
Prostaglandin Transporter and Dipeptidyl Peptidase-4 as New Pharmacological Targets in the Prevention of Acute Kidney Injury in Diabetes: An In Vitro Study

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
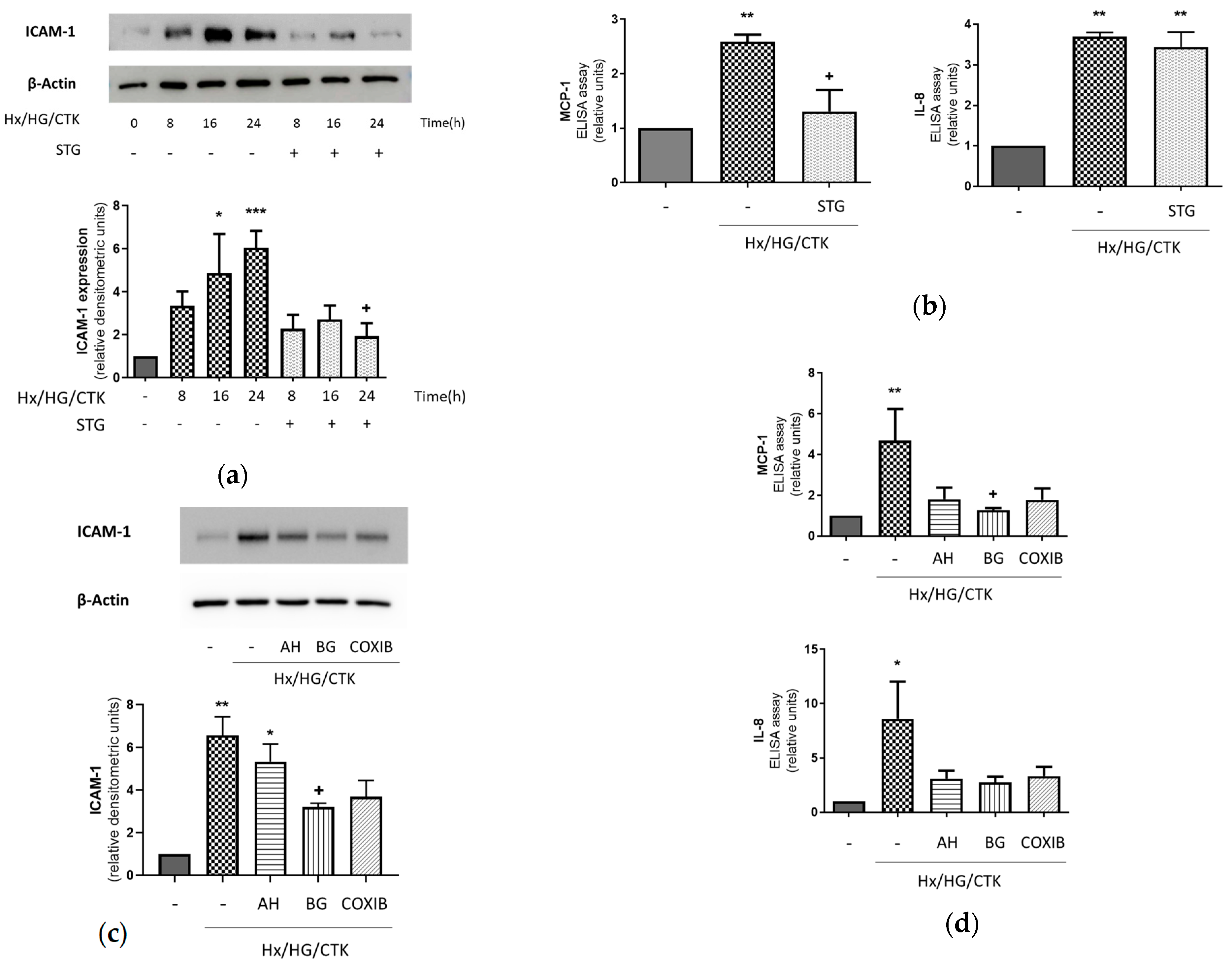
2.1. Hypoxic/Hyperglycemic/Inflammatory Conditions Determine Pro-Inflammatory Responses in Human Renal Proximal Tubular HK-2 Cells: Dependency on DPP-4, COX-2, EP Receptors and PGT
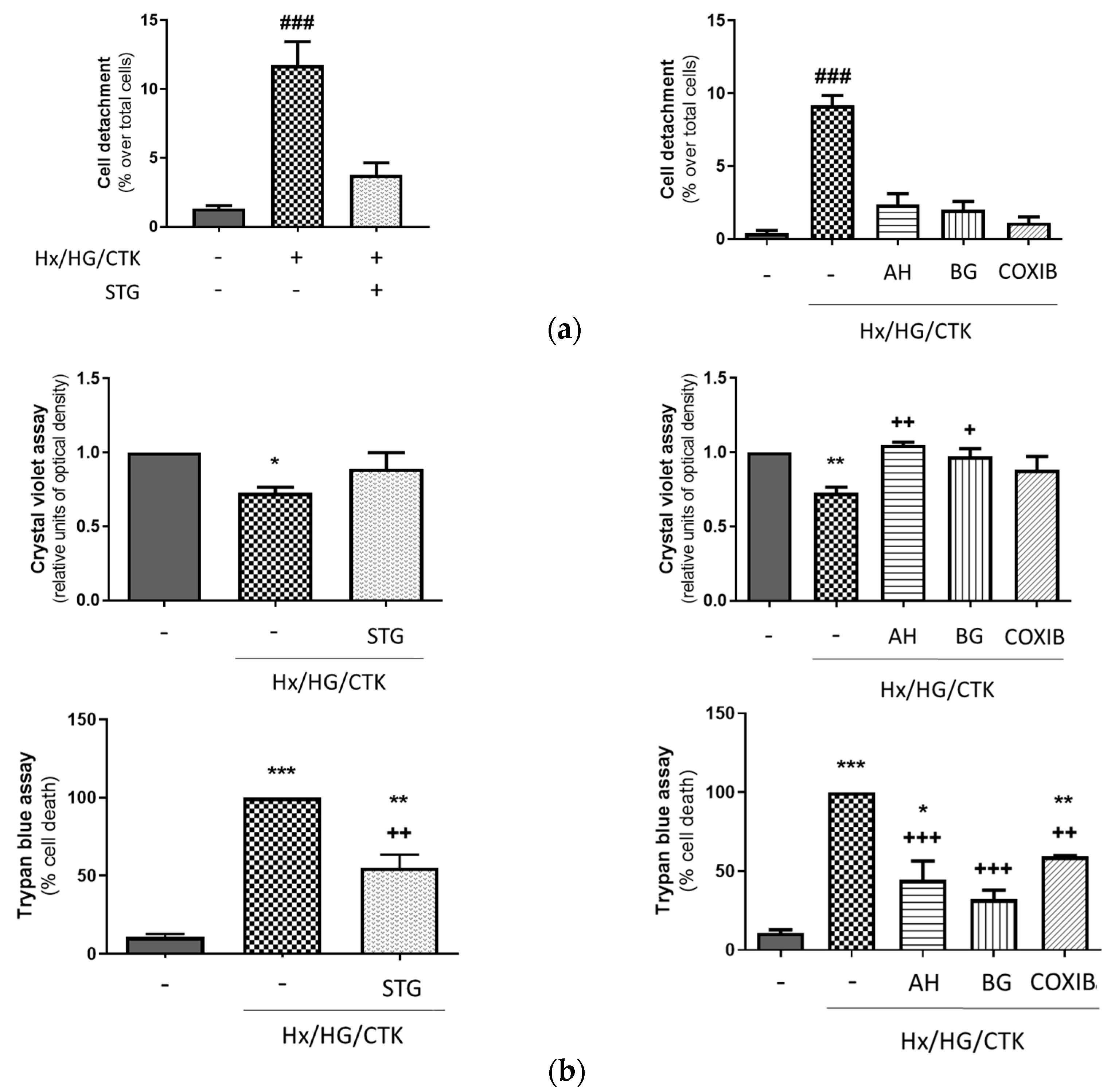
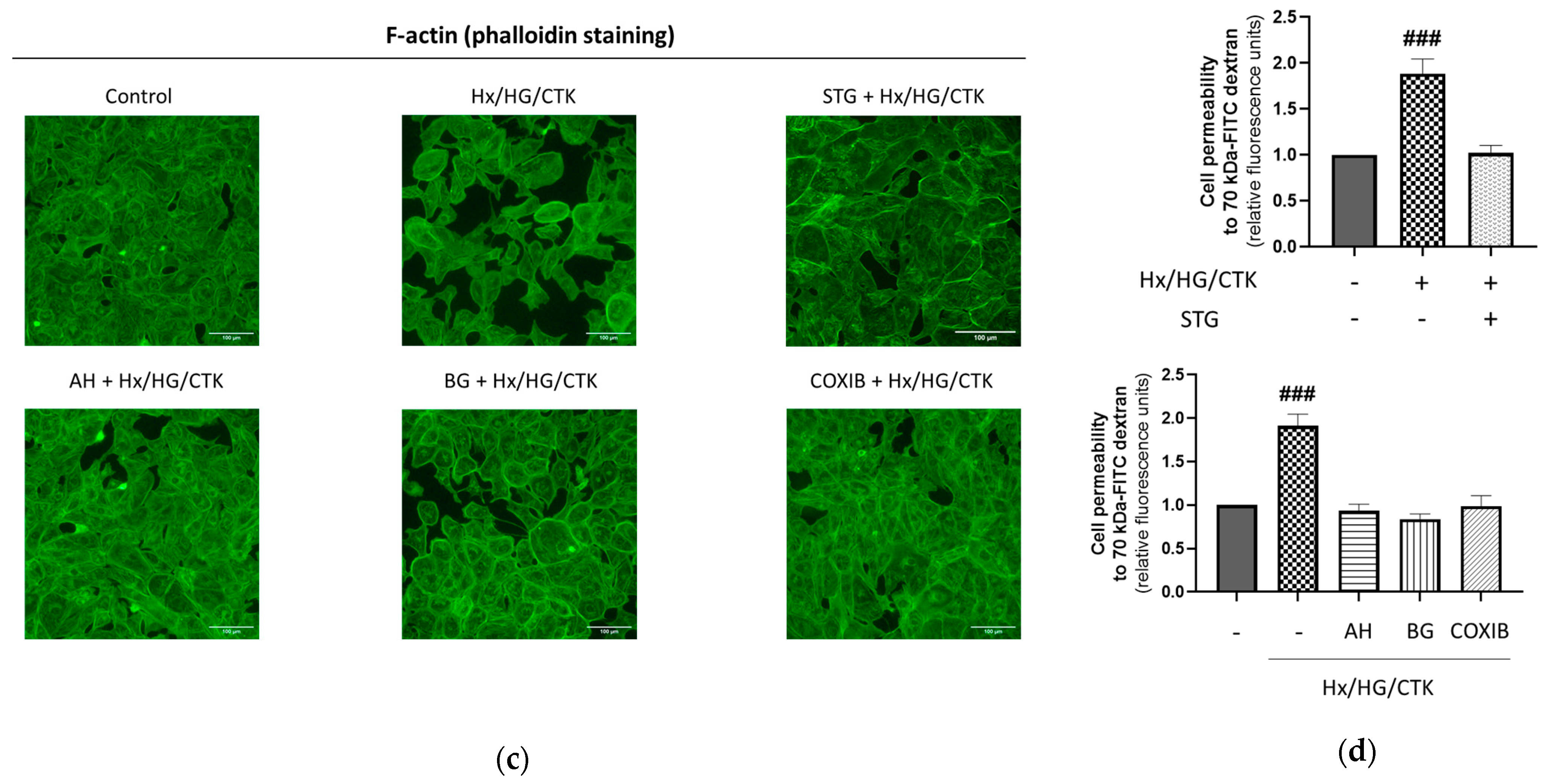
2.2. Hypoxic/Hyperglycemic/Inflammatory Conditions Induce Epithelial Monolayer Injury and Alteration of Paracellular Permeability: Dependency on DPP-4, COX-2, EP Receptors and PGT
2.3. Human Proximal Tubular HK-2 Cells Exposed to Hypoxic/Hyperglycemic/Inflammatory Conditions Exhibit Increased DPP-4 Expression and Activity: Dependency on COX-2, EP Receptors and PGT
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Experimental Conditions
4.3. Protein Extraction and Western Blot Assay
4.4. RNA Isolation and Real-Time PCR (q-RT-PCR) Analysis
4.5. DPP-4 Activity
4.6. MCP-1 and IL-8 Analysis in the Extracellular Medium
4.7. Immunofluorescence Assessment
4.8. Crystal Violet and Trypan Blue Assays
4.9. Assessment of Cell Detachment
4.10. Paracellular Permeability Assay
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Porter, A.W.; Brodsky, J.L.; Buck, T.M. Emerging Links between Endoplasmic Reticulum Stress Responses and Acute Kidney Injury. Am. J. Physiol. Cell Physiol. 2022, 323, C1697–C1703. [Google Scholar] [CrossRef]
- Eckardt, K.-U.; Bernhardt, W.M.; Weidemann, A.; Warnecke, C.; Rosenberger, C.; Wiesener, M.S.; Willam, C. Role of Hypoxia in the Pathogenesis of Renal Disease. Kidney Int. Suppl. 2005, 99, S46–S51. [Google Scholar] [CrossRef]
- Singbartl, K.; Formeck, C.L.; Kellum, J.A. Kidney-Immune System Crosstalk in AKI. Semin. Nephrol. 2019, 39, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Akcay, A.; Nguyen, Q.; Edelstein, C.L. Mediators of Inflammation in Acute Kidney Injury. Mediat. Inflamm. 2009, 2009, 137072. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L.; Lan, H.-Y. Renal Tubule Injury: A Driving Force toward Chronic Kidney Disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Costantini, E.; Carlin, M.; Porta, M.; Brizzi, M.F. Type 2 Diabetes Mellitus and Sepsis: State of the Art, Certainties and Missing Evidence. Acta Diabetol. 2021, 58, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.M.; Morgan, D.J.R. The Proximal Tubule as the Pathogenic and Therapeutic Target in Acute Kidney Injury. Nephron 2022, 146, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Nørregaard, R.; Kwon, T.-H.; Frøkiær, J. Physiology and Pathophysiology of Cyclooxygenase-2 and Prostaglandin E2 in the Kidney. Kidney Res. Clin. Pract. 2015, 34, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Zhang, Y.; Ding, G.; Heiney, K.M.; Huang, S.; Zhang, A. Role of COX-2/mPGES-1/Prostaglandin E2 Cascade in Kidney Injury. Mediat. Inflamm. 2015, 2015, 147894. [Google Scholar] [CrossRef]
- Warner, T.D.; Mitchell, J.A. Cyclooxygenases: New Forms, New Inhibitors, and Lessons from the Clinic. FASEB J. 2004, 18, 790–804. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Cheng, C.-Y.; Chen, Y.-C.; Chen, T.-H.; Sue, Y.-M.; Tsai, W.-L.; Chen, C.-H. Long-Term Leptin Treatment Exerts a pro-Apoptotic Effect on Renal Tubular Cells via Prostaglandin E2 Augmentation. Eur. J. Pharmacol. 2012, 689, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Martínez, A.B.; Benito Martínez, S.; Lucio Cazaña, F.J. Intracellular Prostaglandin E2 Mediates Cisplatin-Induced Proximal Tubular Cell Death. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2016, 1863, 293–302. [Google Scholar] [CrossRef]
- Zhuang, Y.; Zhao, F.; Liang, J.; Deng, X.; Zhang, Y.; Ding, G.; Zhang, A.; Jia, Z.; Huang, S. Activation of COX-2/mPGES-1/PGE2 Cascade via NLRP3 Inflammasome Contributes to Albumin-Induced Proximal Tubule Cell Injury. Cell. Physiol. Biochem. 2017, 42, 797–807. [Google Scholar] [CrossRef]
- Sun, H.-J.; Leng, B.; Wu, Z.-Y.; Bian, J.-S. Polysulfide and Hydrogen Sulfide Ameliorate Cisplatin-Induced Nephrotoxicity and Renal Inflammation through Persulfidating STAT3 and IKKβ. Int. J. Mol. Sci. 2020, 21, 7805. [Google Scholar] [CrossRef]
- García-Pastor, C.; Blázquez-Serra, R.; Bosch, R.J.; Lucio Cazaña, F.J.; Fernández-Martínez, A.B. Apoptosis and Cell Proliferation in Proximal Tubular Cells Exposed to Apoptotic Bodies. Novel Pathophysiological Implications in Cisplatin-Induced Renal Injury. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2019, 1865, 2504–2515. [Google Scholar] [CrossRef] [PubMed]
- García-Pastor, C.; Benito-Martínez, S.; Bosch, R.J.; Fernández-Martínez, A.B.; Lucio-Cazaña, F.J. Intracellular Prostaglandin E2 Contributes to Hypoxia-Induced Proximal Tubular Cell Death. Sci. Rep. 2021, 11, 7047. [Google Scholar] [CrossRef] [PubMed]
- Yago-Ibáñez, J.; Muñoz-Moreno, L.; Gallego-Tamayo, B.; Lucio-Cazaña, F.J.; Fernández-Martínez, A.B. Prostaglandin Transporter PGT as a New Pharmacological Target in the Prevention of Inflammatory Cytokine-Induced Injury in Renal Proximal Tubular HK-2 Cells. Life Sci. 2023, 313, 121260. [Google Scholar] [CrossRef]
- Nakanishi, T.; Tamai, I. Roles of Organic Anion Transporting Polypeptide 2A1 (OATP2A1/SLCO2A1) in Regulating the Pathophysiological Actions of Prostaglandins. AAPS J. 2017, 20, 13. [Google Scholar] [CrossRef]
- Girardi, A.C.; Degray, B.C.; Nagy, T.; Biemesderfer, D.; Aronson, P.S. Association of Na(+)-H(+) Exchanger Isoform NHE3 and Dipeptidyl Peptidase IV in the Renal Proximal Tubule. J. Biol. Chem. 2001, 276, 46671–46677. [Google Scholar] [CrossRef]
- Nicotera, R.; Casarella, A.; Longhitano, E.; Bolignano, D.; Andreucci, M.; De Sarro, G.; Cernaro, V.; Russo, E.; Coppolino, G. Antiproteinuric Effect of DPP-IV Inhibitors in Diabetic and Non-Diabetic Kidney Diseases. Pharmacol. Res. 2020, 159, 105019. [Google Scholar] [CrossRef]
- Wu, T.-J.; Hsieh, Y.-J.; Lu, C.-W.; Lee, C.-J.; Hsu, B.-G. Linagliptin Protects against Endotoxin-Induced Acute Kidney Injury in Rats by Decreasing Inflammatory Cytokines and Reactive Oxygen Species. Int. J. Mol. Sci. 2021, 22, 11190. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, T.; Zhao, Z.; Marschner, J.A.; Devarapu, S.K.; Yasuda, H.; Anders, H.J. Dipeptidyl Peptidase-4 Inhibitor Teneligliptin Accelerates Recovery from Cisplatin-Induced Acute Kidney Injury by Attenuating Inflammation and Promoting Tubular Regeneration. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2019, 34, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Takashi, Y.; Takahashi, H.; Motonaga, R.; Tanabe, M. Renoprotective Effects of DPP-4 Inhibitors. Antioxid. Basel Switz. 2021, 10, 246. [Google Scholar] [CrossRef]
- Kaifu, K.; Ueda, S.; Nakamura, N.; Matsui, T.; Yamada-Obara, N.; Ando, R.; Kaida, Y.; Nakata, M.; Matsukuma-Toyonaga, M.; Higashimoto, Y.; et al. Advanced Glycation End Products Evoke Inflammatory Reactions in Proximal Tubular Cells via Autocrine Production of Dipeptidyl Peptidase-4. Microvasc. Res. 2018, 120, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Slyne, J.; Slattery, C.; McMorrow, T.; Ryan, M.P. New Developments Concerning the Proximal Tubule in Diabetic Nephropathy: In Vitro Models and Mechanisms. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2015, 30 (Suppl. 4), iv60–iv67. [Google Scholar] [CrossRef]
- Zarbock, A.; Nadim, M.K.; Pickkers, P.; Gomez, H.; Bell, S.; Joannidis, M.; Kashani, K.; Koyner, J.L.; Pannu, N.; Meersch, M.; et al. Sepsis-Associated Acute Kidney Injury: Consensus Report of the 28th Acute Disease Quality Initiative Workgroup. Nat. Rev. Nephrol. 2023, 19, 401–417. [Google Scholar] [CrossRef]
- Álvarez Cilleros, D.; López-Oliva, M.E.; Martín, M.Á.; Ramos, S. (-)-Epicatechin and the Colonic Metabolite 2,3-Dihydroxybenzoic Acid Protect against High Glucose and Lipopolysaccharide-Induced Inflammation in Renal Proximal Tubular Cells through NOX-4/P38 Signalling. Food Funct. 2020, 11, 8811–8824. [Google Scholar] [CrossRef]
- Ho, A.W.Y.; Wong, C.K.; Lam, C.W.K. Tumor Necrosis Factor-Alpha up-Regulates the Expression of CCL2 and Adhesion Molecules of Human Proximal Tubular Epithelial Cells through MAPK Signaling Pathways. Immunobiology 2008, 213, 533–544. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, Q.; Shi, Y.; Liu, J.; Zhong, F.; Hao, X.; Li, C.; Chen, N.; Wang, W. SUMOylation of PPARγ by Rosiglitazone Prevents LPS-Induced NCoR Degradation Mediating down Regulation of Chemokines Expression in Renal Proximal Tubular Cells. PLoS ONE 2013, 8, e79815. [Google Scholar] [CrossRef]
- Tang, S.C.W.; Leung, J.C.K.; Chan, L.Y.Y.; Tsang, A.W.L.; Lai, K.N. Activation of Tubular Epithelial Cells in Diabetic Nephropathy and the Role of the Peroxisome Proliferator-Activated Receptor-Gamma Agonist. J. Am. Soc. Nephrol. JASN 2006, 17, 1633–1643. [Google Scholar] [CrossRef]
- Viedt, C.; Dechend, R.; Fei, J.; Hänsch, G.M.; Kreuzer, J.; Orth, S.R. MCP-1 Induces Inflammatory Activation of Human Tubular Epithelial Cells: Involvement of the Transcription Factors, Nuclear Factor-kappaB and Activating Protein-1. J. Am. Soc. Nephrol. JASN 2002, 13, 1534–1547. [Google Scholar] [CrossRef]
- Denker, B.M.; Sabath, E. The Biology of Epithelial Cell Tight Junctions in the Kidney. J. Am. Soc. Nephrol. JASN 2011, 22, 622–625. [Google Scholar] [CrossRef]
- Bhave, G.; Colon, S.; Ferrell, N. The Sulfilimine Cross-Link of Collagen IV Contributes to Kidney Tubular Basement Membrane Stiffness. Am. J. Physiol. Renal Physiol. 2017, 313, F596–F602. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087379. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I. Actin Motors That Drive Formation and Disassembly of Epithelial Apical Junctions. Front. Biosci. J. Virtual Libr. 2008, 13, 6662–6681. [Google Scholar] [CrossRef]
- Kapus, A.; Szászi, K. Coupling between Apical and Paracellular Transport Processes. Biochem. Cell Biol. Biochim. Biol. Cell. 2006, 84, 870–880. [Google Scholar] [CrossRef]
- Valencia, I.; Vallejo, S.; Dongil, P.; Romero, A.; San Hipólito-Luengo, Á.; Shamoon, L.; Posada, M.; García-Olmo, D.; Carraro, R.; Erusalimsky, J.D.; et al. DPP4 Promotes Human Endothelial Cell Senescence and Dysfunction via the PAR2-COX-2-TP Axis and NLRP3 Inflammasome Activation. Hypertension 2022, 79, 1361–1373. [Google Scholar] [CrossRef]
- Li, Y.; Lv, X.; Jiang, M.; Jin, Z. Sitagliptin Ameliorates Hypoxia-Induced Damages in Endometrial Stromal Cells: An Implication in Endometriosis. Bioengineered 2022, 13, 800–809. [Google Scholar] [CrossRef]
- Lin, C.-H.; Lin, C.-C. Sitagliptin Attenuates Inflammatory Responses in Lipopolysaccharide-Stimulated Cardiomyocytes via Nuclear Factor-κB Pathway Inhibition. Exp. Ther. Med. 2016, 11, 2609–2615. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, J.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Xu, A.; Xu, G.; Ng, C.F.; Yao, X.; Gao, Y.; et al. Uncoupling Protein-2 Mediates DPP-4 Inhibitor-Induced Restoration of Endothelial Function in Hypertension through Reducing Oxidative Stress. Antioxid. Redox Signal. 2014, 21, 1571–1581. [Google Scholar] [CrossRef]
- Priante, G.; Gianesello, L.; Ceol, M.; Del Prete, D.; Anglani, F. Cell Death in the Kidney. Int. J. Mol. Sci. 2019, 20, 3598. [Google Scholar] [CrossRef]
- Zoja, C.; Garcia, P.B.; Remuzzi, G. The Role of Chemokines in Progressive Renal Disease. Front. Biosci.-Landmark 2009, 14, 1815–1822. [Google Scholar] [CrossRef]
- Li, B.; Lin, F.; Xia, Y.; Ye, Z.; Yan, X.; Song, B.; Yuan, T.; Li, L.; Zhou, X.; Yu, W.; et al. The Intersection of Acute Kidney Injury and Non-Coding RNAs: Inflammation. Front. Physiol. 2022, 13, 923239. [Google Scholar] [CrossRef]
- LaForge, J.M.; Urso, K.; Day, J.M.; Bourgeois, C.W.; Ross, M.M.; Ahmadzadeh, S.; Shekoohi, S.; Cornett, E.M.; Kaye, A.M.; Kaye, A.D. Non-Steroidal Anti-Inflammatory Drugs: Clinical Implications, Renal Impairment Risks, and AKI. Adv. Ther. 2023, 40, 2082–2096. [Google Scholar] [CrossRef]
- Omori, K.; Kida, T.; Hori, M.; Ozaki, H.; Murata, T. Multiple Roles of the PGE2-EP Receptor Signal in Vascular Permeability. Br. J. Pharmacol. 2014, 171, 4879–4889. [Google Scholar] [CrossRef]
- Tang, L.; Loutzenhiser, K.; Loutzenhiser, R. Biphasic Actions of Prostaglandin E2 on the Renal Afferent Arteriole. Circ. Res. 2000, 86, 663–670. [Google Scholar] [CrossRef]
- Zhao, M.; Sun, S.; Huang, Z.; Wang, T.; Tang, H. Network Meta-Analysis of Novel Glucose-Lowering Drugs on Risk of Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. CJASN 2020, 16, 70–78. [Google Scholar] [CrossRef]
- Yang, S.; He, W.; Zhao, L.; Mi, Y. Association between Use of Sodium-Glucose Cotransporter 2 Inhibitors, Glucagon-like Peptide 1 Agonists, and Dipeptidyl Peptidase 4 Inhibitors with Kidney Outcomes in Patients with Type 2 Diabetes: A Systematic Review and Network Meta-Analysis. PLoS ONE 2022, 17, e0267025. [Google Scholar] [CrossRef]
- Sutton, S.S.; Magagnoli, J.; Cummings, T.H.; Hardin, J.W. Odds of Acute Kidney Injury in Patients Receiving Dipeptidyl Peptidase 4 Inhibitors: A National Cohort Study Within the Department of Veterans Affairs. Clin. Transl. Sci. 2019, 12, 698–703. [Google Scholar] [CrossRef]
- Katagiri, D.; Hamasaki, Y.; Doi, K.; Okamoto, K.; Negishi, K.; Nangaku, M.; Noiri, E. Protection of Glucagon-Like Peptide-1 in Cisplatin-Induced Renal Injury Elucidates Gut-Kidney Connection. J. Am. Soc. Nephrol. JASN 2013, 24, 2034–2043. [Google Scholar] [CrossRef]
- Zhang, Q.; He, L.; Dong, Y.; Fei, Y.; Wen, J.; Li, X.; Guan, J.; Liu, F.; Zhou, T.; Li, Z.; et al. Sitagliptin Ameliorates Renal Tubular Injury in Diabetic Kidney Disease via STAT3-Dependent Mitochondrial Homeostasis through SDF-1α/CXCR4 Pathway. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 7500–7519. [Google Scholar] [CrossRef]
- Coppolino, G.; Leporini, C.; Rivoli, L.; Ursini, F.; di Paola, E.D.; Cernaro, V.; Arturi, F.; Bolignano, D.; Russo, E.; De Sarro, G.; et al. Exploring the Effects of DPP-4 Inhibitors on the Kidney from the Bench to Clinical Trials. Pharmacol. Res. 2018, 129, 274–294. [Google Scholar] [CrossRef]
- Glorie, L.L.F.; Verhulst, A.; Matheeussen, V.; Baerts, L.; Magielse, J.; Hermans, N.; D’Haese, P.C.; De Meester, I.; De Beuf, A. DPP4 Inhibition Improves Functional Outcome after Renal Ischemia-Reperfusion Injury. Am. J. Physiol. Renal Physiol. 2012, 303, F681–F688. [Google Scholar] [CrossRef]
- Tang, Y.; Leng, Y.-F.; Wang, W.; Zhang, J.; Yuan, T.-L.; Wang, J. Protective Effect of Saxagliptin on Diabetic Rats with Renal Ischemia Reperfusion Injury by Targeting Oxidative Stress and Mitochondrial Apoptosis Pathway through Activating Nrf-2/HO-1 Signaling. Transpl. Immunol. 2023, 76, 101762. [Google Scholar] [CrossRef]
- Iwakura, T.; Fukasawa, H.; Kitamura, A.; Ishibuchi, K.; Yasuda, H.; Furuya, R. Effect of Dipeptidyl Peptidase-4 Inhibitors on Cisplatin-Induced Acute Nephrotoxicity in Cancer Patients with Diabetes Mellitus: A Retrospective Study. PLoS ONE 2020, 15, e0229377. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallego-Tamayo, B.; Santos-Aparicio, Á.; Yago-Ibáñez, J.; Muñoz-Moreno, L.; Lucio-Cazaña, F.J.; Fernández-Martínez, A.B. Prostaglandin Transporter and Dipeptidyl Peptidase-4 as New Pharmacological Targets in the Prevention of Acute Kidney Injury in Diabetes: An In Vitro Study. Int. J. Mol. Sci. 2024, 25, 3345. https://doi.org/10.3390/ijms25063345
Gallego-Tamayo B, Santos-Aparicio Á, Yago-Ibáñez J, Muñoz-Moreno L, Lucio-Cazaña FJ, Fernández-Martínez AB. Prostaglandin Transporter and Dipeptidyl Peptidase-4 as New Pharmacological Targets in the Prevention of Acute Kidney Injury in Diabetes: An In Vitro Study. International Journal of Molecular Sciences. 2024; 25(6):3345. https://doi.org/10.3390/ijms25063345
Chicago/Turabian StyleGallego-Tamayo, Beatriz, Ángela Santos-Aparicio, Julia Yago-Ibáñez, Laura Muñoz-Moreno, Francisco Javier Lucio-Cazaña, and Ana B. Fernández-Martínez. 2024. "Prostaglandin Transporter and Dipeptidyl Peptidase-4 as New Pharmacological Targets in the Prevention of Acute Kidney Injury in Diabetes: An In Vitro Study" International Journal of Molecular Sciences 25, no. 6: 3345. https://doi.org/10.3390/ijms25063345
APA StyleGallego-Tamayo, B., Santos-Aparicio, Á., Yago-Ibáñez, J., Muñoz-Moreno, L., Lucio-Cazaña, F. J., & Fernández-Martínez, A. B. (2024). Prostaglandin Transporter and Dipeptidyl Peptidase-4 as New Pharmacological Targets in the Prevention of Acute Kidney Injury in Diabetes: An In Vitro Study. International Journal of Molecular Sciences, 25(6), 3345. https://doi.org/10.3390/ijms25063345