
Microbiome Dysbiosis Is Associated with Castration Resistance and Cancer Stemness in Metastatic Prostate Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Cross-Study Normalization and Contamination Correction
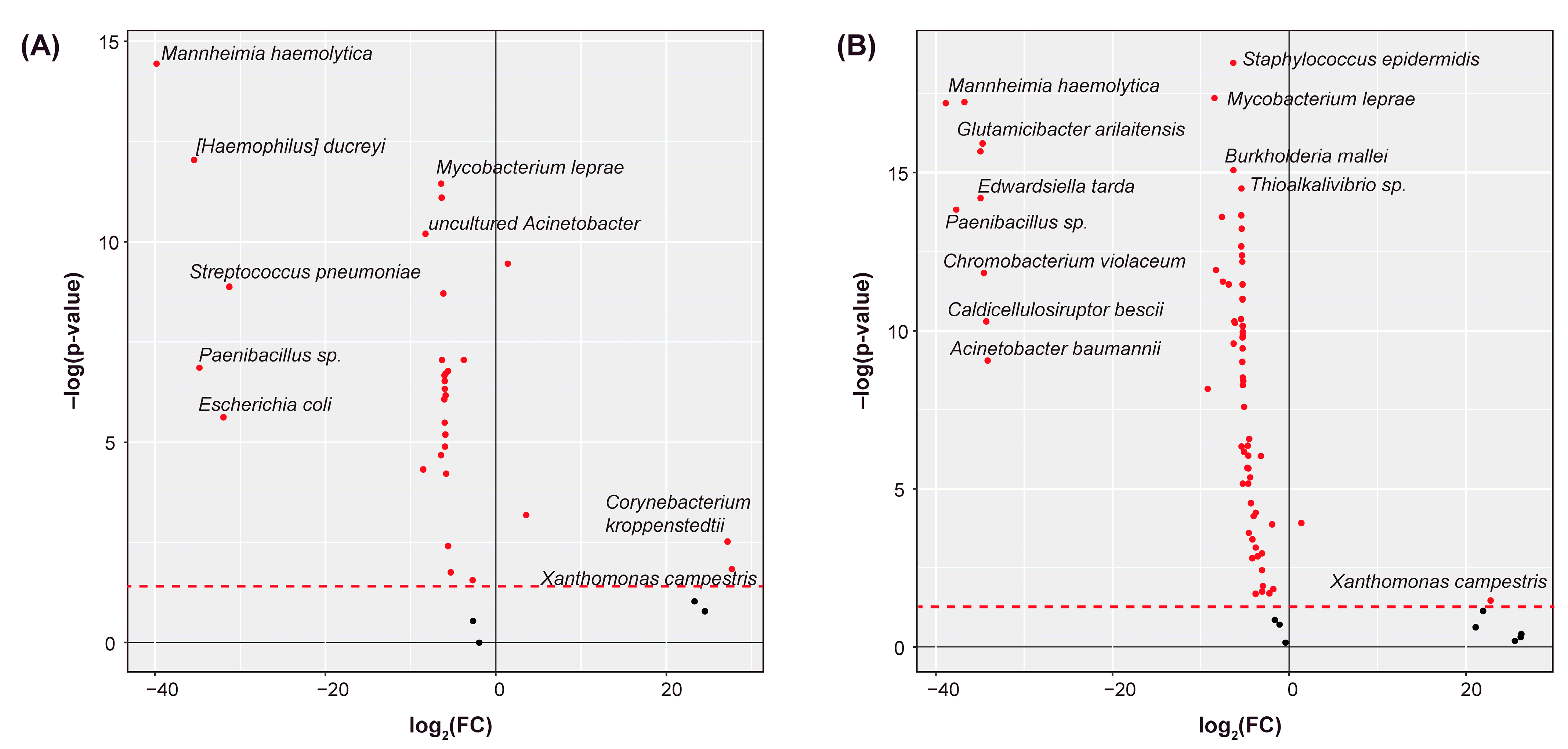
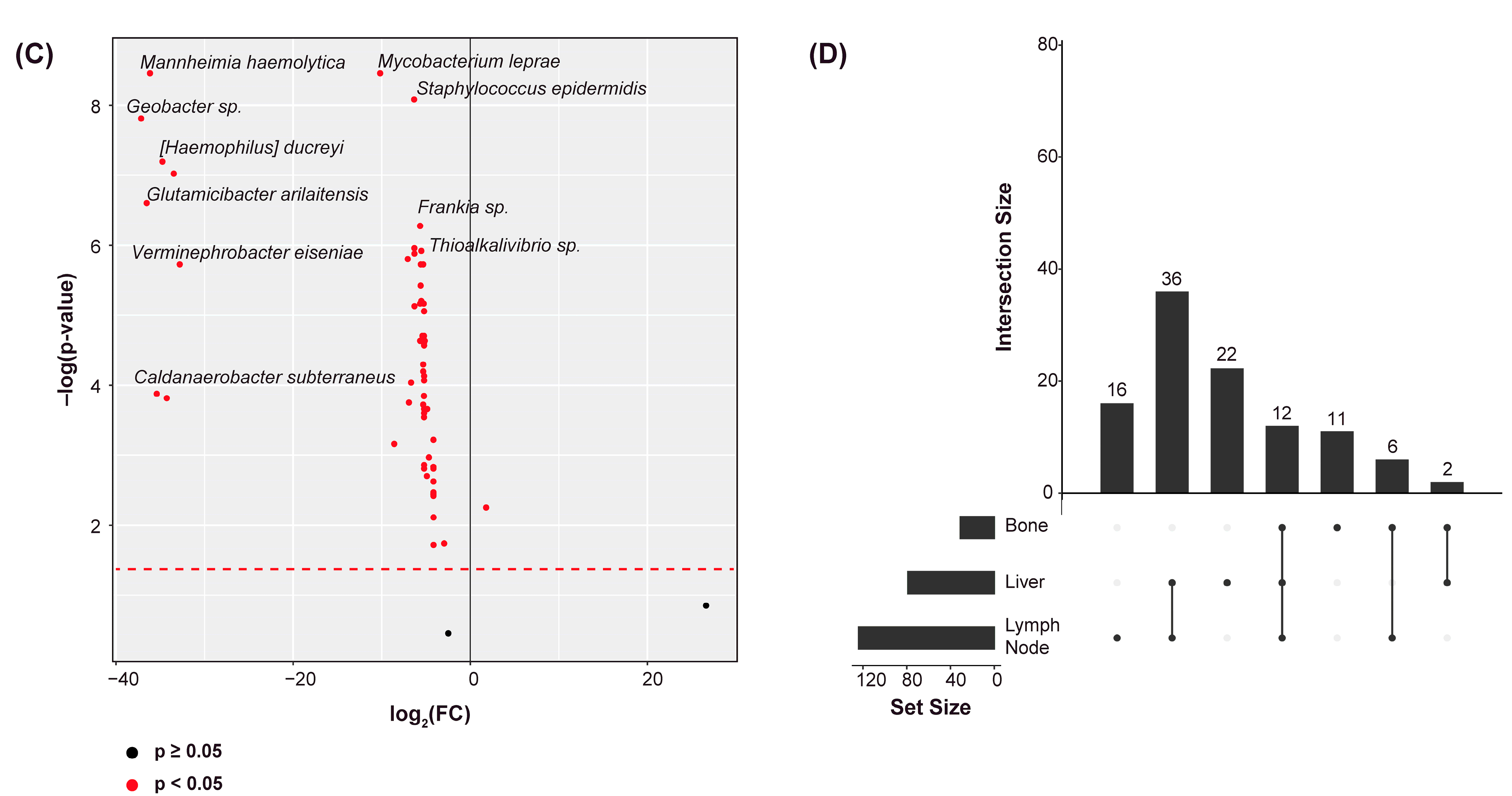
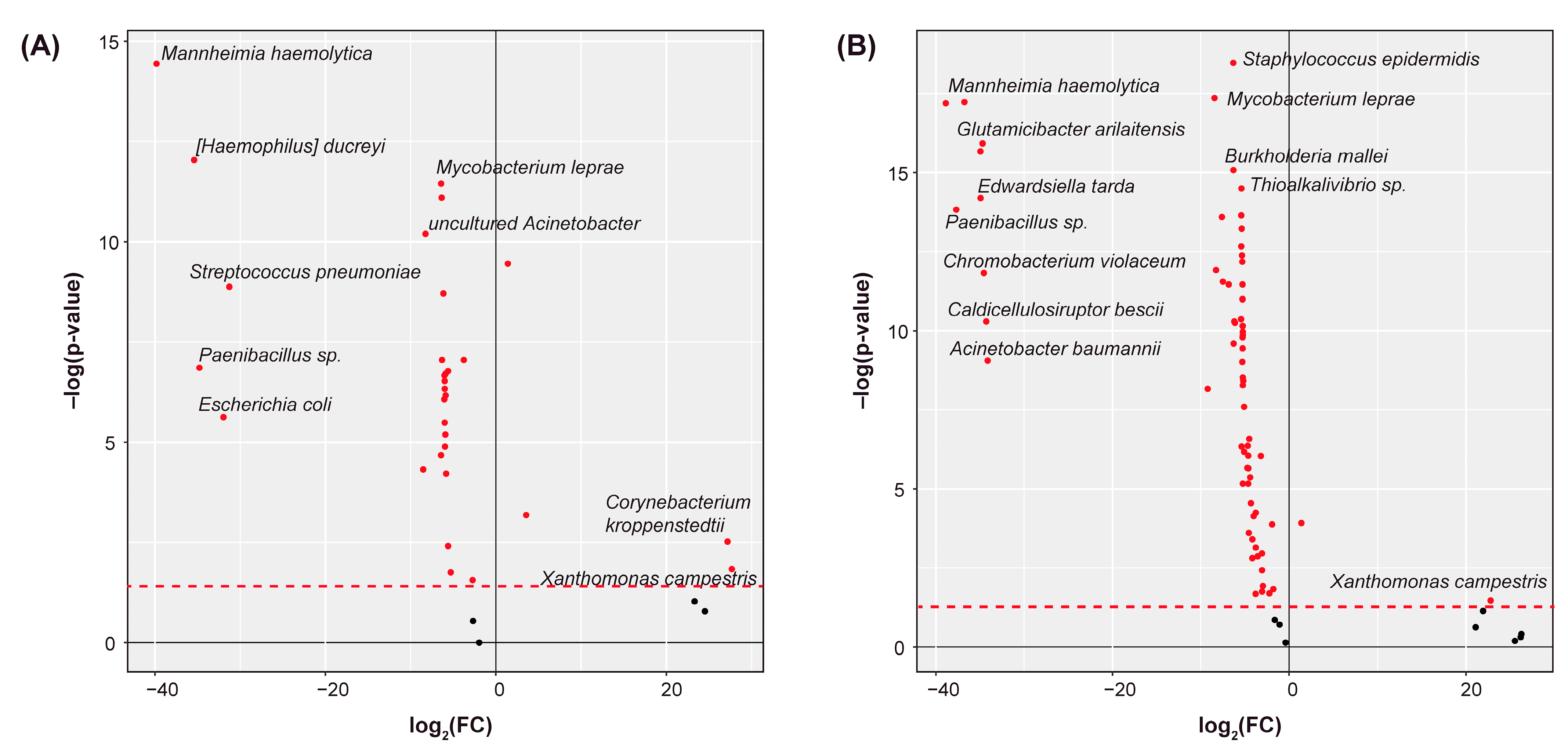
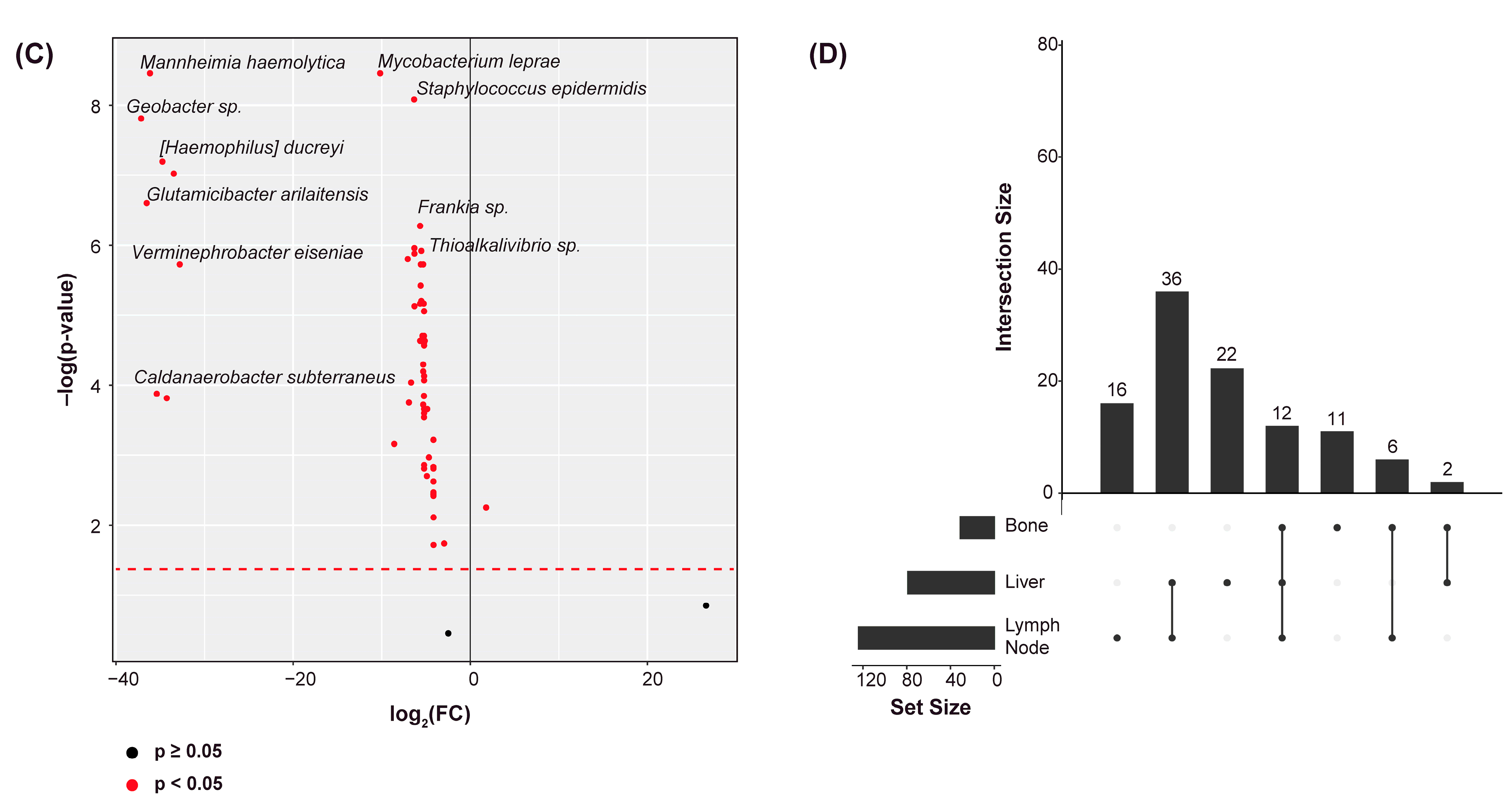
2.2. Differential Abundance Analysis
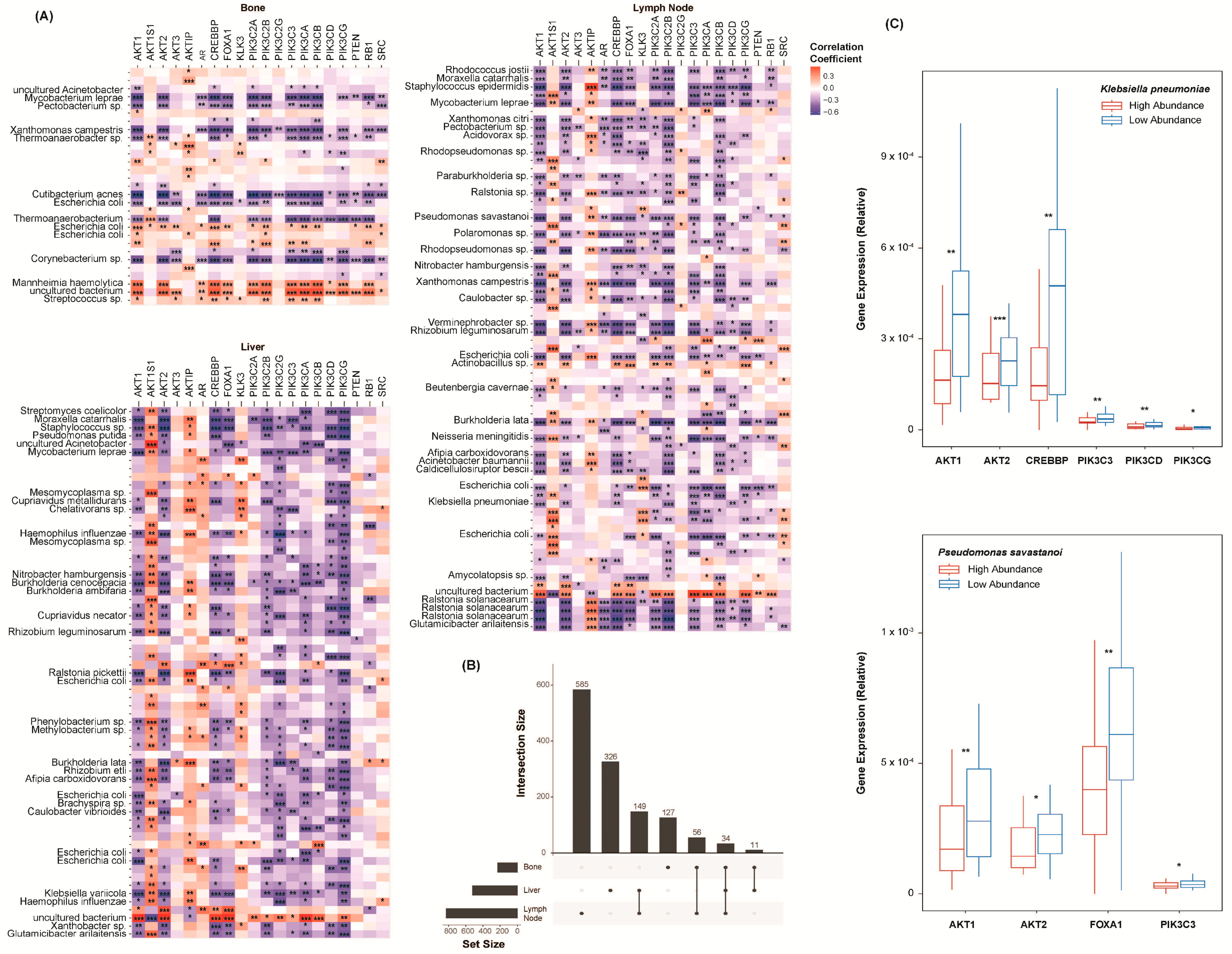
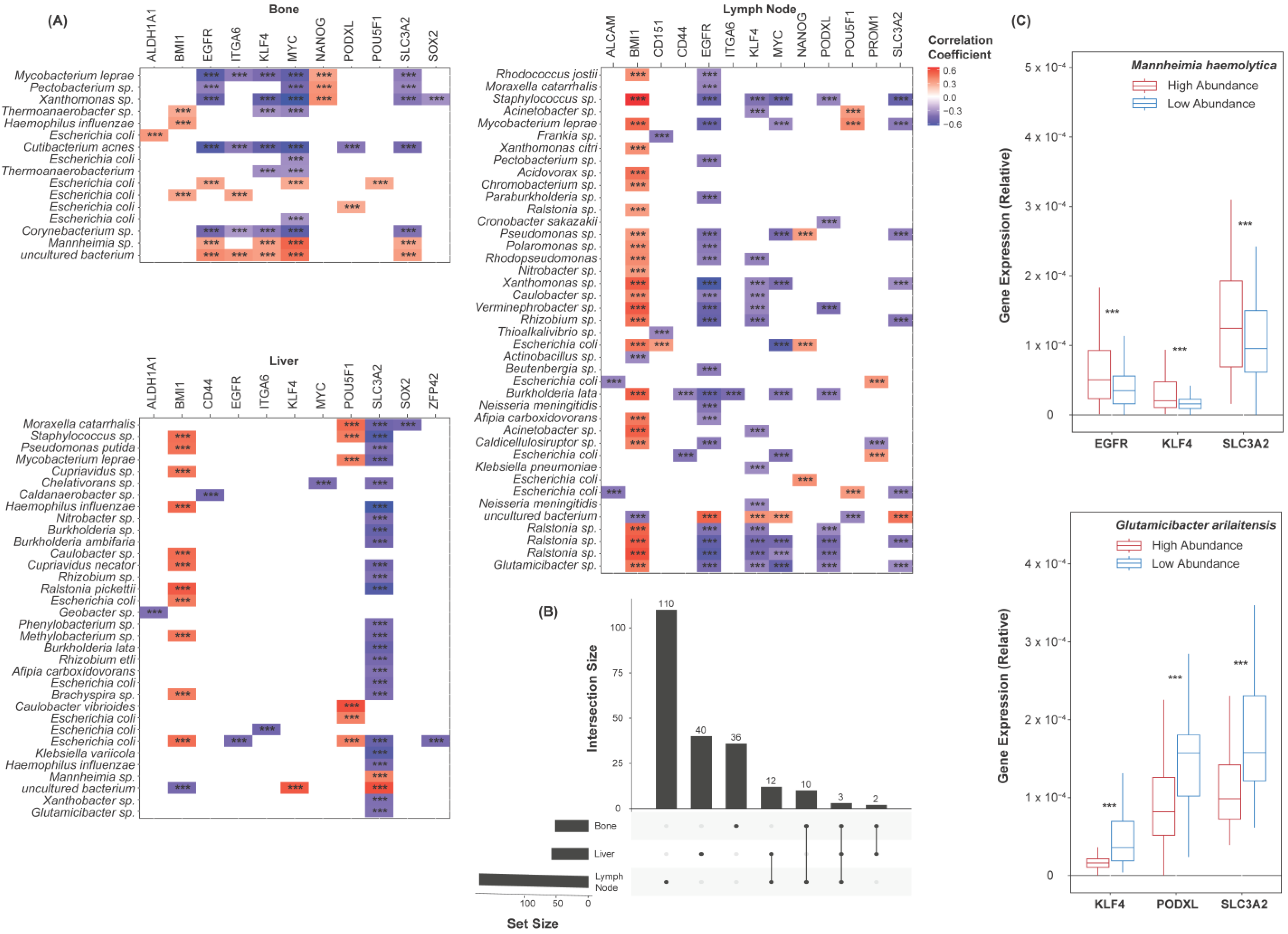
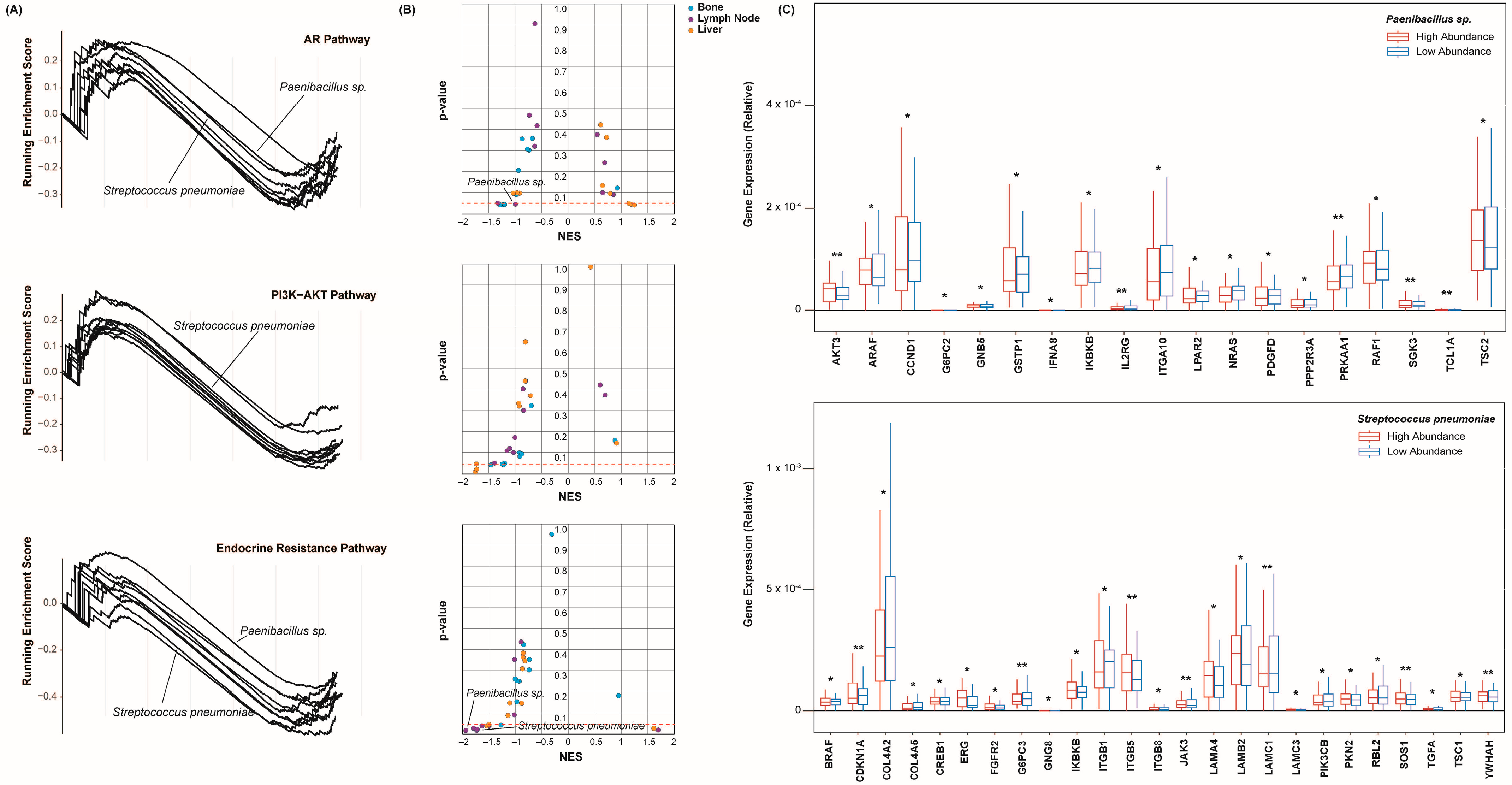
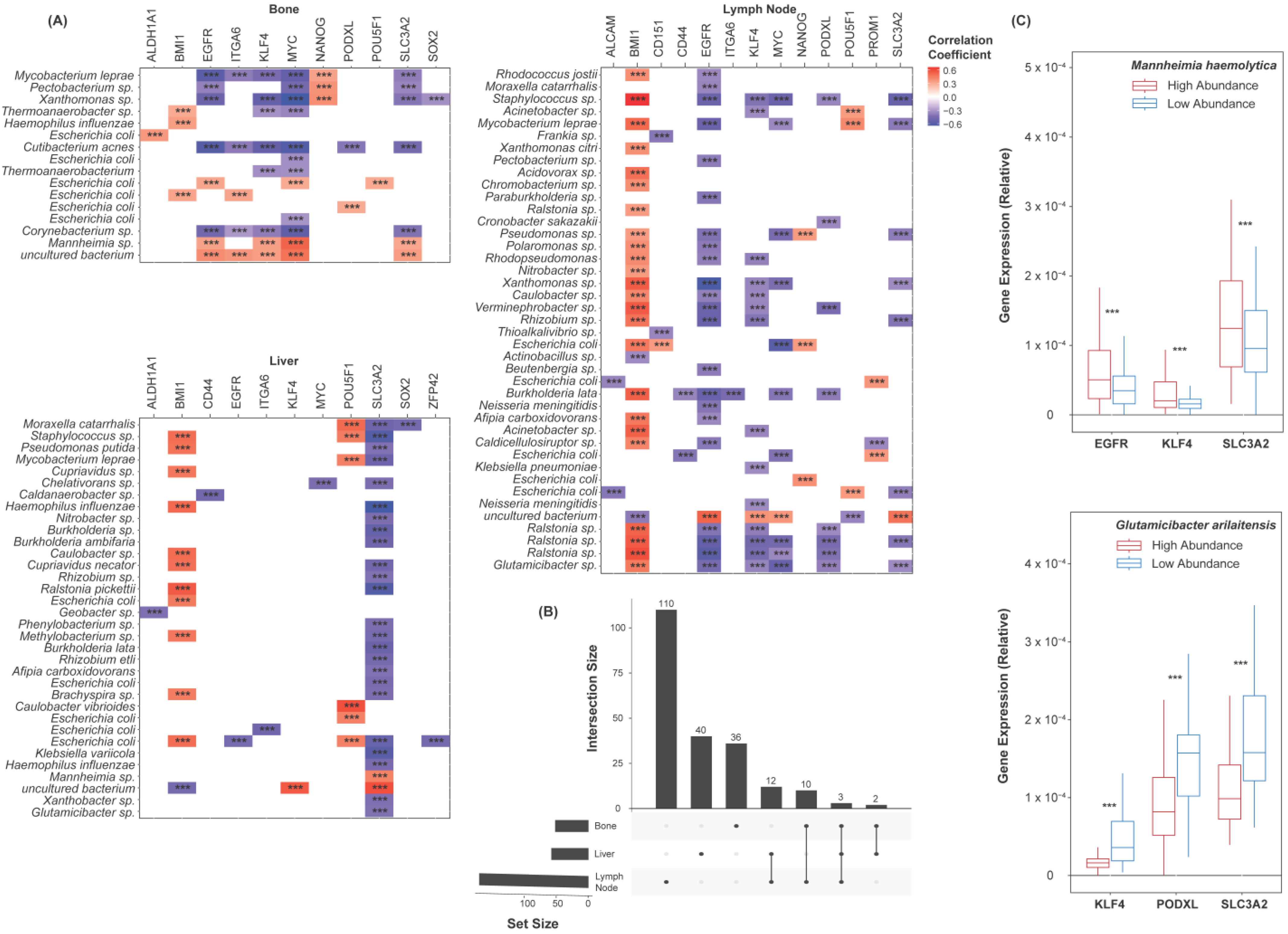
2.3. Species Abundance Correlates with Castration Resistance Marker Expression
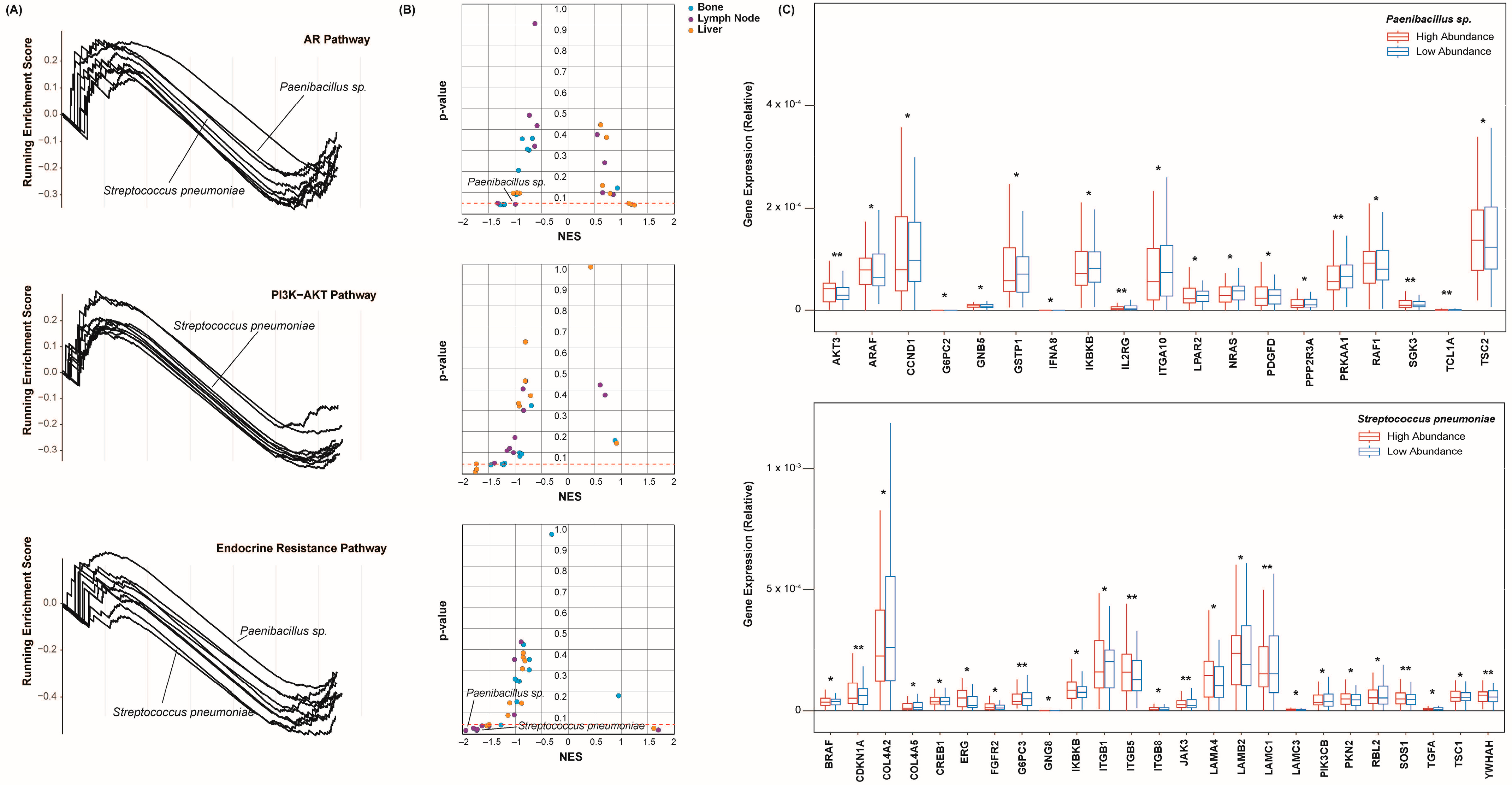
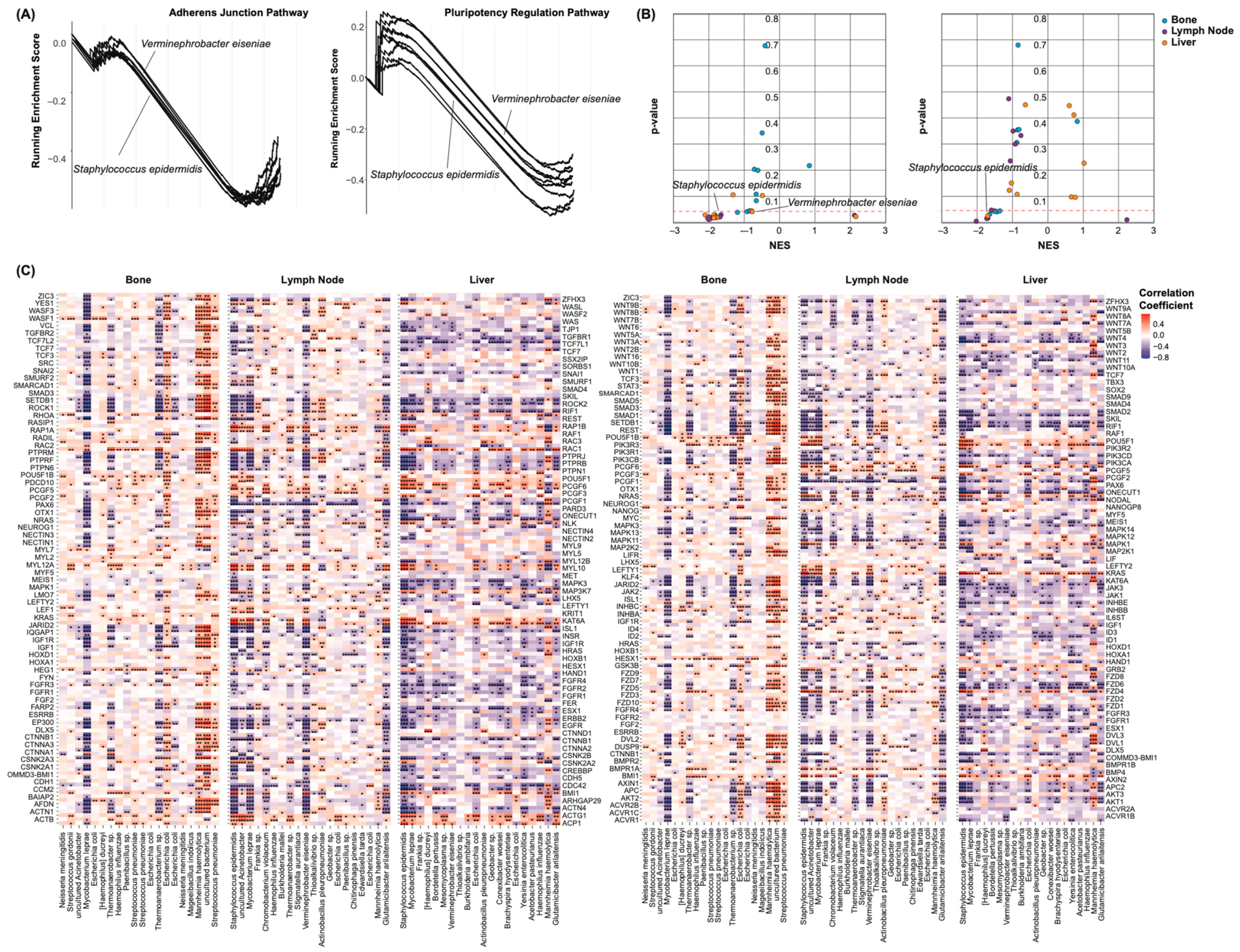
2.4. Castration Resistance Pathway Enrichment
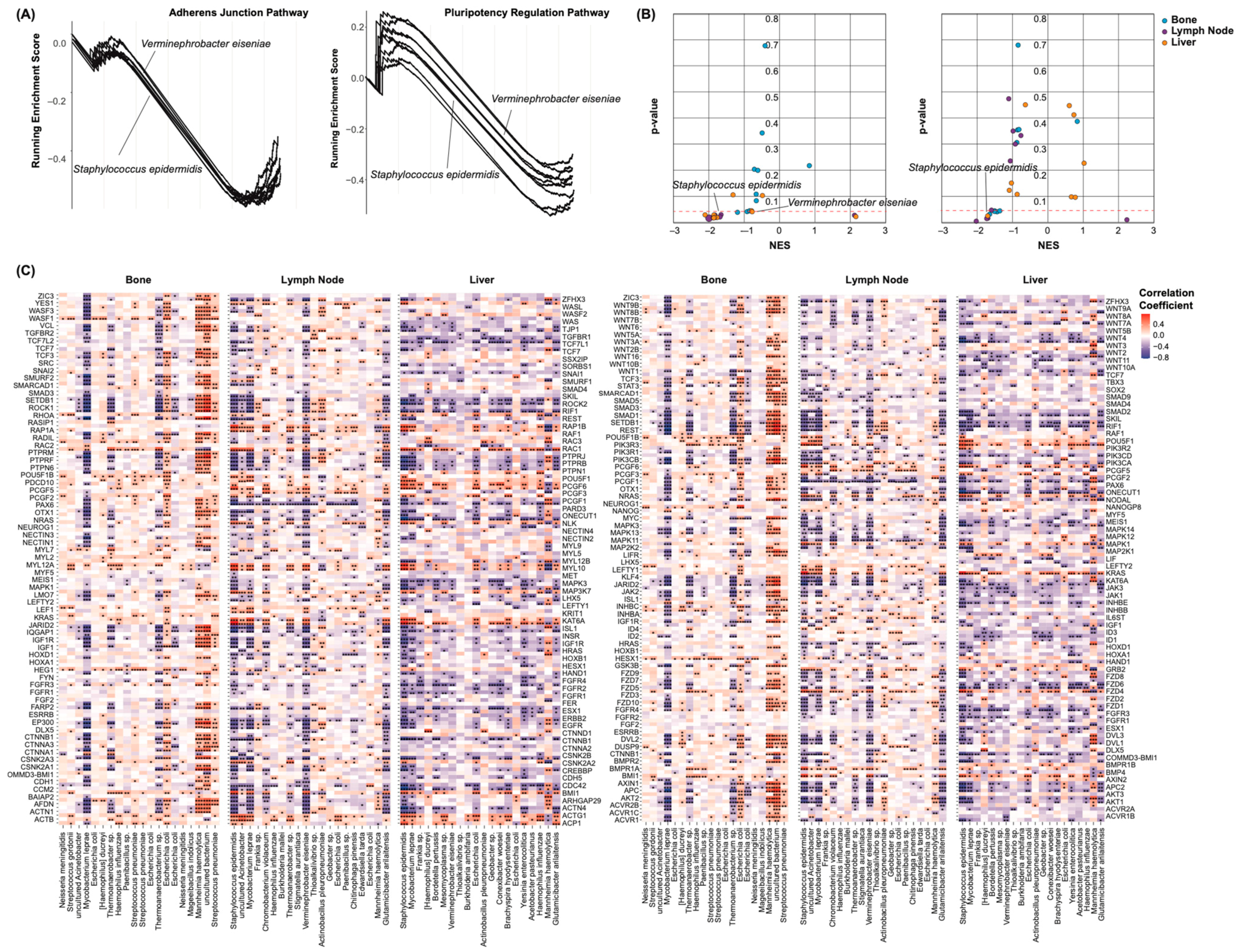
2.5. Species Abundance Correlates with Cancer Stem Cell Marker Expression
2.6. Epithelial–Mesenchymal Transition and Pluripotency Regulation Pathway Enrichment
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Bacterial Read Mapping
4.3. Gene Read Mapping
4.4. Cross-Study Normalization
4.5. Microbial Contamination Correction
4.6. Differential Abundance Analyses
4.7. Expression Correlation Analyses
4.8. Gene Set Enrichment Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Facts & Figures 2023; American Cancer Society: Atlanta, GA, USA, 2023. [Google Scholar]
- National Cancer Institute. SEER Cancer Stat Facts: Prostate Cancer; National Cancer Institute: Bethesda, MD, USA, 2023.
- Gandaglia, G.; Abdollah, F.; Schiffmann, J.; Trudeau, V.; Shariat, S.F.; Kim, S.P.; Perrotte, P.; Montorsi, F.; Briganti, A.; Trinh, Q.D.; et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate 2014, 74, 210–216. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens Health 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Prostate Cancer Treatment—Health Professional Version. Physician Data Query (PDQ); National Cancer Institute: Bethesda, MD, USA, 2019.
- Garje, R.; Chennamadhavuni, A.; Mott, S.L.; Chambers, I.M.; Gellhaus, P.; Zakharia, Y.; Brown, J.A. Utilization and Outcomes of Surgical Castration in Comparison to Medical Castration in Metastatic Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, e157–e166. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [PubMed]
- Alexandra Guerrero, P.; Rodríguez-Zapater, S.; Anel, A.; Mariano Esteban, L.; Camón-Fernandez, A.; Espilez-Ortiz, R.; Jesús Gil-Sanz, M.; Borque-Fernando, Á. Prostate Cancer and the Mevalonate Pathway. Int. J. Mol. Sci. 2024, 25, 2152. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy with Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Hoyle, A.P.; Ali, A.; James, N.D.; Cook, A.; Parker, C.C.; de Bono, J.S.; Attard, G.; Chowdhury, S.; Cross, W.R.; Dearnaley, D.P.; et al. Abiraterone in “High-” and “Low-risk” Metastatic Hormone-sensitive Prostate Cancer. Eur. Urol. 2019, 76, 719–728. [Google Scholar] [CrossRef]
- Gravis, G.; Boher, J.M.; Joly, F.; Soulié, M.; Albiges, L.; Priou, F.; Latorzeff, I.; Delva, R.; Krakowski, I.; Laguerre, B.; et al. Androgen Deprivation Therapy (ADT) Plus Docetaxel Versus ADT Alone in Metastatic Non castrate Prostate Cancer: Impact of Metastatic Burden and Long-term Survival Analysis of the Randomized Phase 3 GETUG-AFU15 Trial. Eur. Urol. 2016, 70, 256–262. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Chen, Y.H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Rajwa, P.; Thibault, C.; Gandaglia, G.; Mori, K.; Kawada, T.; Fukuokaya, W.; Shim, S.R.; Mostafaei, H.; Motlagh, R.S.; et al. Androgen Receptor Signaling Inhibitors in Addition to Docetaxel with Androgen Deprivation Therapy for Metastatic Hormone-sensitive Prostate Cancer: A Systematic Review and Meta-analysis. Eur. Urol. 2022, 82, 584–598. [Google Scholar] [CrossRef]
- Conteduca, V.; Mosca, A.; Brighi, N.; de Giorgi, U.; Rescigno, P. New Prognostic Biomarkers in Metastatic Castration-Resistant Prostate Cancer. Cells 2021, 10, 193. [Google Scholar] [CrossRef]
- Castellón, E.A.; Indo, S.; Contreras, H.R. Cancer Stemness/Epithelial-Mesenchymal Transition Axis Influences Metastasis and Castration Resistance in Prostate Cancer: Potential Therapeutic Target. Int. J. Mol. Sci. 2022, 23, 14917. [Google Scholar] [CrossRef]
- Babaei, G.; Aziz, S.G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Wang, J.; Jiang, L.; James Kang, Y. Cancer and stem cells. Exp. Biol. Med. 2021, 246, 1791–1801. [Google Scholar] [CrossRef]
- Moltzahn, F.R.; Volkmer, J.P.; Rottke, D.; Ackermann, R. “Cancer stem cells”—Lessons from Hercules to fight the Hydra. Urol. Oncol. 2008, 26, 581–589. [Google Scholar] [CrossRef]
- Fang, D.; Kitamura, H. Cancer stem cells and epithelial-mesenchymal transition in urothelial carcinoma: Possible pathways and potential therapeutic approaches. Int. J. Urol. 2018, 25, 7–17. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Barko, P.C.; McMichael, M.A.; Swanson, K.S.; Williams, D.A. The Gastrointestinal Microbiome: A Review. J. Vet. Intern. Med. 2018, 32, 9–25. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Manos, J. The human microbiome in disease and pathology. APMIS 2022, 130, 690–705. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Kayama, H.; Okumura, R.; Takeda, K. Interaction Between the Microbiota, Epithelia, and Immune Cells in the Intestine. Annu. Rev. Immunol. 2020, 38, 23–48. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science 2021, 371, eabc4552. [Google Scholar] [CrossRef]
- Cullin, N.; Azevedo Antunes, C.; Straussman, R.; Stein-Thoeringer, C.K.; Elinav, E. Microbiome and cancer. Cancer Cell 2021, 39, 1317–1341. [Google Scholar] [CrossRef]
- Rebersek, M. Gut microbiome and its role in colorectal cancer. BMC Cancer 2021, 21, 1325. [Google Scholar] [CrossRef]
- Javier-DesLoges, J.; McKay, R.R.; Swafford, A.D.; Sepich-Poore, G.D.; Knight, R.; Parsons, J.K. The microbiome and prostate cancer. Prostate Cancer Prostatic Dis. 2022, 25, 159–164. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, H. Compositional differences of gut microbiome in matched hormone-sensitive and castration-resistant prostate cancer. Transl. Androl. Urol. 2020, 9, 1937–1944. [Google Scholar] [CrossRef]
- Pernigoni, N.; Zagato, E.; Calcinotto, A.; Troiani, M.; Mestre, R.P.; Calì, B.; Attanasio, G.; Troisi, J.; Minini, M.; Mosole, S.; et al. Commensal bacteria promote endocrine resistance in prostate cancer through androgen biosynthesis. Science 2021, 374, 216–224. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef]
- Kure, A.; Tsukimi, T.; Ishii, C.; Aw, W.; Obana, N.; Nakato, G.; Hirayama, A.; Kawano, H.; China, T.; Shimizu, F.; et al. Gut environment changes due to androgen deprivation therapy in patients with prostate cancer. Prostate Cancer Prostatic Dis. 2023, 26, 323–330. [Google Scholar] [CrossRef]
- Wang, L. Changes in the gut microbial profile during long-term androgen deprivation therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2023. [CrossRef]
- Toren, P.; Zoubeidi, A. Targeting the PI3K/Akt pathway in prostate cancer: Challenges and opportunities (review). Int. J. Oncol. 2014, 45, 1793–1801. [Google Scholar] [CrossRef]
- Edlind, M.P.; Hsieh, A.C. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar]
- Zhao, W.; Li, Y.; Zhang, X. Stemness-Related Markers in Cancer. Cancer Transl. Med. 2017, 3, 87–95. [Google Scholar]
- Day, K.C.; Lorenzatti Hiles, G.; Kozminsky, M.; Dawsey, S.J.; Paul, A.; Broses, L.J.; Shah, R.; Kunja, L.P.; Hall, C.; Palanisamy, N.; et al. HER2 and EGFR Overexpression Support Metastatic Progression of Prostate Cancer to Bone. Cancer Res. 2017, 77, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Dubrovska, A. CD98 heavy chain as a prognostic biomarker and target for cancer treatment. Front. Oncol. 2023, 13, 1251100. [Google Scholar] [CrossRef]
- Mei, W.; Lin, X.; Kapoor, A.; Gu, Y.; Zhao, K.; Tang, D. The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis. Cancers 2019, 11, 434. [Google Scholar] [CrossRef]
- Marzano, M.; Fosso, B.; Piancone, E.; Defazio, G.; Pesole, G.; De Robertis, M. Stem Cell Impairment at the Host-Microbiota Interface in Colorectal Cancer. Cancers 2021, 13, 996. [Google Scholar] [CrossRef]
- Gupta, I.; Pedersen, S.; Vranic, S.; Al Moustafa, A.E. Implications of Gut Microbiota in Epithelial-Mesenchymal Transition and Cancer Progression: A Concise Review. Cancers 2022, 14, 2964. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef]
- Cavarretta, I.; Ferrarese, R.; Cazzaniga, W.; Saita, D.; Lucianò, R.; Ceresola, E.R.; Locatelli, I.; Visconti, L.; Lavorgna, G.; Briganti, A.; et al. The Microbiome of the Prostate Tumor Microenvironment. Eur. Urol. 2017, 72, 625–631. [Google Scholar] [CrossRef]
- Salachan, P.V.; Rasmussen, M.; Fredsøe, J.; Ulhøi, B.; Borre, M.; Sørensen, K.D. Microbiota of the prostate tumor environment investigated by whole-transcriptome profiling. Genome Med. 2022, 14, 9. [Google Scholar] [CrossRef]
- Mohseni, A.H.; Casolaro, V.; Bermúdez-Humarán, L.G.; Keyvani, H.; Taghinezhad, S.S. Modulation of the PI3K/Akt/mTOR signaling pathway by probiotics as a fruitful target for orchestrating the immune response. Gut Microbes 2021, 13, 1886844. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Labanca, E.; Vazquez, E.S.; Corn, P.G.; Roberts, J.M.; Wang, F.; Logothetis, C.J.; Navone, N.M. Fibroblast growth factors signaling in bone metastasis. Endocr. Relat. Cancer 2020, 27, R255–R265. [Google Scholar] [CrossRef]
- Labanca, E.; Yang, J.; Shepherd, P.D.A.; Wan, X.; Starbuck, M.W.; Guerra, L.D.; Anselmino, N.; Bizzotto, J.A.; Dong, J.; Chinnaiyan, A.M.; et al. Fibroblast Growth Factor Receptor 1 Drives the Metastatic Progression of Prostate Cancer. Eur. Urol. Oncol. 2022, 5, 164–175. [Google Scholar] [CrossRef]
- Hong, C.; Manimaran, S.; Shen, Y.; Perez-Rogers, J.F.; Byrd, A.L.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. PathoScope 2.0: A complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2014, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Maimaiti, M.; Sakamoto, S.; Sugiura, M.; Kanesaka, M.; Fujimoto, A.; Matsusaka, K.; Xu, M.; Ando, K.; Saito, S.; Wakai, K.; et al. The heavy chain of 4F2 antigen promote prostate cancer progression via SKP-2. Sci. Rep. 2021, 11, 11478. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uzelac, M.; Xin, R.; Ongkeko, W.M. Microbiome Dysbiosis Is Associated with Castration Resistance and Cancer Stemness in Metastatic Prostate Cancer. Int. J. Mol. Sci. 2024, 25, 3291. https://doi.org/10.3390/ijms25063291
Uzelac M, Xin R, Ongkeko WM. Microbiome Dysbiosis Is Associated with Castration Resistance and Cancer Stemness in Metastatic Prostate Cancer. International Journal of Molecular Sciences. 2024; 25(6):3291. https://doi.org/10.3390/ijms25063291
Chicago/Turabian StyleUzelac, Matthew, Ruomin Xin, and Weg M. Ongkeko. 2024. "Microbiome Dysbiosis Is Associated with Castration Resistance and Cancer Stemness in Metastatic Prostate Cancer" International Journal of Molecular Sciences 25, no. 6: 3291. https://doi.org/10.3390/ijms25063291
APA StyleUzelac, M., Xin, R., & Ongkeko, W. M. (2024). Microbiome Dysbiosis Is Associated with Castration Resistance and Cancer Stemness in Metastatic Prostate Cancer. International Journal of Molecular Sciences, 25(6), 3291. https://doi.org/10.3390/ijms25063291