
Exploring the Role of Platelets in Virus-Induced Inflammatory Demyelinating Disease and Myocarditis


 ,
,  , ,
, ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
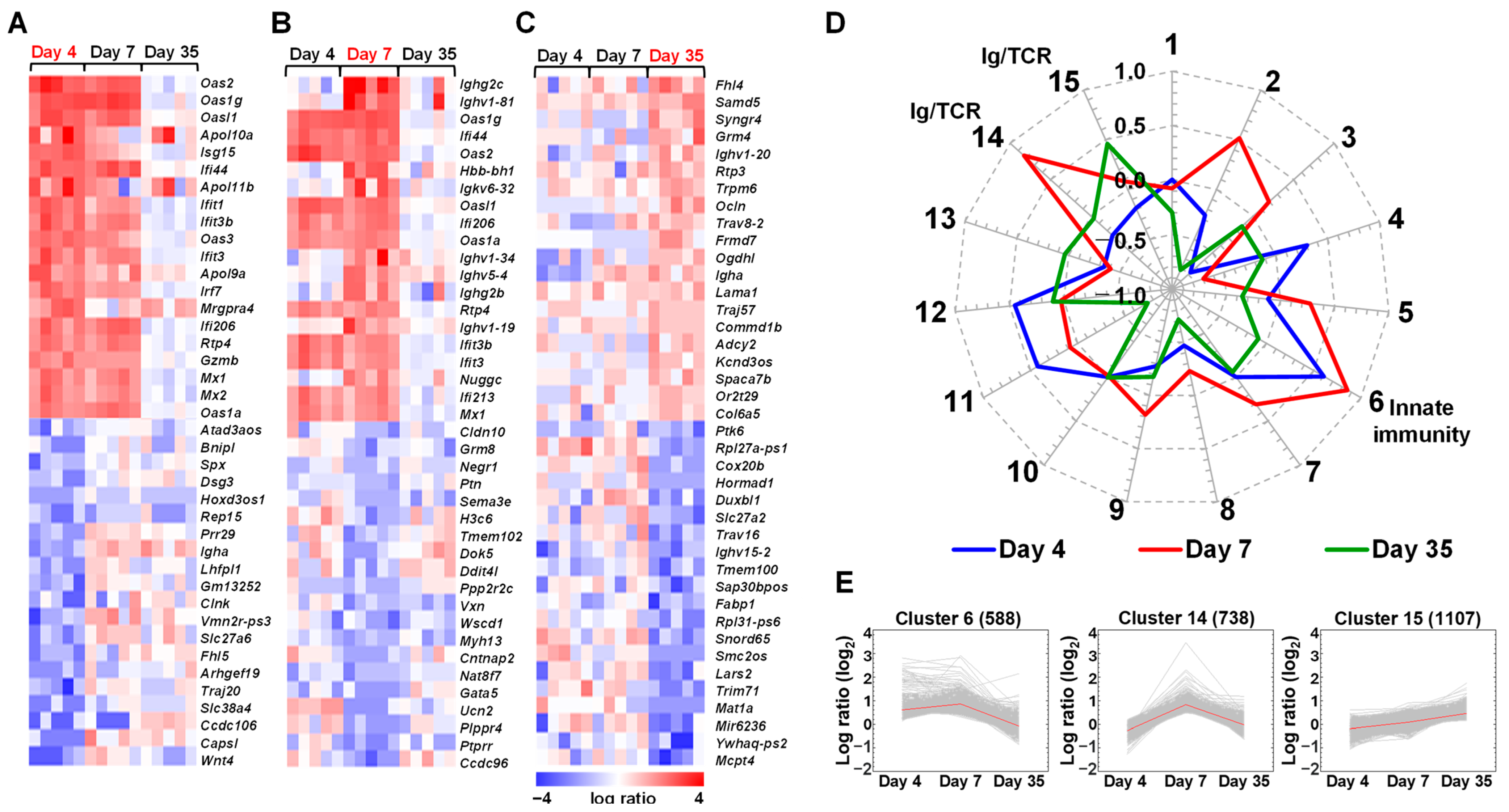
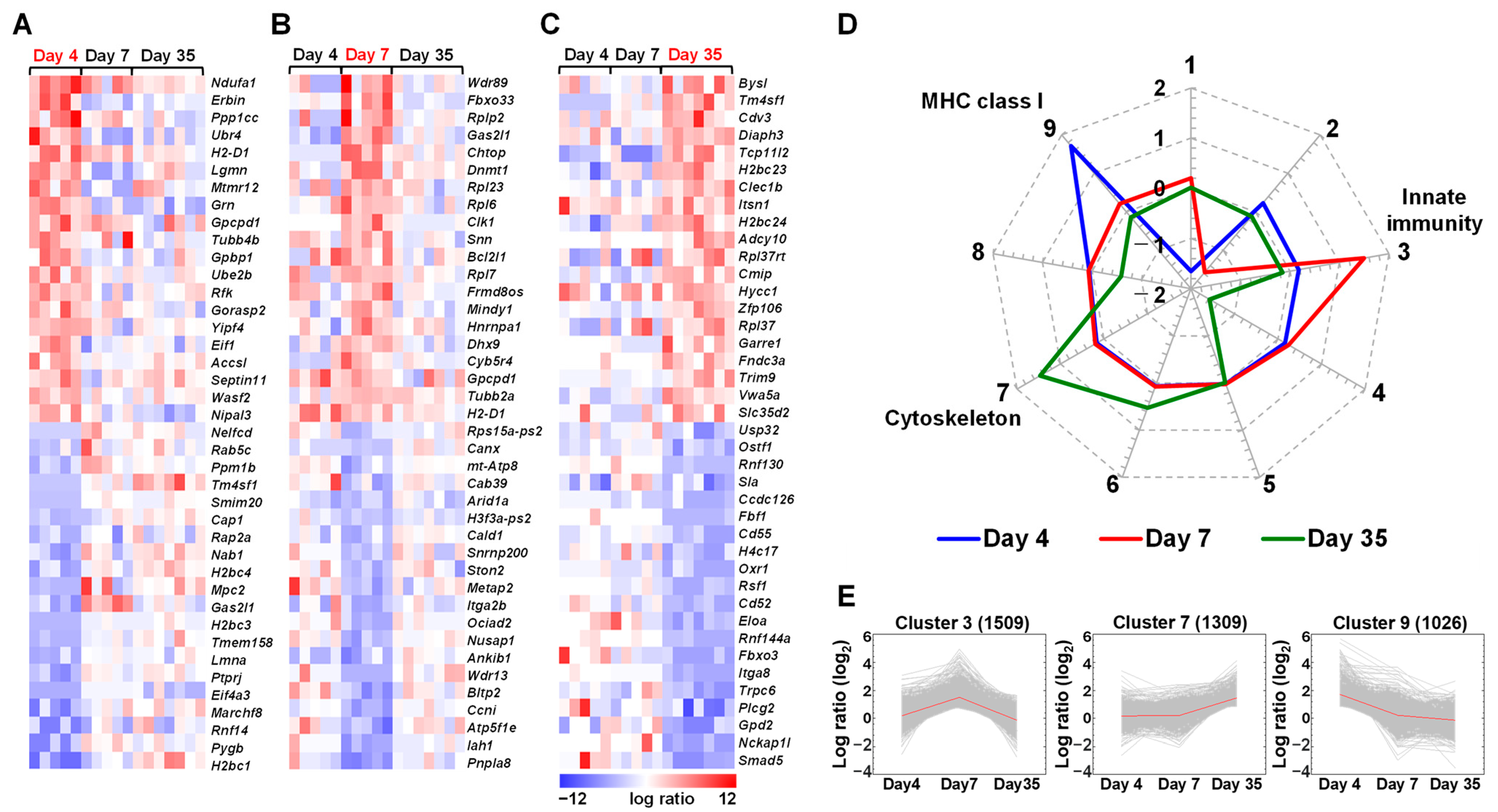
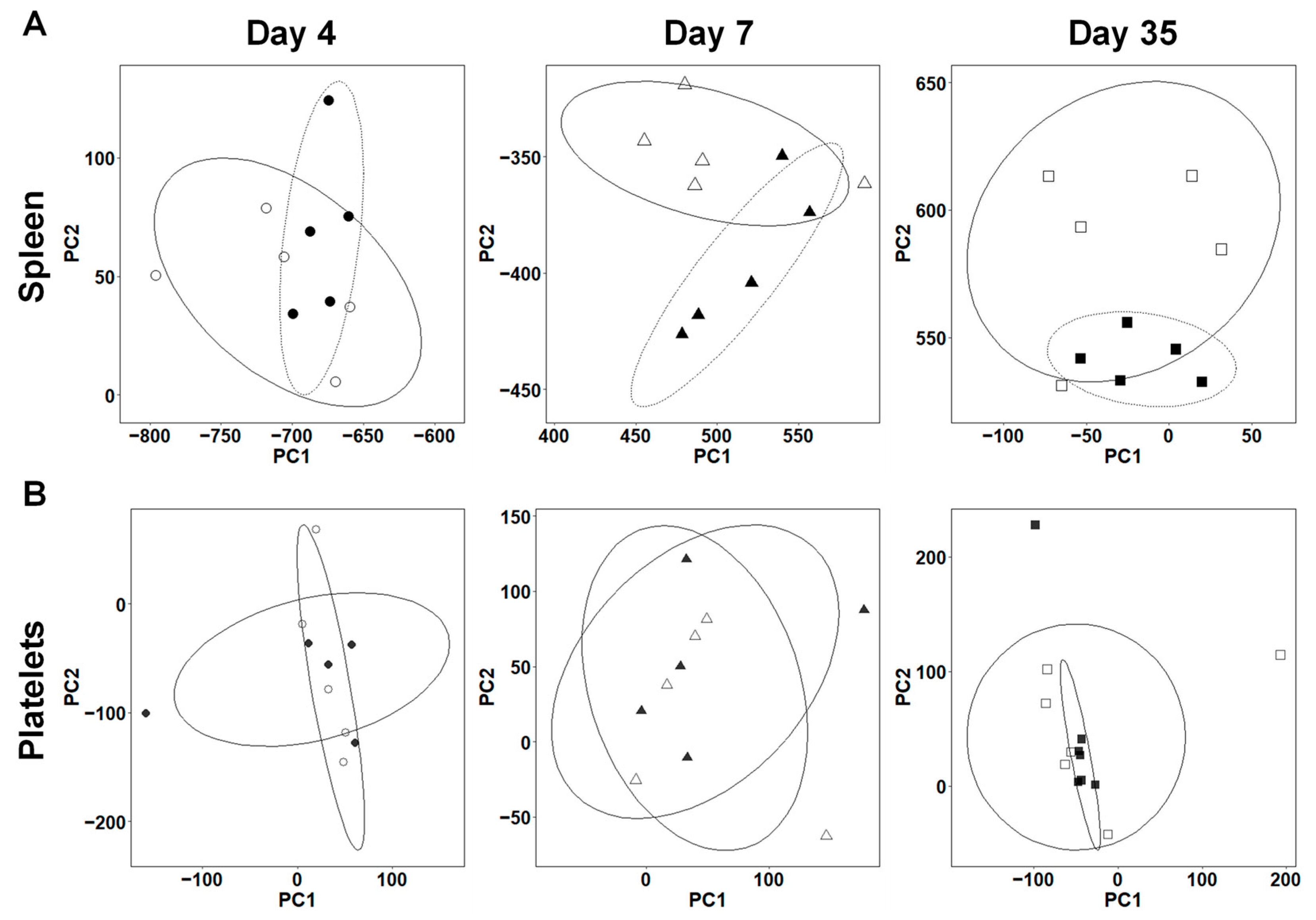
2.1. Expressions of Immune Genes Are Altered in the Spleen of TMEV-Infected Mice
2.2. Distinct Gene Expressions, Including Immune-Related Genes, in Platelets of TMEV-Infected Mice
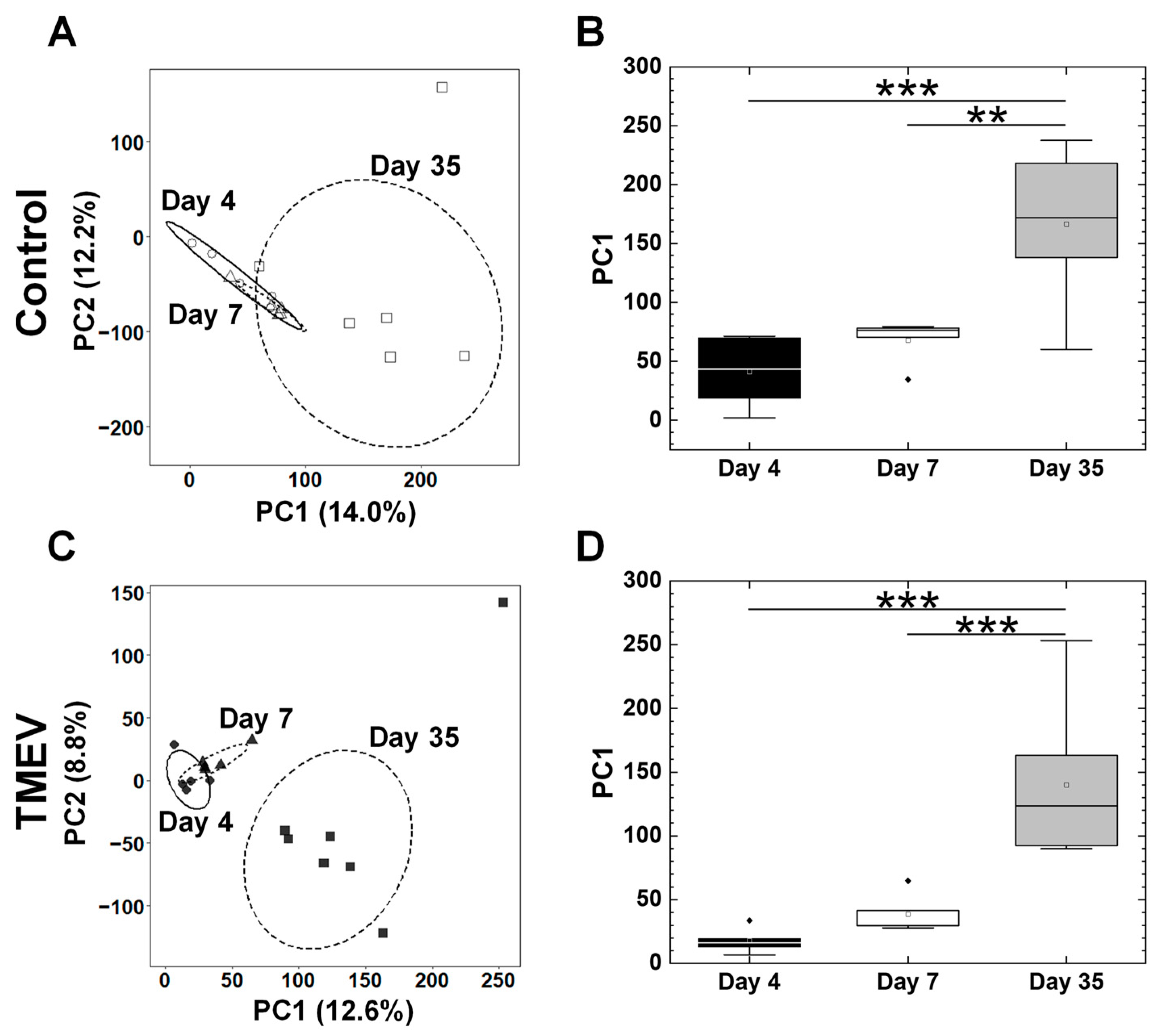
2.3. Overall Gene Expression Profiles Differ in the Spleens, but Not in the Platelets between TMEV-Infected and Control Mice
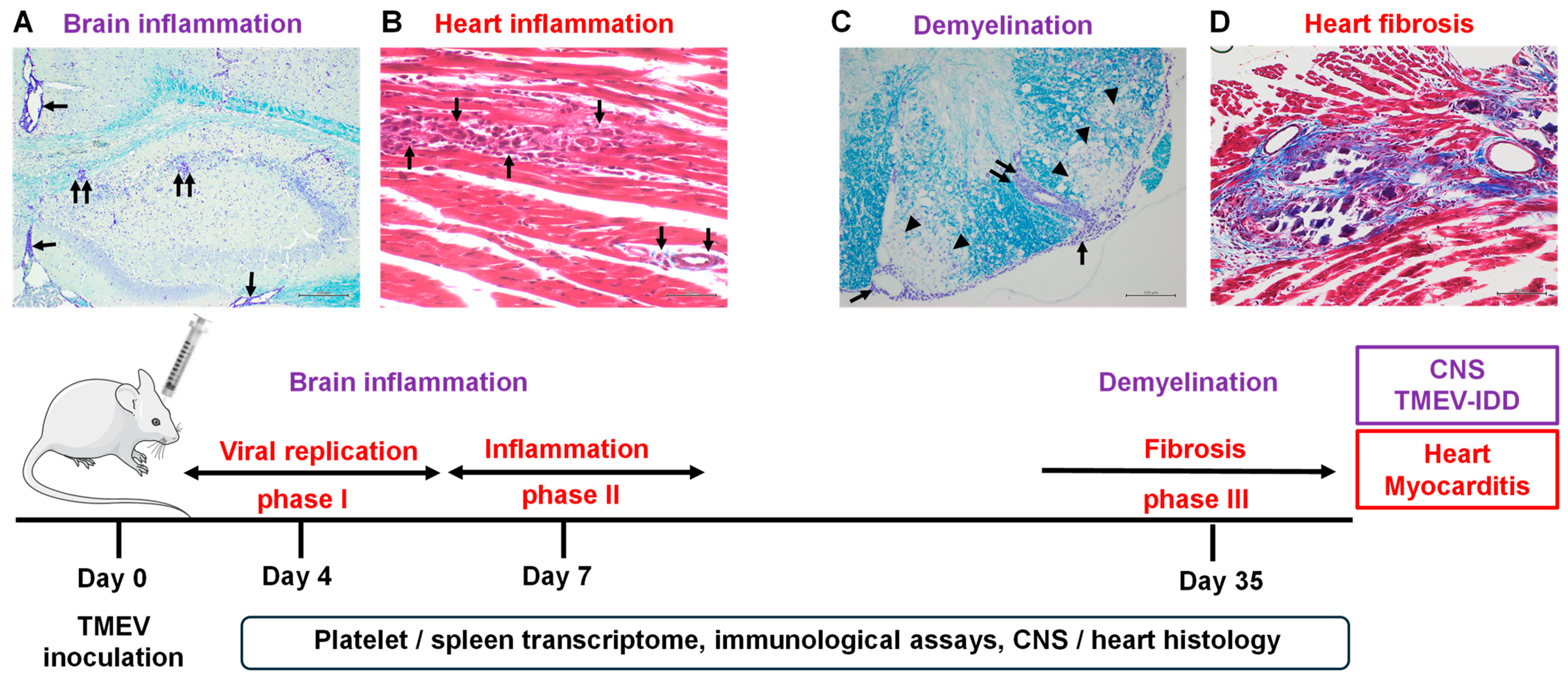
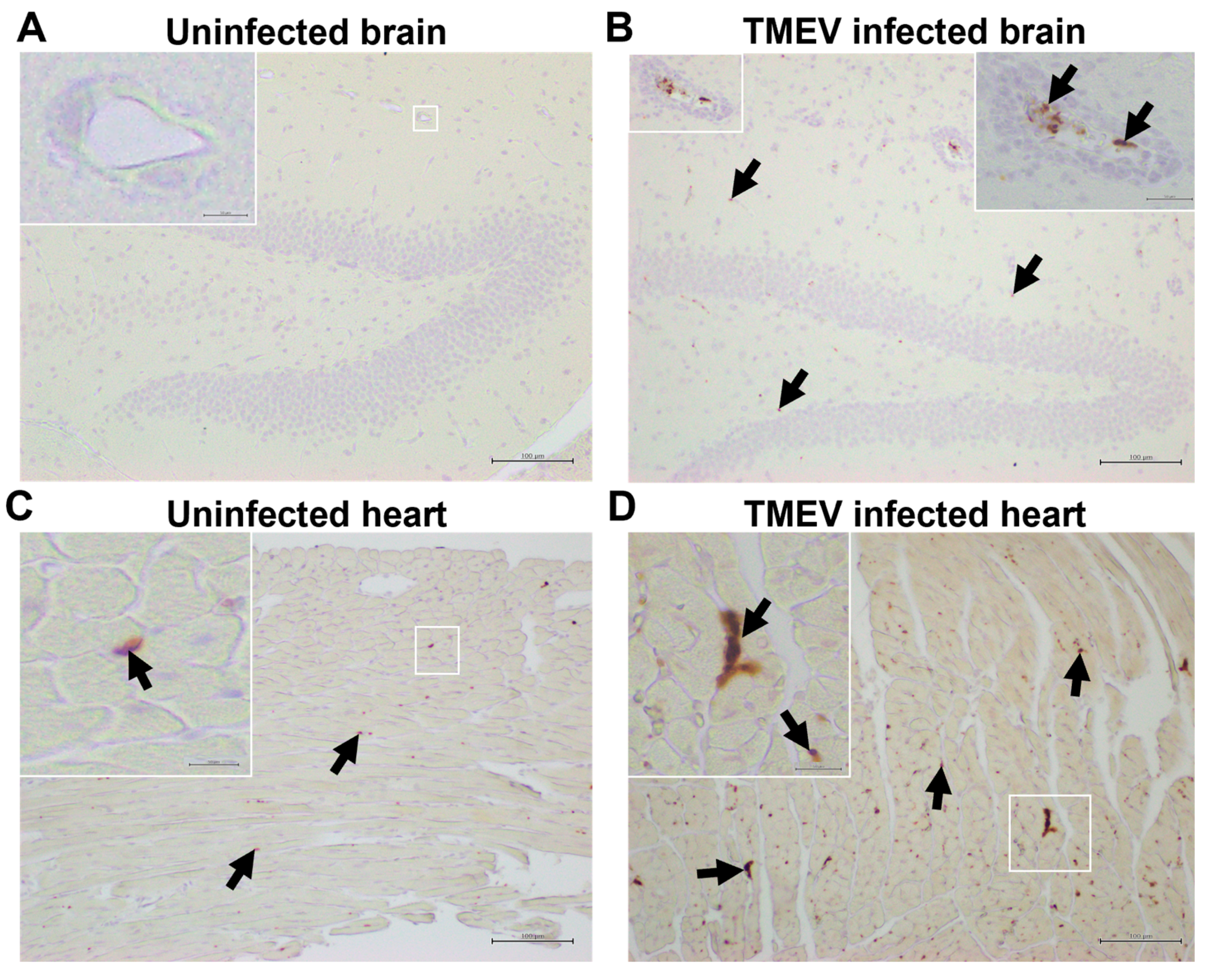
2.4. Platelets Are Present in the CNS and Heart of TMEV-Infected Mice
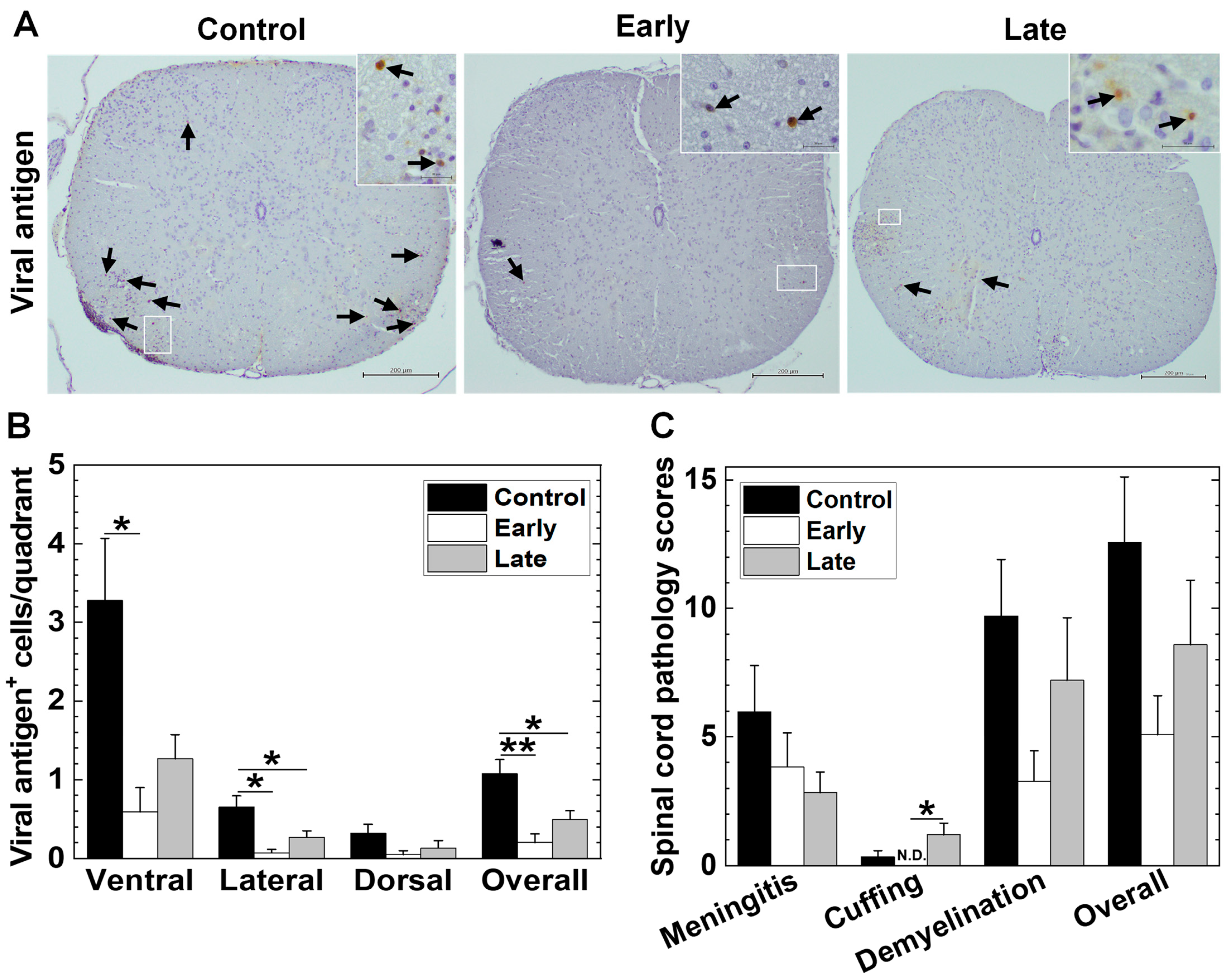
2.5. Platelets’ Depletion Reduces Viral Persistence in TMEV-IDD
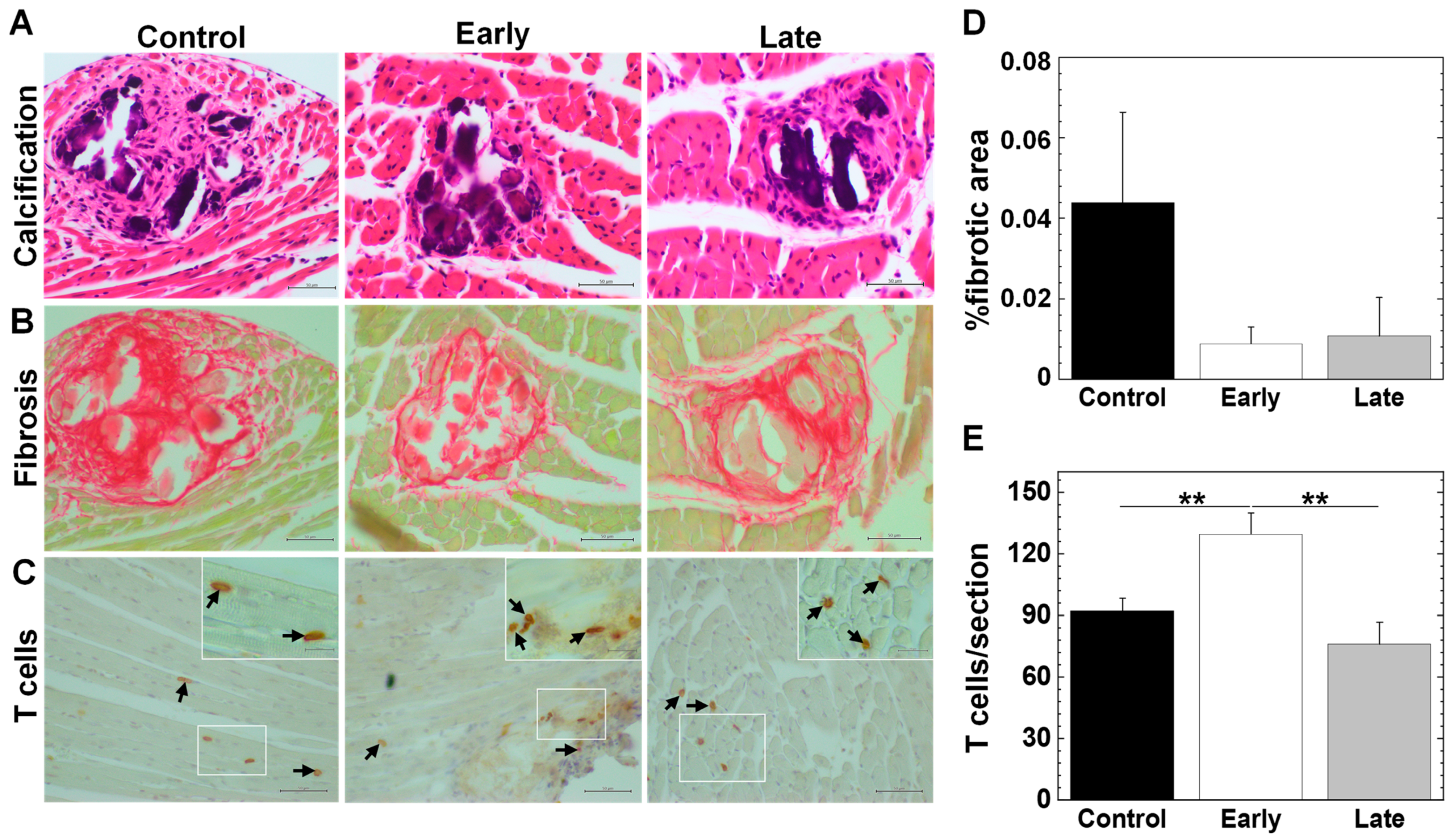
2.6. Platelets Contribute to the Pathogenesis of Myocarditis
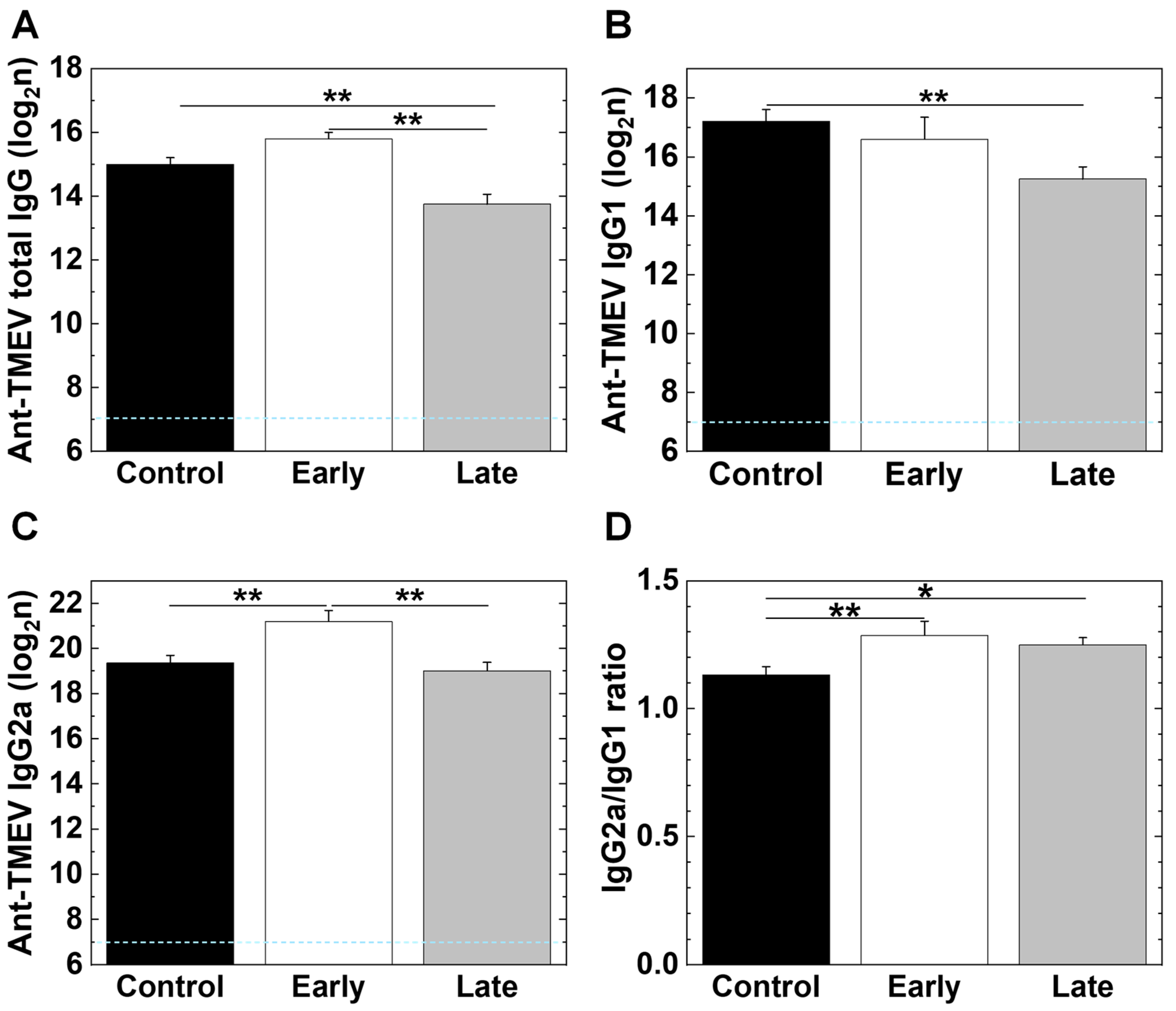
2.7. Platelet Depletion Alters Antibody Isotype Responses
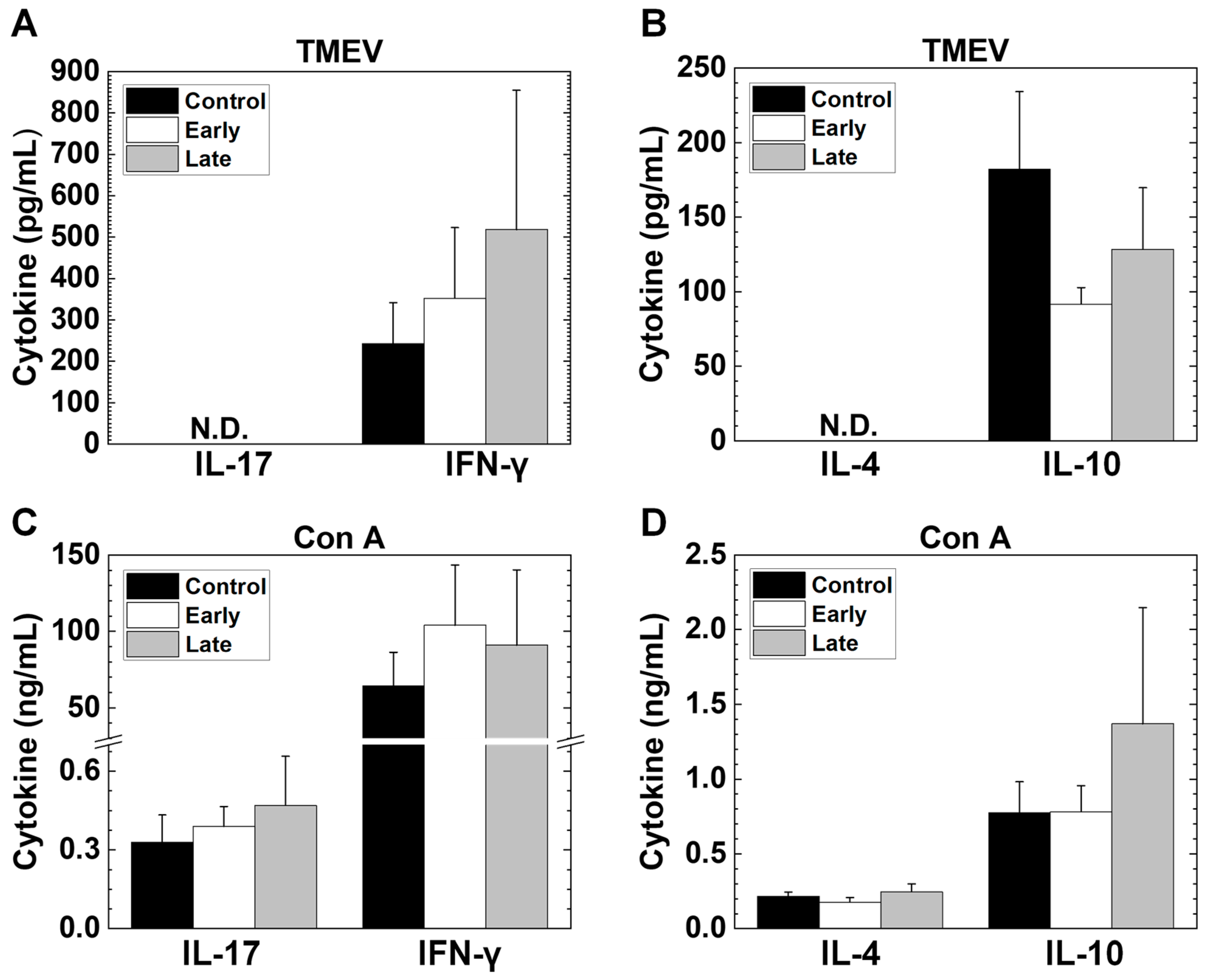
2.8. Platelet Depletion Alters Lymphoproliferative Responses and Cytokine Production
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Platelet Isolation
4.3. RNA-Seq
4.4. Bioinformatics Analyses
4.5. Platelet Depletion In Vivo
4.6. Neuropathology and Cardiac Pathology
4.7. Immunohistochemistry
4.8. Anti-TMEV Isotype Antibody ELISAs
4.9. Lymphoproliferative Responses and Cytokine ELISAs
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flint, S.J.; Racaniello, V.R.; Rall, G.F.; Hatziioannou, T.; Skalka, A.M. Patterns and pathogenesis. In Principles of Virology, Volume 2: Pathogenesis and Control, 5th ed.; Flint, S.J., Racaniello, V.R., Rall, G.F., Hatziioannou, T., Skalka, A.M., Eds.; ASM Press: Washington, DC, USA, 2020; pp. 146–186. [Google Scholar]
- Fairweather, D.; Rose, N.R. Coxsackievirus-induced myocarditis in mice: A model of autoimmune disease for studying immunotoxicity. Methods 2007, 41, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Liu, Q.; Yao, Q.; Wang, X.; Zhang, H.; Chen, R.; Ren, L.; Min, J.; Deng, F.; et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov. 2021, 7, 17. [Google Scholar] [CrossRef]
- Sato, F.; Tanaka, H.; Hasanovic, F.; Tsunoda, I. Theiler’s virus infection: Pathophysiology of demyelination and neurodegeneration. Pathophysiology 2011, 18, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Theiler, M.; Gard, S. Encephalomyelitis of mice: I. Characteristics and pathogenesis of the virus. J. Exp. Med. 1940, 72, 49–67. [Google Scholar] [CrossRef] [PubMed]
- DePaula-Silva, A.B. The contribution of microglia and brain-infiltrating macrophages to the pathogenesis of neuroinflammatory and neurodegenerative diseases during TMEV infection of the central nervous system. Viruses 2024, 16, 119. [Google Scholar] [CrossRef]
- Tsunoda, I.; McCright, I.J.; Kuang, L.-Q.; Zurbriggen, A.; Fujinami, R.S. Hydrocephalus in mice infected with a Theiler’s murine encephalomyelitis virus variant. J. Neuropathol. Exp. Neurol. 1997, 56, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Kurtz, C.I.B.; Fujinami, R.S. Apoptosis in acute and chronic central nervous system disease induced by Theiler’s murine encephalomyelitis virus. Virology 1997, 228, 388–393. [Google Scholar] [CrossRef]
- Oleszak, E.L.; Chang, J.R.; Friedman, H.; Katsetos, C.D.; Platsoucas, C.D. Theiler’s virus infection: A model for multiple sclerosis. Clin. Microbiol. Rev. 2004, 17, 174–207. [Google Scholar] [CrossRef]
- Tsunoda, I.; Kuang, L.-Q.; Fujinami, R.S. Induction of autoreactive CD8+ cytotoxic T cells during Theiler’s murine encephalomyelitis virus infection: Implications for autoimmunity. J. Virol. 2002, 76, 12834–12844. [Google Scholar] [CrossRef]
- Kriesel, J.D.; White, A.; Hayden, F.G.; Spruance, S.L.; Petajan, J. Multiple sclerosis attacks are associated with picornavirus infections. Mult. Scler. 2004, 10, 145–148. [Google Scholar] [CrossRef]
- Ascherio, A. Environmental factors in multiple sclerosis. Expert. Rev. Neurother. 2013, 13, 3–9. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Münz, C. EBV in MS: Guilty by association? Trends Immunol. 2009, 30, 243–248. [Google Scholar] [CrossRef]
- Sato, F.; Omura, S.; Kawai, E.; Martinez, N.E.; Acharya, M.M.; Reddy, P.C.; Chaitanya, G.V.; Alexander, J.S.; Tsunoda, I. Distinct kinetics of viral replication, T cell infiltration, and fibrosis in three phases of myocarditis following Theiler’s virus infection. Cell Immunol. 2014, 292, 85–93. [Google Scholar] [CrossRef]
- Goetzke, C.C.; Althof, N.; Neumaier, H.L.; Heuser, A.; Kaya, Z.; Kespohl, M.; Klingel, K.; Beling, A. Mitigated viral myocarditis in A/J mice by the immunoproteasome inhibitor ONX 0914 depends on inhibition of systemic inflammatory responses in CoxsackievirusB3 infection. Basic Res. Cardiol. 2021, 116, 7. [Google Scholar] [CrossRef]
- Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 743–800. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T., Jr. Myocarditis. N. Engl. J. Med. 2009, 360, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A.; Gauntt, C.J.; Sakkinen, P. Enteroviruses and myocarditis: Viral pathogenesis through replication, cytokine induction, and immunopathogenicity. Adv. Virus Res. 1998, 51, 35–80. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Shiwani, H.A.; Elfaki, M.Y.; Hamid, M.; Pharithi, R.; Kamgang, R.; Egom, C.B.; Oyono, J.L.E.; Egom, E.E.-A. COVID-19 and myocarditis: A review of literature. Egypt. Heart J. 2022, 74, 23. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Stafford, K.A.; Sung, Y.K. Update on coxsackievirus B3 myocarditis. Curr. Opin. Rheumatol. 2012, 24, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Mason, J.W. Advances in the understanding of myocarditis. Circulation 2001, 104, 1076–1082. [Google Scholar] [CrossRef]
- Gómez, R.M.; Rinehart, J.E.; Wollmann, R.; Roos, R.P. Theiler’s murine encephalomyelitis virus-induced cardiac and skeletal muscle disease. J. Virol. 1996, 70, 8926–8933. [Google Scholar] [CrossRef]
- Rames, D.S. The Etiopathogenesis of Theiler’s Murine Encephalomyelitis Virus (TMEV)-Induced Cardiomyopathy, Including Characterization of New Strains of TMEV; Texas A&M University: College Station, TX, USA, 1995. [Google Scholar]
- Omura, S.; Kawai, E.; Sato, F.; Martinez, N.E.; Chaitanya, G.V.; Rollyson, P.A.; Cvek, U.; Trutschl, M.; Alexander, J.S.; Tsunoda, I. Bioinformatics multivariate analysis determined a set of phase-specific biomarker candidates in a novel mouse model for viral myocarditis. Circ. Cardiovasc. Genet. 2014, 7, 444–454. [Google Scholar] [CrossRef]
- Omura, S.; Tsunoda, I. Cutting-edge research about phathomechanisms of multiple sclerosis. Pharma Medica 2021, 39, 67–71. [Google Scholar]
- Mehmood, A.; Song, S.; Du, X.; Yan, H.; Wang, X.; Guo, L.; Li, B. mRNA expression profile reveals differentially expressed genes in splenocytes of experimental autoimmune encephalomyelitis model. Int. J. Exp. Pathol. 2023, 104, 247–257. [Google Scholar] [CrossRef]
- Vögtle, T.; Sharma, S.; Mori, J.; Nagy, Z.; Semeniak, D.; Scandola, C.; Geer, M.J.; Smith, C.W.; Lane, J.; Pollack, S.; et al. Heparan sulfates are critical regulators of the inhibitory megakaryocyte-platelet receptor G6b-B. Elife 2019, 8, e46840. [Google Scholar] [CrossRef]
- Liu, Z.; Li, L.; Zhang, H.; Pang, X.; Qiu, Z.; Xiang, Q.; Cui, Y. Platelet factor 4(PF4) and its multiple roles in diseases. Blood Rev. 2023, 64, 101155. [Google Scholar] [CrossRef]
- Omura, S.; Sato, F.; Martinez, N.E.; Park, A.-M.; Fujita, M.; Kennett, N.J.; Cvek, U.; Minagar, A.; Alexander, J.S.; Tsunoda, I. Bioinformatics analyses determined the distinct CNS and peripheral surrogate biomarker candidates between two mouse models for progressive multiple sclerosis. Front. Immunol. 2019, 10, 516. [Google Scholar] [CrossRef] [PubMed]
- Jeimy, S.B.; Tasneem, S.; Cramer, E.M.; Hayward, C.P.M. Multimerin 1. Platelets 2008, 19, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Choi, E.Y.; Zhou, H.; Schleicher, R.; Chung, K.-J.; Tang, Z.; Göbel, K.; Bdeir, K.; Chatzigeorgiou, A.; Wong, C.; et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ. Res. 2012, 110, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Schrottmaier, W.C.; Schmuckenschlager, A.; Pirabe, A.; Assinger, A. Platelets in viral infections—Brave soldiers or trojan horses. Front. Immunol. 2022, 13, 856713. [Google Scholar] [CrossRef] [PubMed]
- Terada, H.; Baldini, M.; Ebbe, S.; Madoff, M.A. Interaction of influenza virus with blood platelets. Blood 1966, 28, 213–228. [Google Scholar] [CrossRef]
- Page, M.J.; Pretorius, E. A champion of host defense: A generic large-scale cause for platelet dysfunction and depletion in infection. Semin. Thromb. Hemost. 2020, 46, 302–319. [Google Scholar] [CrossRef]
- Youssefian, T.; Drouin, A.; Massé, J.-M.; Guichard, J.; Cramer, E.M. Host defense role of platelets: Engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 2002, 99, 4021–4029. [Google Scholar] [CrossRef]
- Flaujac, C.; Boukour, S.; Cramer-Bordé, E. Platelets and viruses: An ambivalent relationship. Cell Mol. Life Sci. 2010, 67, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Zahn, A.; Jennings, N.; Ouwehand, W.H.; Allain, J.-P. Hepatitis C virus interacts with human platelet glycoprotein VI. J. Gen. Virol. 2006, 87, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Chabert, A.; Hamzeh-Cognasse, H.; Pozzetto, B.; Cognasse, F.; Schattner, M.; Gomez, R.M.; Garraud, O. Human platelets and their capacity of binding viruses: Meaning and challenges? BMC Immunol. 2015, 16, 26. [Google Scholar] [CrossRef]
- Negrotto, S.; Jaquenod de Giusti, C.; Rivadeneyra, L.; Ure, A.E.; Mena, H.A.; Schattner, M.; Gomez, R.M. Platelets interact with Coxsackieviruses B and have a critical role in the pathogenesis of virus-induced myocarditis. J. Thromb. Haemost. 2015, 13, 271–282. [Google Scholar] [CrossRef]
- Elzey, B.D.; Tian, J.; Jensen, R.J.; Swanson, A.K.; Lees, J.R.; Lentz, S.R.; Stein, C.S.; Nieswandt, B.; Wang, Y.; Davidson, B.L.; et al. Platelet-mediated modulation of adaptive immunity: A communication link between innate and adaptive immune compartments. Immunity 2003, 19, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Italiano, J.E., Jr.; Shivdasani, R.A. Megakaryocytes and beyond: The birth of platelets. J. Thromb. Haemost. 2003, 1, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; McEver, R.P.; Weyrich, A.S.; Morrell, C.N.; Hoffman, M.R.; Arepally, G.M.; French, P.A.; Dauerman, H.L.; Becker, R.C.; 2009 Platelet Colloquium Participants. Platelet functions beyond hemostasis. J. Thromb. Haemost. 2009, 7, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Herter, J.M.; Rossaint, J.; Zarbock, A. Platelets in inflammation and immunity. J. Thromb. Haemost. 2014, 12, 1764–1775. [Google Scholar] [CrossRef]
- Jenne, C.N.; Urrutia, R.; Kubes, P. Platelets: Bridging hemostasis, inflammation, and immunity. Int. J. Lab. Hematol. 2013, 35, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Pulavendran, S.; Rudd, J.M.; Maram, P.; Thomas, P.G.; Akhilesh, R.; Malayer, J.R.; Chow, V.T.K.; Teluguakula, N. Combination therapy targeting platelet activation and virus replication protects mice against lethal influenza pneumonia. Am. J. Respir. Cell Mol. Biol. 2019, 61, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Lê, V.B.; Schneider, J.G.; Boergeling, Y.; Berri, F.; Ducatez, M.; Guerin, J.-L.; Adrian, I.; Errazuriz-Cerda, E.; Frasquilho, S.; Antunes, L.; et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am. J. Respir. Crit. Care Med. 2015, 191, 804–819. [Google Scholar] [CrossRef]
- Ohtsuka, M.; Inoko, H.; Kulski, J.K.; Yoshimura, S. Major histocompatibility complex (Mhc) class Ib gene duplications, organization and expression patterns in mouse strain C57BL/6. BMC Genom. 2008, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- White, J.G.; Rao, G.H. Microtubule coils versus the surface membrane cytoskeleton in maintenance and restoration of platelet discoid shape. Am. J. Pathol. 1998, 152, 597–609. [Google Scholar] [PubMed]
- Marek, I.; Hilgers, K.F.; Rascher, W.; Woelfle, J.; Hartner, A. A role for the alpha-8 integrin chain (itga8) in glomerular homeostasis of the kidney. Mol. Cell Pediatr. 2020, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Hridi, S.U.; Barbour, M.; Wilson, C.; Franssen, A.J.P.M.; Harte, T.; Bushell, T.J.; Jiang, H.-R. Increased levels of IL-16 in the central nervous system during neuroinflammation are associated with infiltrating immune cells and resident glial cells. Biology 2021, 10, 472. [Google Scholar] [CrossRef]
- Tanaka, H.; Horioka, K.; Yamamoto, M.; Asari, M.; Okuda, K.; Yamazaki, K.; Shimizu, K.; Ogawa, K. Overproduction of thrombopoietin by BRAFV600E-mutated mouse hepatocytes and contribution of thrombopoietin to hepatocarcinogenesis. Cancer Sci. 2019, 110, 2748–2759. [Google Scholar] [CrossRef]
- Tsunoda, I.; Tolley, N.D.; Theil, D.J.; Whitton, J.L.; Kobayashi, H.; Fujinami, R.S. Exacerbation of viral and autoimmune animal models for multiple sclerosis by bacterial DNA. Brain Pathol. 1999, 9, 481–493. [Google Scholar] [CrossRef]
- Tsunoda, I.; Iwasaki, Y.; Terunuma, H.; Sako, K.; Ohara, Y. A comparative study of acute and chronic diseases induced by two subgroups of Theiler’s murine encephalomyelitis virus. Acta Neuropathol. 1996, 91, 595–602. [Google Scholar] [CrossRef]
- Zucker-Franklin, D.; Seremetis, S.; Zheng, Z.Y. Internalization of human immunodeficiency virus type I and other retroviruses by megakaryocytes and platelets. Blood 1990, 75, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets contribute to disease severity in COVID-19. J. Thromb. Haemost. 2021, 19, 3139–3153. [Google Scholar] [CrossRef] [PubMed]
- Trugilho, M.R.d.; Hottz, E.D.; Brunoro, G.V.F.; Teixeira-Ferreira, A.; Carvalho, P.C.; Salazar, G.A.; Zimmerman, G.A.; Bozza, F.A.; Bozza, P.T.; Perales, J. Platelet proteome reveals novel pathways of platelet activation and platelet-mediated immunoregulation in dengue. PLoS Pathog. 2017, 13, e1006385. [Google Scholar] [CrossRef]
- Kopeikina, E.; Ponomarev, E.D. The role of platelets in the stimulation of neuronal synaptic plasticity, electric activity, and oxidative phosphorylation: Possibilities for new therapy of neurodegenerative diseases. Front. Cell Neurosci. 2021, 15, 680126. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Braig, M.; Jakob, K.; Bienert, T.; Schäper, M.; Merkle, A.; Wadle, C.; Menza, M.; Neudorfer, I.; Bojti, I.; et al. Molecular magnetic resonance imaging of activated platelets allows noninvasive detection of early myocarditis in mice. Sci. Rep. 2020, 10, 13211. [Google Scholar] [CrossRef] [PubMed]
- McCright, I.J.; Fujinami, R.S. Lack of correlation of Theiler’s virus binding to cells with infection. J. Neurovirol. 1997, 3 (Suppl. S1), S68–S70. [Google Scholar] [PubMed]
- Takeda, K.; Kaifu, T.; Michihata, R.; Kinugawa, N.; Fujioka, A.; Tateno, A.; Toshima, K.; Kanoh, H.; Inamori, K.; Kamijo, K.; et al. Chronic encephalomyelitis virus exhibits cellular tropism and evades pDCs by binding to sialylated integrins as the cell surface receptors. Eur. J. Immunol. 2023, 53, e2350452. [Google Scholar] [CrossRef]
- Lipton, H.L.; Kumar, A.S.M.; Hertzler, S.; Reddi, H.V. Differential usage of carbohydrate co-receptors influences cellular tropism of Theiler’s murine encephalomyelitis virus infection of the central nervous system. Glycoconj. J. 2006, 23, 39–49. [Google Scholar] [CrossRef]
- Orian, J.M.; D’Souza, C.S.; Kocovski, P.; Krippner, G.; Hale, M.W.; Wang, X.; Peter, K. Platelets in multiple sclerosis: Early and central mediators of inflammation and neurodegeneration and attractive targets for molecular imaging and site-directed therapy. Front. Immunol. 2021, 12, 620963. [Google Scholar] [CrossRef]
- Saluk-Bijak, J.; Dziedzic, A.; Bijak, M. Pro-thrombotic activity of blood platelets in multiple sclerosis. Cells 2019, 8, 110. [Google Scholar] [CrossRef]
- Krishnamurti, C.; Peat, R.A.; Cutting, M.A.; Rothwell, S.W. Platelet adhesion to dengue-2 virus-infected endothelial cells. Am. J. Trop. Med. Hyg. 2002, 66, 435–441. [Google Scholar] [CrossRef]
- Kapur, R.; Zufferey, A.; Boilard, E.; Semple, J.W. Nouvelle cuisine: Platelets served with inflammation. J. Immunol. 2015, 194, 5579–5587. [Google Scholar] [CrossRef]
- Rowley, J.W.; Schwertz, H.; Weyrich, A.S. Platelet mRNA: The meaning behind the message. Curr. Opin. Hematol. 2012, 19, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Gawaz, M. Platelets and inflammation. In Platelet Function: Assessment, Diagnosis, and Treatment, 1st ed.; Quinn, M., Fitzgerald, D., Eds.; Humana Press: Totowa, NJ, USA, 2005; pp. 115–147. [Google Scholar]
- Olson, J.K. Effect of the innate immune response on development of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Neurovirol. 2014, 20, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. The role of T cells and the innate immune system in the pathogenesis of Theiler’s virus demyelinating disease. In Experimental Models of Multiple Sclerosis, 1st ed.; Lavi, E., Constantinescu, C.S., Eds.; Springer: New York, NY, USA, 2005; pp. 645–657. [Google Scholar]
- Argilaguet, J.; Pedragosa, M.; Esteve-Codina, A.; Riera, G.; Vidal, E.; Peligero-Cruz, C.; Casella, V.; Andreu, D.; Kaisho, T.; Bocharov, G.; et al. Systems analysis reveals complex biological processes during virus infection fate decisions. Genome Res. 2019, 29, 907–919. [Google Scholar] [CrossRef]
- Omura, S.; Sato, F.; Park, A.-M.; Fujita, M.; Khadka, S.; Nakamura, Y.; Katsuki, A.; Nishio, K.; Gavins, F.N.E.; Tsunoda, I. Bioinformatics analysis of gut microbiota and CNS transcriptome in virus-induced acute myelitis and chronic inflammatory demyelination; potential association of distinct bacteria with CNS IgA upregulation. Front. Immunol. 2020, 11, 1138. [Google Scholar] [CrossRef] [PubMed]
- Arad, I.D.; Alpan, G.; Sznajderman, S.D.; Eldor, A. The mean platelet volume (MPV) in the neonatal period. Am. J. Perinatol. 1986, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Saxonhouse, M.A.; Garner, R.; Mammel, L.; Li, Q.; Muller, K.E.; Greywoode, J.; Miller, C.; Sola-Visner, M. Closure times measured by the platelet function analyzer PFA-100® are longer in neonatal blood compared to cord blood samples. Neonatology 2010, 97, 242–249. [Google Scholar] [CrossRef]
- Sitaru, A.G.; Holzhauer, S.; Speer, C.P.; Singer, D.; Obergfell, A.; Walter, U.; Grossmann, R. Neonatal platelets from cord blood and peripheral blood. Platelets 2005, 16, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, D.; Kestin, A.S.; Bednarek, F.J.; Ellis, P.A.; Barnard, M.R.; Michelson, A.D. Neonatal platelets are less reactive than adult platelets to physiological agonists in whole blood. Thromb. Haemost. 1994, 72, 957–963. [Google Scholar] [CrossRef]
- Baker-Groberg, S.M.; Lattimore, S.; Recht, M.; McCarty, O.J.T.; Haley, K.M. Assessment of neonatal platelet adhesion, activation, and aggregation. J. Thromb. Haemost. 2016, 14, 815–827. [Google Scholar] [CrossRef]
- Caparrós-Pérez, E.; Teruel-Montoya, R.; López-Andreo, M.J.; Llanos, M.C.; Rivera, J.; Palma-Barqueros, V.; Blanco, J.E.; Vicente, V.; Martinez, C.; Ferrer-Marin, F. Comprehensive comparison of neonate and adult human platelet transcriptomes. PLoS ONE 2017, 12, e0183042. [Google Scholar] [CrossRef]
- Stolla, M.C.; Catherman, S.C.; Kingsley, P.D.; Rowe, R.G.; Koniski, A.D.; Fegan, K.; Vit, L.; McGrath, K.E.; Daley, G.Q.; Palis, J. Lin28b regulates age-dependent differences in murine platelet function. Blood Adv. 2019, 3, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Davenport, P.; Sola-Visner, M. Platelets in the neonate: Not just a small adult. Res. Pract. Thromb. Haemost. 2022, 6, e12719. [Google Scholar] [CrossRef] [PubMed]
- Chapman, L.M.; Aggrey, A.A.; Field, D.J.; Srivastava, K.; Ture, S.; Yui, K.; Topham, D.J.; Baldwin, W.M., III; Morrell, C.N. Platelets present antigen in the context of MHC class I. J. Immunol. 2012, 189, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Shen, S.; Rowley, J.W.; Tolley, N.D.; Jia, W.; Manne, B.K.; McComas, K.N.; Bolingbroke, B.; Kosaka, Y.; Krauel, K.; et al. Platelet MHC class I mediates CD8+ T-cell suppression during sepsis. Blood 2021, 138, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Quiñones, C.; McGavern, D.; Schmelzer, J.D.; Hunter, S.F.; Low, P.A.; Rodriguez, M. Absence of neurological deficits following extensive demyelination in a class I-deficient murine model of multiple sclerosis. Nat. Med. 1998, 4, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Fujinami, R.S. Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J. Neuroimmune Pharmacol. 2010, 5, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Huang, D.; Nacion, K.; Bu, H.; Lin, F. Augmenting DAF levels in vivo ameliorates experimental autoimmune encephalomyelitis. Mol. Immunol. 2009, 46, 2885–2891. [Google Scholar] [CrossRef][Green Version]
- Ge, X.; Yu, Z.; Guo, X.; Li, L.; Ye, L.; Ye, M.; Yuan, J.; Zhu, C.; Hu, W.; Hou, Y. Complement and complement regulatory proteins are upregulated in lungs of COVID-19 patients. Pathol. Res. Pract. 2023, 247, 154519. [Google Scholar] [CrossRef]
- Sadoul, K. New explanations for old observations: Marginal band coiling during platelet activation. J. Thromb. Haemost. 2015, 13, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Zamora, E.J.; Ferrer-Marín, F.; Rivera, J.; Teruel-Montoya, R. Tubulin in Platelets: When the Shape Matters. Int. J. Mol. Sci. 2019, 20, 3484. [Google Scholar] [CrossRef] [PubMed]
- Diagouraga, B.; Grichine, A.; Fertin, A.; Wang, J.; Khochbin, S.; Sadoul, K. Motor-driven marginal band coiling promotes cell shape change during platelet activation. J. Cell Biol. 2014, 204, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Reddel, C.J.; Pennings, G.J.; Chen, V.M.; Gnanenthiran, S.R.; Kritharides, L. Colchicine as a modulator of platelet function: A systematic review. Semin. Thromb. Hemost. 2022, 48, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.Y.; Koyama-Nasu, R.; Yagi, R.; Nakayama, T. A new therapeutic target: The CD69-Myl9 system in immune responses. Semin. Immunopathol. 2019, 41, 349–358. [Google Scholar] [CrossRef]
- Reusswig, F.; Polzin, A.; Klier, M.; Dille, M.A.; Ayhan, A.; Benkhoff, M.; Lersch, C.; Prinz, A.; Gorressen, S.; Fischer, J.W.; et al. Only acute but not chronic thrombocytopenia protects mice against left ventricular dysfunction after acute myocardial infarction. Cells 2022, 11, 3500. [Google Scholar] [CrossRef]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef]
- D’Souza, C.S.; Li, Z.; Maxwell, D.L.; Trusler, O.; Murphy, M.; Crewther, S.; Peter, K.; Orian, J.M. Platelets drive inflammation and target gray matter and the retina in autoimmune-mediated encephalomyelitis. J. Neuropathol. Exp. Neurol. 2018, 77, 567–576. [Google Scholar] [CrossRef]
- Martinez, N.E.; Karlsson, F.; Sato, F.; Kawai, E.; Omura, S.; Minagar, A.; Grisham, M.B.; Tsunoda, I. Protective and detrimental roles for regulatory T cells in a viral model for multiple sclerosis. Brain Pathol. 2014, 24, 436–451. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Coffman, R.L. Heterogeneity of cytokine secretion patterns and functions of helper T cells. Adv. Immunol. 1989, 46, 111–147. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Zoecklein, L.J.; Howe, C.L.; Pavelko, K.D.; Gamez, J.D.; Nakane, S.; Papke, L.M. Gamma interferon is critical for neuronal viral clearance and protection in a susceptible mouse strain following early intracranial Theiler’s murine encephalomyelitis virus infection. J. Virol. 2003, 77, 12252–12265. [Google Scholar] [CrossRef] [PubMed]
- Binder, G.K.; Griffin, D.E. Interferon-γ-mediated site-specific clearance of alphavirus from CNS neurons. Science 2001, 293, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.E.; Lawrence, D.M.P.; Echols, L.A.; Rall, G.F. Immune-mediated protection from measles virus-induced central nervous system disease is noncytolytic and gamma interferon dependent. J. Virol. 2002, 76, 4497–4506. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Frisancho-Kiss, S.; Yusung, S.A.; Barrett, M.A.; Davis, S.E.; Gatewood, S.J.; Njoku, D.B.; Rose, N.R. Interferon-γ protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-β1, interleukin-1β, and interleukin-4 in the heart. Am. J. Pathol. 2004, 165, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Shibamori, M.; Ogura, M.; Seko, Y.; Kikuchi, M. Effects of intranasal administration of recombinant murine interferon-γ on murine acute myocarditis caused by encephalomyocarditis virus. Circulation 1998, 97, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Park, A.-M.; Tsunoda, I. Forensic luminol reaction for detecting fecal occult blood in experimental mice. Biotechniques 2018, 65, 227–230. [Google Scholar] [CrossRef]
- Harrington, R.A.; Stone, G.W.; McNulty, S.; White, H.D.; Lincoff, A.M.; Gibson, C.M.; Pollack, C.V., Jr.; Montalescot, G.; Mahaffey, K.W.; Kleiman, N.S.; et al. Platelet inhibition with cangrelor in patients undergoing PCI. N. Engl. J. Med. 2009, 361, 2318–2329. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Stone, G.W.; Mahaffey, K.W.; Gibson, C.M.; Steg, P.G.; Hamm, C.W.; Price, M.J.; Leonardi, S.; Gallup, D.; Bramucci, E.; et al. Effect of platelet inhibition with cangrelor during PCI on ischemic events. N. Engl. J. Med. 2013, 368, 1303–1313. [Google Scholar] [CrossRef]
- Becker, R.C.; Bassand, J.P.; Budaj, A.; Wojdyla, D.M.; James, S.K.; Cornel, J.H.; French, J.; Held, C.; Horrow, J.; Husted, S.; et al. Bleeding complications with the P2Y12 receptor antagonists clopidogrel and ticagrelor in the PLATelet inhibition and patient Outcomes (PLATO) trial. Eur. Heart J. 2011, 32, 2933–2944. [Google Scholar] [CrossRef]
- Baurand, A.; Raboisson, P.; Freund, M.; Léon, C.; Cazenave, J.-P.; Bourguignon, J.-J.; Gachet, C. Inhibition of platelet function by administration of MRS2179, a P2Y1 receptor antagonist. Eur. J. Pharmacol. 2001, 412, 213–221. [Google Scholar] [CrossRef]
- Dunne, H.; Cowman, J.; Kenny, D. MRS2179: A novel inhibitor of platelet function. BMC Proc. 2013, 9, A2. [Google Scholar] [CrossRef]
- The Epic Investigators. Use of a monoclonal antibody directed against the platelet glycoprotein IIb/IIIa receptor in high-risk coronary angioplasty. N. Engl. J. Med. 1994, 330, 956–961. [Google Scholar] [CrossRef]
- Moeckel, D.; Jeong, S.S.; Sun, X.; Broekman, M.J.; Nguyen, A.; Drosopoulos, J.H.F.; Marcus, A.J.; Robson, S.C.; Chen, R.; Abendschein, D. Optimizing human apyrase to treat arterial thrombosis and limit reperfusion injury without increasing bleeding risk. Sci. Transl. Med. 2014, 6, 248ra105. [Google Scholar] [CrossRef] [PubMed]
- Daniels, J.B.; Pappenheimer, A.M.; Richardson, S. Observations on encephalomyelitis of mice (DA strain). J. Exp. Med. 1952, 96, 517–530. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Senchenkova, E.Y.; Ansari, J.; Becker, F.; Vital, S.A.; Al-Yafeai, Z.; Sparkenbaugh, E.M.; Pawlinski, R.; Stokes, K.Y.; Carroll, J.L.; Dragoi, A.-M.; et al. Novel role for the AnxA1-Fpr2/ALX signaling axis as a key regulator of platelet function to promote resolution of inflammation. Circulation 2019, 140, 319–335. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Sun, J.; Nishiyama, T.; Shimizu, K.; Kadota, K. TCC: An R package for comparing tag count data with robust normalization strategies. BMC Bioinform. 2013, 14, 219. [Google Scholar] [CrossRef] [PubMed]
- Ringnér, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Loke, P.; Favre, D.; Hunt, P.W.; Leung, J.M.; Kanwar, B.; Martin, J.N.; Deeks, S.G.; McCune, J.M. Correlating cellular and molecular signatures of mucosal immunity that distinguish HIV controllers from noncontrollers. Blood 2010, 115, e20–e32. [Google Scholar] [CrossRef][Green Version]
- Davies, D.L.; Bouldin, D.W. A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell. 1979, 1, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Sholl, L.; Isermann, B.; Zogg, M.; Coughlin, S.R.; Weiler, H. Maternal Par4 and platelets contribute to defective placenta formation in mouse embryos lacking thrombomodulin. Blood 2008, 112, 585–591. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sato, F.; Nakamura, Y.; Katsuki, A.; Khadka, S.; Ahmad, I.; Omura, S.; Martinez, N.E.; Tsunoda, I. Curdlan, a microbial β-glucan, has contrasting effects on autoimmune and viral models of multiple sclerosis. Front. Cell Infect. Microbiol. 2022, 12, 805302. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Kawai, E.; Martinez, N.E.; Omura, S.; Park, A.-M.; Takahashi, S.; Yoh, K.; Tsunoda, I. T-bet, but not Gata3, overexpression is detrimental in a neurotropic viral infection. Sci. Rep. 2017, 7, 10496. [Google Scholar] [CrossRef]
- Nitayaphan, S.; Toth, M.M.; Roos, R.P. Neutralizing monoclonal antibodies to Theiler’s murine encephalomyelitis viruses. J. Virol. 1985, 53, 651–657. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, I.; Omura, S.; Sato, F.; Park, A.-M.; Khadka, S.; Gavins, F.N.E.; Tanaka, H.; Kimura, M.Y.; Tsunoda, I. Exploring the Role of Platelets in Virus-Induced Inflammatory Demyelinating Disease and Myocarditis. Int. J. Mol. Sci. 2024, 25, 3460. https://doi.org/10.3390/ijms25063460
Ahmad I, Omura S, Sato F, Park A-M, Khadka S, Gavins FNE, Tanaka H, Kimura MY, Tsunoda I. Exploring the Role of Platelets in Virus-Induced Inflammatory Demyelinating Disease and Myocarditis. International Journal of Molecular Sciences. 2024; 25(6):3460. https://doi.org/10.3390/ijms25063460
Chicago/Turabian StyleAhmad, Ijaz, Seiichi Omura, Fumitaka Sato, Ah-Mee Park, Sundar Khadka, Felicity N. E. Gavins, Hiroki Tanaka, Motoko Y. Kimura, and Ikuo Tsunoda. 2024. "Exploring the Role of Platelets in Virus-Induced Inflammatory Demyelinating Disease and Myocarditis" International Journal of Molecular Sciences 25, no. 6: 3460. https://doi.org/10.3390/ijms25063460
APA StyleAhmad, I., Omura, S., Sato, F., Park, A.-M., Khadka, S., Gavins, F. N. E., Tanaka, H., Kimura, M. Y., & Tsunoda, I. (2024). Exploring the Role of Platelets in Virus-Induced Inflammatory Demyelinating Disease and Myocarditis. International Journal of Molecular Sciences, 25(6), 3460. https://doi.org/10.3390/ijms25063460