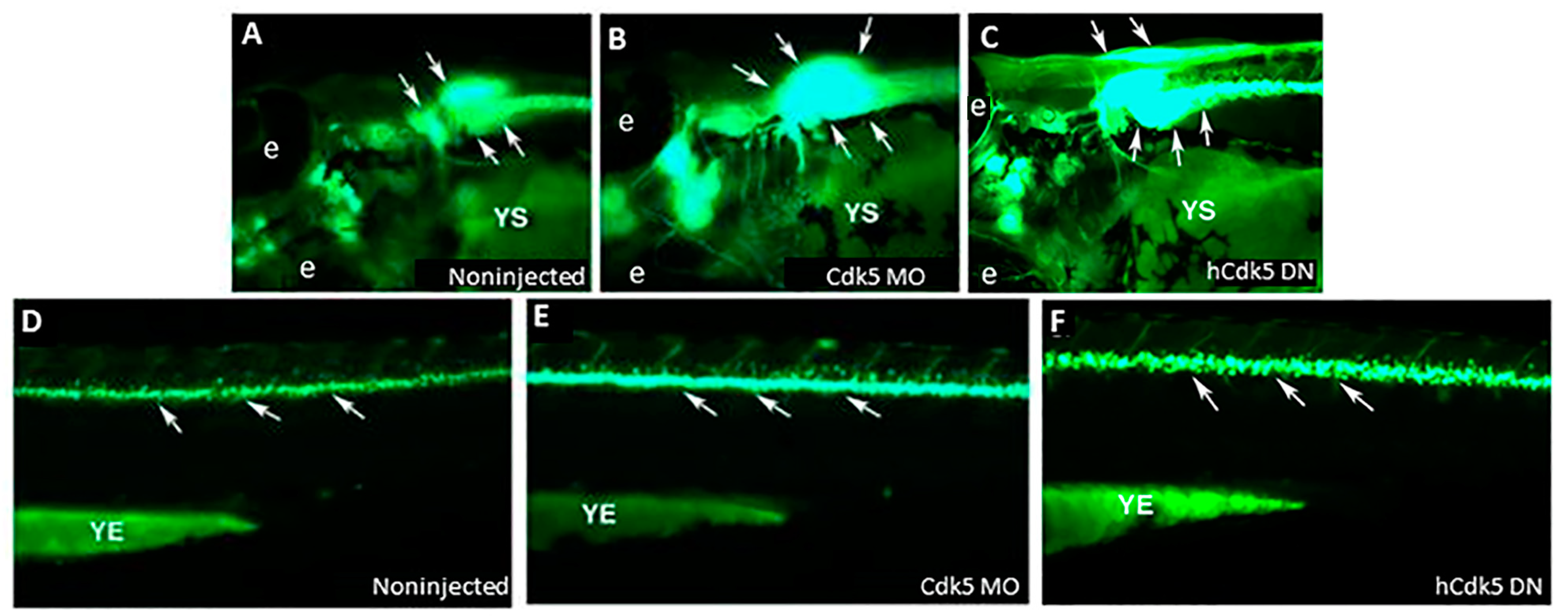
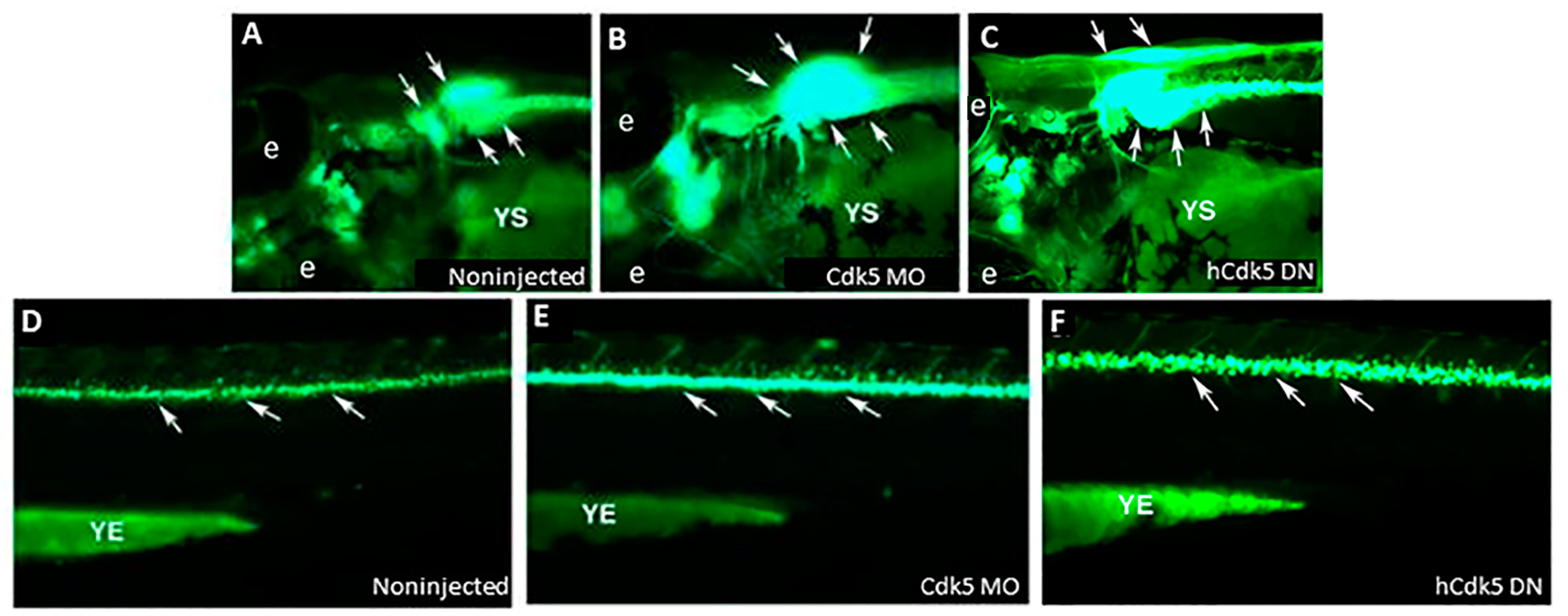
5. Cdk5 Knockdown and Zebrafish Motor Neurons: Relevance to Autism
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASD | Autism spectrum disorder |
| Cdk5 | Cyclin-dependent kinase 5 |
| CHD8 | Chromodomain helicase DNA binding protein 8 |
| FMR1 | Fragile X Messenger Ribonucleoprotein 1 |
| GFP | Green fluorescent protein |
| Gli | Glioma-associated oncogene, a zinc finger protein |
| MECP2 | Methyl CpG binding protein 2 |
| Mib1 | Mindbomb E3 ubiquitin protein ligase 1 |
| NLGN3 | Neuroligin-3 |
| NLGN4 | Neuroligin-4 |
| NRXN1 | Neurexin 1 |
| SHANK3 | SH3 and multiple ankyrin repeat domains 3 (also known as proline-rich synapse-associated protein 2) |
| TSC1/2 | Tuberous sclerosis complex 1/2 |
| UBE3A | Ubiquitin-protein ligase E3A (also known as E6AP ubiquitin-protein ligase) |
References
- Pierce, K.; Gazestani, V.H.; Bacon, E.; Barnes, C.C.; Cha, D.; Nalabolu, S.; Lopez, L.; Moore, A.; Pence-Stophaeros, S.; Courchesne, E. Evaluation of the Diagnostic Stability of the Early Autism Spectrum Disorder Phenotype in the General Population Starting at 12 Months. JAMA Pediatr. 2019, 173, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Rea, V.; Van Raay, T.J. Using Zebrafish to Model Autism Spectrum Disorder: A Comparison of ASD Risk Genes between Zebrafish and Their Mammalian Counterparts. Front. Mol. Neurosci. 2020, 13, 575575. [Google Scholar] [CrossRef] [PubMed]
- Dyches, T.T.; Wilder, L.K.; Sudweeks, R.R.; Obiakor, F.E.; Algozzine, B. Multicultural issues in autism. J. Autism Dev. Disord. 2004, 34, 211–222. [Google Scholar] [CrossRef]
- Szatmari, P.; Georgiades, S.; Bryson, S.; Zwaigenbaum, L.; Roberts, W.; Mahoney, W.; Goldberg, J.; Tuff, L. Investigating the structure of the restricted, repetitive behaviours and interests domain of autism. J. Child Psychol. Psychiatry 2006, 47, 582–590. [Google Scholar] [CrossRef]
- Sharma, A.; Bhalla, S.; Mehan, S. PI3K/AKT/mTOR signalling inhibitor chrysophanol ameliorates neurobehavioural and neurochemical defects in propionic acid-induced experimental model of autism in adult rats. Metab. Brain Dis. 2022, 37, 1909–1929. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.R.; Gonda, X.; Tarazi, F.I. Autism Spectrum Disorder: Classification, diagnosis and therapy. Pharmacol. Ther. 2018, 190, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Morales Hidalgo, P.; Voltas Moreso, N.; Canals Sans, J. Autism spectrum disorder prevalence and associated sociodemographic factors in the school population: EPINED study. Autism 2021, 25, 1999–2011. [Google Scholar] [CrossRef]
- CDC. Autism Prevalence Higher, According to Data from 11 ADDM Communities; Center for Disease Control, CDC News: Atlanta, GA, USA, 2023. [Google Scholar]
- Fakhoury, M. Autistic spectrum disorders: A review of clinical features, theories and diagnosis. Int. J. Dev. Neurosci. 2015, 43, 70–77. [Google Scholar] [CrossRef]
- Mandy, W.; Lai, M.C. Annual Research Review: The role of the environment in the developmental psychopathology of autism spectrum condition. J. Child Psychol. Psychiatry 2016, 57, 271–292. [Google Scholar] [CrossRef]
- Yasuda, H.; Tsutsui, T. Assessment of infantile mineral imbalances in autism spectrum disorders (ASDs). Int. J. Environ. Res. Public Health 2013, 10, 6027–6043. [Google Scholar] [CrossRef]
- Huang, J.Y.; Tian, Y.; Wang, H.J.; Shen, H.; Wang, H.; Long, S.; Liao, M.H.; Liu, Z.R.; Wang, Z.M.; Li, D.; et al. Functional Genomic Analyses Identify Pathways Dysregulated in Animal Model of Autism. CNS Neurosci. Ther. 2016, 22, 845–853. [Google Scholar] [CrossRef]
- Jiang, C.C.; Lin, L.S.; Long, S.; Ke, X.Y.; Fukunaga, K.; Lu, Y.M.; Han, F. Signalling pathways in autism spectrum disorder: Mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2022, 7, 229. [Google Scholar] [CrossRef]
- Ding, M.; Shi, S.; Qie, S.; Li, J.; Xi, X. Association between heavy metals exposure (cadmium, lead, arsenic, mercury) and child autistic disorder: A systematic review and meta-analysis. Front. Pediatr. 2023, 11, 1169733. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Flieger, W.; Flieger, M.; Forma, A.; Sitarz, E.; Skórzyńska-Dziduszko, K.; Grochowski, C.; Maciejewski, R.; Karakuła-Juchnowicz, H. Autism spectrum disorder: Trace elements imbalances and the pathogenesis and severity of autistic symptoms. Neurosci. Biobehav. Rev. 2021, 129, 117–132. [Google Scholar] [CrossRef]
- Skogheim, T.S.; Weyde, K.V.F.; Engel, S.M.; Aase, H.; Surén, P.; Øie, M.G.; Biele, G.; Reichborn-Kjennerud, T.; Caspersen, I.H.; Hornig, M.; et al. Metal and essential element concentrations during pregnancy and associations with autism spectrum disorder and attention-deficit/hyperactivity disorder in children. Environ. Int. 2021, 152, 106468. [Google Scholar] [CrossRef]
- Dickerson, A.S.; Rahbar, M.H.; Han, I.; Bakian, A.V.; Bilder, D.A.; Harrington, R.A.; Pettygrove, S.; Durkin, M.; Kirby, R.S.; Wingate, M.S.; et al. Autism spectrum disorder prevalence and proximity to industrial facilities releasing arsenic, lead or mercury. Sci. Total Environ. 2015, 536, 245–251. [Google Scholar] [CrossRef]
- Rodríguez-Barranco, M.; Gil, F.; Hernández, A.F.; Alguacil, J.; Lorca, A.; Mendoza, R.; Gómez, I.; Molina-Villalba, I.; González-Alzaga, B.; Aguilar-Garduño, C.; et al. Postnatal arsenic exposure and attention impairment in school children. Cortex 2016, 74, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Liu, Z.; Gao, T.; Yin, Y.; Wang, Z.; Hou, Y.; Fu, J.; Liu, S.; Wang, H.; Xu, Y.; et al. Prolonged inorganic arsenic exposure via drinking water impairs brown adipose tissue function in mice. Sci. Total Environ. 2019, 668, 310–317. [Google Scholar] [CrossRef]
- Rezaei, M.; Rezaei, A.; Esmaeili, A.; Nakhaee, S.; Azadi, N.A.; Mansouri, B. A case-control study on the relationship between urine trace element levels and autism spectrum disorder among Iranian children. Environ. Sci. Pollut. Res. Int. 2022, 29, 57287–57295. [Google Scholar] [CrossRef] [PubMed]
- Shiani, A.; Sharafi, K.; Omer, A.K.; Kiani, A.; Karamimatin, B.; Massahi, T.; Ebrahimzadeh, G. A systematic literature review on the association between exposures to toxic elements and an autism spectrum disorder. Sci. Total Environ. 2023, 857 Pt 2, 159246. [Google Scholar] [CrossRef]
- Nabgha, E.A.; Eqani, S.; Khuram, F.; Alamdar, A.; Tahir, A.; Shah, S.T.A.; Nasir, A.; Javed, S.; Bibi, N.; Hussain, A.; et al. Environmental exposure pathway analysis of trace elements and autism risk in Pakistani children population. Sci. Total Environ. 2020, 712, 136471. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, D.; Shamberger, R.J.; Audhya, T. Treatment of autism spectrum children with thiamine tetrahydrofurfuryl disulfide: A pilot study. Neuro Endocrinol. Lett. 2002, 23, 303–308. [Google Scholar]
- Amadi, C.N.; Orish, C.N.; Frazzoli, C.; Orisakwe, O.E. Association of autism with toxic metals: A systematic review of case-control studies. Pharmacol. Biochem. Behav. 2022, 212, 173313. [Google Scholar] [CrossRef]
- Mochizuki, H.; Phyu, K.P.; Aung, M.N.; Zin, P.W.; Yano, Y.; Myint, M.Z.; Thit, W.M.; Yamamoto, Y.; Hishikawa, Y.; Thant, K.Z.; et al. Peripheral neuropathy induced by drinking water contaminated with low-dose arsenic in Myanmar. Environ. Health Prev. Med. 2019, 24, 23. [Google Scholar] [CrossRef]
- Vibol, S.; Hashim, J.H.; Sarmani, S. Neurobehavioral effects of arsenic exposure among secondary school children in the Kandal Province, Cambodia. Environ. Res. 2015, 137, 329–337. [Google Scholar] [CrossRef]
- Aung, K.H.; Kyi-Tha-Thu, C.; Sano, K.; Nakamura, K.; Tanoue, A.; Nohara, K.; Kakeyama, M.; Tohyama, C.; Tsukahara, S.; Maekawa, F. Prenatal Exposure to Arsenic Impairs Behavioral Flexibility and Cortical Structure in Mice. Front. Neurosci. 2016, 10, 137. [Google Scholar] [CrossRef]
- Sanchez-Diaz, G.; Escobar, F.; Badland, H.; Arias-Merino, G.; Posada de la Paz, M.; Alonso-Ferreira, V. Geographic Analysis of Motor Neuron Disease Mortality and Heavy Metals Released to Rivers in Spain. Int. J. Environ. Res. Public Health 2018, 15, 2522. [Google Scholar] [CrossRef]
- Moncini, S.; Castronovo, P.; Murgia, A.; Russo, S.; Bedeschi, M.F.; Lunghi, M.; Selicorni, A.; Bonati, M.T.; Riva, P.; Venturin, M. Functional characterization of CDK5 and CDK5R1 mutations identified in patients with non-syndromic intellectual disability. J. Hum. Genet. 2016, 61, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Tan, C.; Mettlach, G.; Pozo, K.; Plattner, F.; Bibb, J.A. Cdk5 Modulates Long-Term Synaptic Plasticity and Motor Learning in Dorsolateral Striatum. Sci. Rep. 2016, 6, 29812. [Google Scholar] [CrossRef]
- Chomiak, T.; Hu, B. Alterations of neocortical development and maturation in autism: Insight from valproic acid exposure and animal models of autism. Neurotoxicology Teratol. 2013, 36, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lei, L.; Tian, L.; Hou, F.; Roper, C.; Ge, X.; Zhao, Y.; Chen, Y.; Dong, Q.; Tanguay, R.L.; et al. Developmental and behavioral alterations in zebrafish embryonically exposed to valproic acid (VPA): An aquatic model for autism. Neurotoxicology Teratol. 2018, 66, 8–16. [Google Scholar] [CrossRef]
- Takahashi, M.; Ishida, M.; Saito, T.; Ohshima, T.; Hisanaga, S. Valproic acid downregulates Cdk5 activity via the transcription of the p35 mRNA. Biochem. Biophys. Res. Commun. 2014, 447, 678–682. [Google Scholar] [CrossRef]
- Uchino, S.; Waga, C. SHANK3 as an autism spectrum disorder-associated gene. Brain Dev. 2013, 35, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.M.; Nguyen, M.; Wong, K.; Poudel, M.K.; Kalueff, A.V. Developing zebrafish models of autism spectrum disorder (ASD). Prog. Psychopharmacol. Biol. Psychiatry 2014, 50, 27–36. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Wang, Y.-Q.; Yu, X.-G.; Lin, Y.; Liu, J.-X.; Wang, W.-Y.; Yan, C.-H. Chronic environmental inorganic arsenic exposure causes social behavioral changes in juvenile zebrafish (Danio rerio). Sci. Total. Environ. 2023, 867, 161296. [Google Scholar] [CrossRef] [PubMed]
- Abu Bakar, N.; Ibrahim, W.N.W.; Abdullah, C.A.C.; Ramlan, N.F.; Shaari, K.; Shohaimi, S.; Mediani, A.; Nasruddin, N.S.; Kim, C.-H.; Faudzi, S.M.M. Embryonic Arsenic Exposure Triggers Long-Term Behavioral Impairment with Metabolite Alterations in Zebrafish. Toxics 2022, 10, 493. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.; Hernández-Sánchez, J.; Dipp, V.R.; Huerta-González, D.; Olivares-Bañuelos, T.N.; González-Fraga, J.; Bardullas, U. Exposure to low doses of inorganic arsenic induces transgenerational changes on behavioral and epigenetic markers in zebrafish (Danio rerio). Toxicol. Appl. Pharmacol. 2020, 396, 115002. [Google Scholar] [CrossRef] [PubMed]
- Olivares, C.I.; Field, J.A.; Simonich, M.; Tanguay, R.L.; Sierra-Alvarez, R. Arsenic (III, V), indium (III), and gallium (III) toxicity to zebrafish embryos using a high-throughput multi-endpoint in vivo developmental and behavioral assay. Chemosphere 2016, 148, 361–368. [Google Scholar] [CrossRef]
- Baldissarelli, L.A.; Capiotti, K.M.; Bogo, M.R.; Ghisleni, G.; Bonan, C.D. Arsenic alters behavioral parameters and brain ectonucleotidases activities in zebrafish (Danio rerio). Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2012, 155, 566–572. [Google Scholar] [CrossRef]
- Elamin, M.; Dumarchey, A.; Stoddard, C.; Robinson, T.M.; Cowie, C.; Gorka, D.; Chamberlain, S.J.; Levine, E.S. The role of UBE3A in the autism and epilepsy-related Dup15q syndrome using patient-derived, CRISPR-corrected neurons. Stem Cell Rep. 2023, 18, 884–898. [Google Scholar] [CrossRef]
- Kalkman, H.O. Potential opposite roles of the extracellular signal-regulated kinase (ERK) pathway in autism spectrum and bipolar disorders. Neurosci. Biobehav. Rev. 2012, 36, 2206–2213. [Google Scholar] [CrossRef]
- Bludau, A.; Royer, M.; Meister, G.; Neumann, I.D.; Menon, R. Epigenetic Regulation of the Social Brain. Trends Neurosci. 2019, 42, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Weinschutz Mendes, H.; Neelakantan, U.; Liu, Y.; Fitzpatrick, S.E.; Chen, T.; Wu, W.; Pruitt, A.; Jin, D.S.; Jamadagni, P.; Carlson, M.; et al. High-throughput functional analysis of autism genes in zebrafish identifies convergence in dopaminergic and neuroimmune pathways. Cell Rep. 2023, 42, 112243. [Google Scholar] [CrossRef] [PubMed]
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4468–E4477. [Google Scholar] [CrossRef]
- Medishetti, R.; Rani, R.; Kavati, S.; Mahilkar, A.; Akella, V.; Saxena, U.; Kulkarni, P.; Sevilimedu, A. A DNAzyme based knockdown model for Fragile-X syndrome in zebrafish reveals a critical window for therapeutic intervention. J. Pharmacol. Toxicol. Methods 2020, 101, 106656. [Google Scholar] [CrossRef]
- Dwivedi, S.; Medishetti, R.; Rani, R.; Sevilimedu, A.; Kulkarni, P.; Yogeeswari, P. Larval zebrafish model for studying the effects of valproic acid on neurodevelopment: An approach towards modeling autism. J. Pharmacol. Toxicol. Methods 2019, 95, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Pietri, T.; Roman, A.C.; Guyon, N.; Romano, S.A.; Washbourne, P.; Moens, C.B.; de Polavieja, G.G.; Sumbre, G. The first mecp2-null zebrafish model shows altered motor behaviors. Front. Neural Circuits 2013, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Bu, Y.; Wu, Q.; Wang, X.; Chang, N.; Lei, L.; Chen, S.; Liu, D.; Zhu, X.; Hu, K.; et al. Mecp2 regulates neural cell differentiation by suppressing the Id1 to Her2 axis in zebrafish. J. Cell Sci. 2015, 128, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.J.; Turner, K.J.; Fernandez, J.M.; Cifuentes, D.; Ghosh, M.; Ijaz, S.; Jain, R.A.; Kubo, F.; Bill, B.R.; Baier, H.; et al. Estrogens Suppress a Behavioral Phenotype in Zebrafish Mutants of the Autism Risk Gene, CNTNAP2. Neuron 2016, 89, 725–733. [Google Scholar] [CrossRef]
- Kim, O.H.; Cho, H.J.; Han, E.; Hong, T.I.; Ariyasiri, K.; Choi, J.H.; Hwang, K.S.; Jeong, Y.M.; Yang, S.Y.; Yu, K.; et al. Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 2017, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 2013, 4, 2410. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.X.; Li, C.Y.; Hu, C.C.; Wang, Y.; Lin, J.; Jiang, Y.H.; Li, Q.; Xu, X. CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism 2018, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Kozol, R.A.; Cukier, H.N.; Zou, B.; Mayo, V.; De Rubeis, S.; Cai, G.; Griswold, A.J.; Whitehead, P.L.; Haines, J.L.; Gilbert, J.R.; et al. Two knockdown models of the autism genes SYNGAP1 and SHANK3 in zebrafish produce similar behavioral phenotypes associated with embryonic disruptions of brain morphogenesis. Hum. Mol. Genet. 2015, 24, 4006–4023. [Google Scholar] [CrossRef]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.Q.; Chen, W.W.; Jiang, L.; Liu, K.; Yung, W.H.; Fu, A.K.Y.; Ip, N.Y. Overproduction of upper-layer neurons in the neocortex leads to autism-like features in mice. Cell Rep. 2014, 9, 1635–1643. [Google Scholar] [CrossRef] [PubMed]
- Avino, T.A.; Barger, N.; Vargas, M.V.; Carlson, E.L.; Amaral, D.G.; Bauman, M.D.; Schumann, C.M. Neuron numbers increase in the human amygdala from birth to adulthood, but not in autism. Proc. Natl. Acad. Sci. USA 2018, 115, 3710–3715. [Google Scholar] [CrossRef]
- Contractor, A.; Ethell, I.M.; Portera-Cailliau, C. Cortical interneurons in autism. Nat. Neurosci. 2021, 24, 1648–1659. [Google Scholar] [CrossRef]
- Kang, Y.; Zhou, Y.; Li, Y.; Han, Y.; Xu, J.; Niu, W.; Li, Z.; Liu, S.; Feng, H.; Huang, W.; et al. A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat. Neurosci. 2021, 24, 1377–1391. [Google Scholar] [CrossRef]
- Casanova, M.F. Neuropathological and genetic findings in autism: The significance of a putative minicolumnopathy. Neuroscientist 2006, 12, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Schumann, C.M.; Barnes, C.C.; Lord, C.; Courchesne, E. Amygdala enlargement in toddlers with autism related to severity of social and communication impairments. Biol. Psychiatry 2009, 66, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Flory, M.; Kuchna, I.; Nowicki, K.; Ma, S.Y.; Imaki, H.; Wegiel, J.; Cohen, I.L.; London, E.; Wisniewski, T.; et al. Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol. Commun. 2014, 2, 141. [Google Scholar] [CrossRef] [PubMed]
- Avino, T.; Hutsler, J.J. Supernumerary neurons within the cerebral cortical subplate in autism spectrum disorders. Brain Res. 2021, 1760, 147350. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.; Gabriele, S.; Persico, A.M. Head circumference and brain size in autism spectrum disorder: A systematic review and meta-analysis. Psychiatry Res. 2015, 234, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Marini, C. Genetic malformations of cortical development. Exp. Brain Res. 2006, 173, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Schwartzkroin, P.A.; Walsh, C.A. Cortical malformations and epilepsy. Ment. Retard. Dev. Disabil. Res. Rev. 2000, 6, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Flory, M.; Kuchna, I.; Nowicki, K.; Ma, S.Y.; Imaki, H.; Wegiel, J.; Cohen, I.L.; London, E.; Brown, W.T.; et al. Brain-region-specific alterations of the trajectories of neuronal volume growth throughout the lifespan in autism. Acta Neuropathol. Commun. 2014, 2, 28. [Google Scholar] [CrossRef]
- Sah, P. Fear, Anxiety, and the Amygdala. Neuron 2017, 96, 1–2. [Google Scholar] [CrossRef]
- Hertz-Picciotto, I.; Croen, L.A.; Hansen, R.; Jones, C.R.; van de Water, J.; Pessah, I.N. The CHARGE study: An epidemiologic investigation of genetic and environmental factors contributing to autism. Environ. Health Perspect. 2006, 114, 1119–1125. [Google Scholar] [CrossRef]
- Dietert, R.R.; Dietert, J.M.; Dewitt, J.C. Environmental risk factors for autism. Emerg. Health Threat. J. 2011, 4, 7111. [Google Scholar] [CrossRef]
- Juliandi, B.; Tanemura, K.; Igarashi, K.; Tominaga, T.; Furukawa, Y.; Otsuka, M.; Moriyama, N.; Ikegami, D.; Abematsu, M.; Sanosaka, T.; et al. Reduced Adult Hippocampal Neurogenesis and Cognitive Impairments following Prenatal Treatment of the Antiepileptic Drug Valproic Acid. Stem Cell Rep. 2015, 5, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.C.; Calder, B.J.; Lilya, S.M.; Davies, B.M.; Martin, A.; Peterson, M.; Hansen, J.M.; Suli, A. Valproic acid affects neurogenesis during early optic tectum development in zebrafish. Biol. Open 2023, 12, bio059567. [Google Scholar] [CrossRef]
- Muhsen, M.; Youngs, J.; Riu, A.; Gustafsson, J.; Kondamadugu, V.S.; Garyfalidis, E.; Bondesson, M. Folic acid supplementation rescues valproic acid-induced developmental neurotoxicity and behavioral alterations in zebrafish embryos. Epilepsia 2021, 62, 1689–1700. [Google Scholar] [CrossRef]
- Kanungo, J.; Twaddle, N.C.; Silva, C.; Robinson, B.; Wolle, M.; Conklin, S.; MacMahon, S.; Gu, Q.; Edhlund, I.; Benjamin, L.; et al. Inorganic arsenic alters the development of dopaminergic neurons but not serotonergic neurons and induces motor neuron development via Sonic hedgehog pathway in zebrafish. Neurosci. Lett. 2023, 795, 137042. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A. Gli proteins and Hedgehog signaling: Development and cancer. Trends Genet. 1999, 15, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Fei, D.L.; Li, H.; Kozul, C.D.; Black, K.E.; Singh, S.; Gosse, J.A.; DiRenzo, J.; Martin, K.A.; Wang, B.; Hamilton, J.W.; et al. Activation of Hedgehog signaling by the environmental toxicant arsenic may contribute to the etiology of arsenic-induced tumors. Cancer Res. 2010, 70, 1981–1988. [Google Scholar] [CrossRef]
- Peterson, R.; Turnbull, J. Sonic hedgehog is cytoprotective against oxidative challenge in a cellular model of amyotrophic lateral sclerosis. J. Mol. Neurosci. 2012, 47, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J. Agonizing hedgehog. Nat. Chem. Biol. 2006, 2, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Petrov, K.; Watanabe, M.; Salic, A. Mechanism of inhibition of the tumor suppressor Patched by Sonic Hedgehog. Proc. Natl. Acad. Sci. USA 2016, 113, E5866–E5875. [Google Scholar] [CrossRef]
- Robbins, D.J.; Hebrok, M. Hedgehogs: La dolce vita. Workshop on Hedgehog-Gli Signaling in Cancer and Stem Cells. EMBO Rep. 2007, 8, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A.; Mas, C.; Stecca, B. The Gli code: An information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007, 17, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.B.; Joyner, A.L. Gli1 can rescue the in vivo function of Gli2. Development 2001, 128, 5161–5172. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.B.; Stephen, D.; Joyner, A.L. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell 2004, 6, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A. Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development 1998, 125, 2203–2212. [Google Scholar] [CrossRef]
- Silva, C.S.; Kudlyk, T.; Tryndyak, V.P.; Twaddle, N.C.; Robinson, B.; Gu, Q.; Beland, F.A.; Fitzpatrick, S.C.; Kanungo, J. Gene expression analyses reveal potential mechanism of inorganic arsenic-induced apoptosis in zebrafish. J. Appl. Toxicol. 2023, 43, 1872–1882. [Google Scholar] [CrossRef]
- Andrews, M.G.; Kong, J.; Novitch, B.G.; Butler, S.J. New perspectives on the mechanisms establishing the dorsal-ventral axis of the spinal cord. Curr. Top. Dev. Biol. 2019, 132, 417–450. [Google Scholar] [CrossRef]
- Yang, C.; Li, S.; Li, X.; Li, H.; Li, Y.; Zhang, C.; Lin, J. Effect of sonic hedgehog on motor neuron positioning in the spinal cord during chicken embryonic development. J. Cell. Mol. Med. 2019, 23, 3549–3562. [Google Scholar] [CrossRef]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Krauss, S.; Concordet, J.P.; Ingham, P.W. A functionally conserved homolog of the Drosophila segment polarity gene hh is expressed in tissues with polarizing activity in zebrafish embryos. Cell 1993, 75, 1431–1444. [Google Scholar] [CrossRef]
- Roelink, H.; Augsburger, A.; Heemskerk, J.; Korzh, V.; Norlin, S.; Ruiz i Altaba, A.; Tanabe, Y.; Placzek, M.; Edlund, T.; Jessell, T.M.; et al. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell 1994, 76, 761–775. [Google Scholar] [CrossRef]
- Catela, C.; Kratsios, P. Transcriptional mechanisms of motor neuron development in vertebrates and invertebrates. Dev. Biol. 2021, 475, 193–204. [Google Scholar] [CrossRef]
- Dhar, P.; Jaitley, M.; Kalaivani, M.; Mehra, R.D. Preliminary morphological and histochemical changes in rat spinal cord neurons following arsenic ingestion. Neurotoxicology 2005, 26, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Al-Ayadhi, L.Y. Relationship between Sonic hedgehog protein, brain-derived neurotrophic factor and oxidative stress in autism spectrum disorders. Neurochem. Res. 2012, 37, 394–400. [Google Scholar] [CrossRef]
- Rahi, S.; Mehan, S. Understanding Abnormal SMO-SHH Signaling in Autism Spectrum Disorder: Potential Drug Target and Therapeutic Goals. Cell. Mol. Neurobiol. 2022, 42, 931–953. [Google Scholar] [CrossRef] [PubMed]
- Chirila, A.M.; Rankin, G.; Tseng, S.Y.; Emanuel, A.J.; Chavez-Martinez, C.L.; Zhang, D.; Harvey, C.D.; Ginty, D.D. Mechanoreceptor signal convergence and transformation in the dorsal horn flexibly shape a diversity of outputs to the brain. Cell 2022, 185, 4541–4559.e23. [Google Scholar] [CrossRef] [PubMed]
- Orefice, L.L.; Mosko, J.R.; Morency, D.T.; Wells, M.F.; Tasnim, A.; Mozeika, S.M.; Ye, M.; Chirila, A.M.; Emanuel, A.J.; Rankin, G.; et al. Targeting Peripheral Somatosensory Neurons to Improve Tactile-Related Phenotypes in ASD Models. Cell 2019, 178, 867–886.e24. [Google Scholar] [CrossRef] [PubMed]
- Orefice, L.L.; Zimmerman, A.L.; Chirila, A.M.; Sleboda, S.J.; Head, J.P.; Ginty, D.D. Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 2016, 166, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, A.; Alkislar, I.; Hakim, R.; Turecek, J.; Abdelaziz, A.; Orefice, L.L.; Ginty, D.D. The developmental timing of spinal touch processing alterations predicts behavioral changes in genetic mouse models of autism spectrum disorders. Nat. Neurosci. 2024, 27, 484–496. [Google Scholar] [CrossRef]
- Zimmerman, A.L.; Kovatsis, E.M.; Pozsgai, R.Y.; Tasnim, A.; Zhang, Q.; Ginty, D.D. Distinct Modes of Presynaptic Inhibition of Cutaneous Afferents and Their Functions in Behavior. Neuron 2019, 102, 420–434.e8. [Google Scholar] [CrossRef]
- Pao, P.C.; Tsai, L.H. Three decades of Cdk5. J. Biomed. Sci. 2021, 28, 79. [Google Scholar] [CrossRef]
- Cheung, Z.H.; Fu, A.K.; Ip, N.Y. Synaptic roles of Cdk5: Implications in higher cognitive functions and neurodegenerative diseases. Neuron 2006, 50, 13–18. [Google Scholar] [CrossRef]
- Dhavan, R.; Tsai, L.H. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, M.; Dudek, H.; Kwon, Y.T.; Ramos, Y.F.; Tsai, L.H. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996, 10, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Ward, J.M.; Huh, C.G.; Longenecker, G.; Veeranna; Pant, H.C.; Brady, R.O.; Martin, L.J.; Kulkarni, A.B. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. USA 1996, 93, 11173–11178. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, J.; Zheng, Y.L.; Amin, N.D.; Kaur, S.; Ramchandran, R.; Pant, H.C. Specific inhibition of cyclin-dependent kinase 5 activity induces motor neuron development in vivo. Biochem. Biophys. Res. Commun. 2009, 386, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Ao, C.; Li, C.; Chen, J.; Tan, J.; Zeng, L. The role of Cdk5 in neurological disorders. Front. Cell. Neurosci. 2022, 16, 951202. [Google Scholar] [CrossRef] [PubMed]
- Drerup, J.M.; Hayashi, K.; Cui, H.; Mettlach, G.L.; Long, M.A.; Marvin, M.; Sun, X.; Goldberg, M.S.; Lutter, M.; Bibb, J.A. Attention-deficit/hyperactivity phenotype in mice lacking the cyclin-dependent kinase 5 cofactor p35. Biol. Psychiatry 2010, 68, 1163–1171. [Google Scholar] [CrossRef]
- Liu, X.X.; Yang, L.; Shao, L.X.; He, Y.; Wu, G.; Bao, Y.H.; Lu, N.N.; Gong, D.M.; Lu, Y.P.; Cui, T.T.; et al. Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis. J. Exp. Med. 2020, 217, e20180992. [Google Scholar] [CrossRef]
- Engmann, O.; Hortobágyi, T.; Pidsley, R.; Troakes, C.; Bernstein, H.G.; Kreutz, M.R.; Mill, J.; Nikolic, M.; Giese, K.P. Schizophrenia is associated with dysregulation of a Cdk5 activator that regulates synaptic protein expression and cognition. Brain 2011, 134, 2408–2421. [Google Scholar] [CrossRef]
- Costa, L.; Tempio, A.; Lacivita, E.; Leopoldo, M.; Ciranna, L. Serotonin 5-HT7 receptors require cyclin-dependent kinase 5 to rescue hippocampal synaptic plasticity in a mouse model of Fragile X Syndrome. Eur. J. Neurosci. 2021, 54, 4124–4132. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Jia, M.; Yue, W.; Ruan, Y.; Lu, T.; Zhang, J.; Liu, J.; Zhang, D. No association of polymorphisms in the CDK5, NDEL1, and LIS1 with autism in Chinese Han population. Psychiatry Res. 2011, 190, 369–371. [Google Scholar] [CrossRef]
- Tao, J.; Van Esch, H.; Hagedorn-Greiwe, M.; Hoffmann, K.; Moser, B.; Raynaud, M.; Sperner, J.; Fryns, J.P.; Schwinger, E.; Gécz, J.; et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 2004, 75, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.T.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef] [PubMed]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P.; et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef]
- Kanungo, J.; Goswami, M.T.; Pant, H.C. Notch and Cdk5 in Zebrafish Mindbomb Mutant: Co-regulation or Coincidence? Folia Biol. 2018, 64, 35–40. [Google Scholar] [CrossRef]
- Itoh, M.; Kim, C.H.; Palardy, G.; Oda, T.; Jiang, Y.J.; Maust, D.; Yeo, S.Y.; Lorick, K.; Wright, G.J.; Ariza-McNaughton, L.; et al. Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev. Cell 2003, 4, 67–82. [Google Scholar] [CrossRef]
- Weinmaster, G.; Kintner, C. Modulation of notch signaling during somitogenesis. Annu. Rev. Cell Dev. Biol. 2003, 19, 367–395. [Google Scholar] [CrossRef]
- Koo, B.K.; Lim, H.S.; Song, R.; Yoon, M.J.; Yoon, K.J.; Moon, J.S.; Kim, Y.W.; Kwon, M.C.; Yoo, K.W.; Kong, M.P.; et al. Mind bomb 1 is essential for generating functional Notch ligands to activate Notch. Development 2005, 132, 3459–3470. [Google Scholar] [CrossRef]
- Jiang, Y.J.; Aerne, B.L.; Smithers, L.; Haddon, C.; Ish-Horowicz, D.; Lewis, J. Notch signalling and the synchronization of the somite segmentation clock. Nature 2000, 408, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Lee, D.; Hong, S.; Park, S.G.; Song, M.R. The E3 ligase Mind bomb-1 (Mib1) modulates Delta-Notch signaling to control neurogenesis and gliogenesis in the developing spinal cord. J. Biol. Chem. 2013, 288, 2580–2592. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Scheer, N.; Pham, V.N.; Kim, C.H.; Chitnis, A.B.; Campos-Ortega, J.A.; Weinstein, B.M. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 2001, 128, 3675–3683. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yu, L.; Liu, D.; Yang, X.; Zheng, Y.; Gui, Y.; Wang, H. MIB1 mutations reduce Notch signaling activation and contribute to congenital heart disease. Clin. Sci. 2018, 132, 2483–2491. [Google Scholar] [CrossRef] [PubMed]
- Hortopan, G.A.; Baraban, S.C. Aberrant expression of genes necessary for neuronal development and Notch signaling in an epileptic mind bomb zebrafish. Dev. Dyn. 2011, 240, 1964–1976. [Google Scholar] [CrossRef] [PubMed]
- Hortopan, G.A.; Dinday, M.T.; Baraban, S.C. Spontaneous seizures and altered gene expression in GABA signaling pathways in a mind bomb mutant zebrafish. J. Neurosci. 2010, 30, 13718–13728. [Google Scholar] [CrossRef]
- Muhle, R.; Trentacoste, S.V.; Rapin, I. The genetics of autism. Pediatrics 2004, 113, e472–e486. [Google Scholar] [CrossRef]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef]
- Choe, E.A.; Liao, L.; Zhou, J.Y.; Cheng, D.; Duong, D.M.; Jin, P.; Tsai, L.H.; Peng, J. Neuronal morphogenesis is regulated by the interplay between cyclin-dependent kinase 5 and the ubiquitin ligase mind bomb 1. J. Neurosci. 2007, 27, 9503–9512. [Google Scholar] [CrossRef]
- Kwon, D.Y.; Dimitriadi, M.; Terzic, B.; Cable, C.; Hart, A.C.; Chitnis, A.; Fischbeck, K.H.; Burnett, B.G. The E3 ubiquitin ligase mind bomb 1 ubiquitinates and promotes the degradation of survival of motor neuron protein. Mol. Biol. Cell 2013, 24, 1863–1871. [Google Scholar] [CrossRef]
- Yang, M.H.; Chang, K.J.; Li, B.; Chen, W.S. Arsenic Trioxide Suppresses Tumor Growth through Antiangiogenesis via Notch Signaling Blockade in Small-Cell Lung Cancer. BioMed Res. Int. 2019, 2019, 4647252. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Li, B.; Chang, K.J. Notch pathway inhibition mediated by arsenic trioxide depletes tumor initiating cells in small cell lung cancer. Mol. Biol. Rep. 2022, 49, 2245–2253. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Voineagu, I.; Paşca, S.P.; Won, H.; Chandran, V.; Horvath, S.; Dolmetsch, R.E.; Geschwind, D.H. Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy syndrome. Genome Med. 2014, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, J.; Cuevas, E.; Ali, S.F.; Paule, M.G. Zebrafish model in drug safety assessment. Curr. Pharm. Des. 2014, 20, 5416–5429. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, Q.; Kanungo, J. Neurogenic Effects of Inorganic Arsenic and Cdk5 Knockdown in Zebrafish Embryos: A Perspective on Modeling Autism. Int. J. Mol. Sci. 2024, 25, 3459. https://doi.org/10.3390/ijms25063459
Gu Q, Kanungo J. Neurogenic Effects of Inorganic Arsenic and Cdk5 Knockdown in Zebrafish Embryos: A Perspective on Modeling Autism. International Journal of Molecular Sciences. 2024; 25(6):3459. https://doi.org/10.3390/ijms25063459
Chicago/Turabian StyleGu, Qiang, and Jyotshna Kanungo. 2024. "Neurogenic Effects of Inorganic Arsenic and Cdk5 Knockdown in Zebrafish Embryos: A Perspective on Modeling Autism" International Journal of Molecular Sciences 25, no. 6: 3459. https://doi.org/10.3390/ijms25063459
APA StyleGu, Q., & Kanungo, J. (2024). Neurogenic Effects of Inorganic Arsenic and Cdk5 Knockdown in Zebrafish Embryos: A Perspective on Modeling Autism. International Journal of Molecular Sciences, 25(6), 3459. https://doi.org/10.3390/ijms25063459