

A Novel Mutation in the INSR Gene Causes Severe Insulin Resistance and Rabson–Mendenhall Syndrome in a Paraguayan Patient
 , ,
, , 
Abstract
1. Introduction
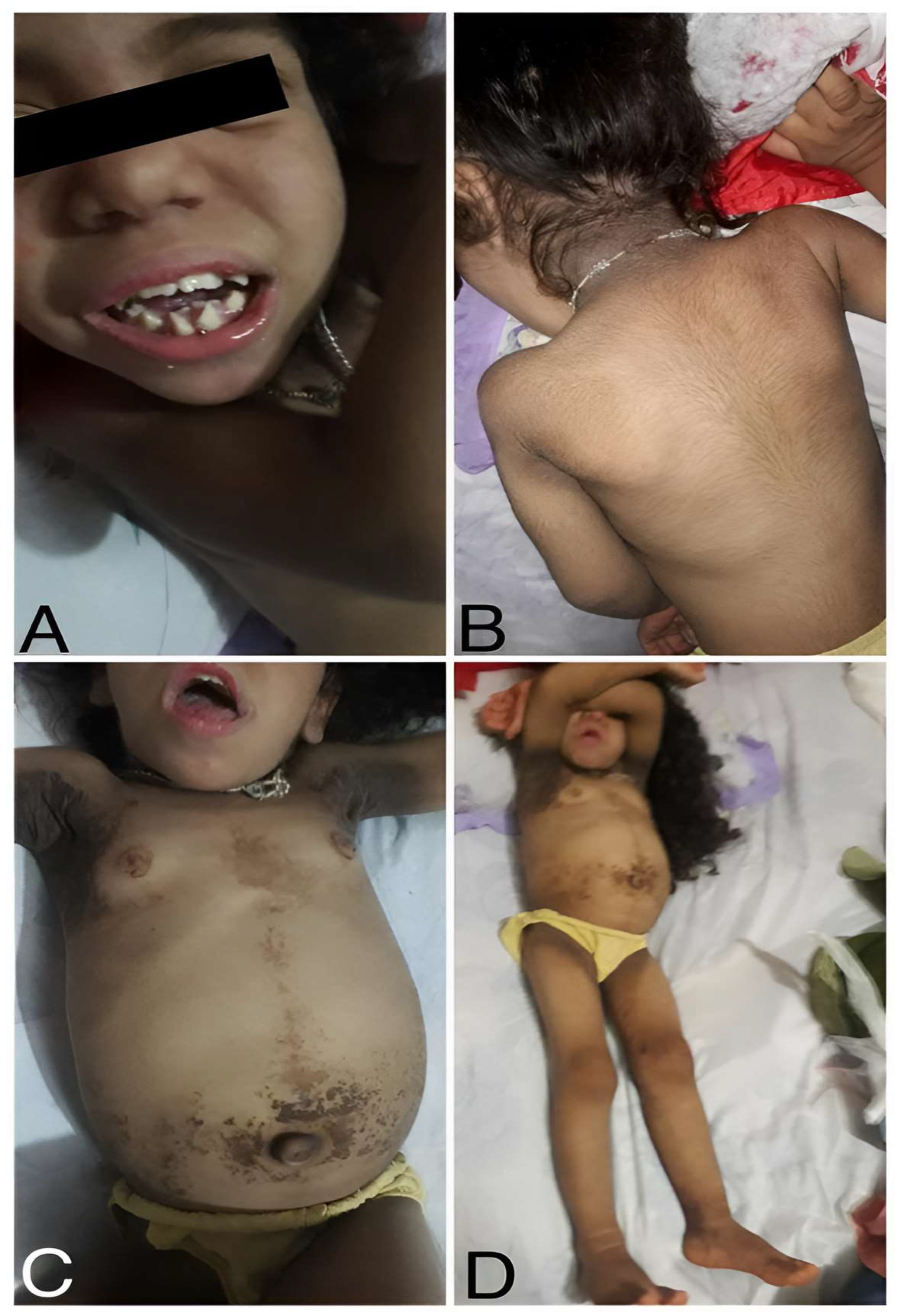
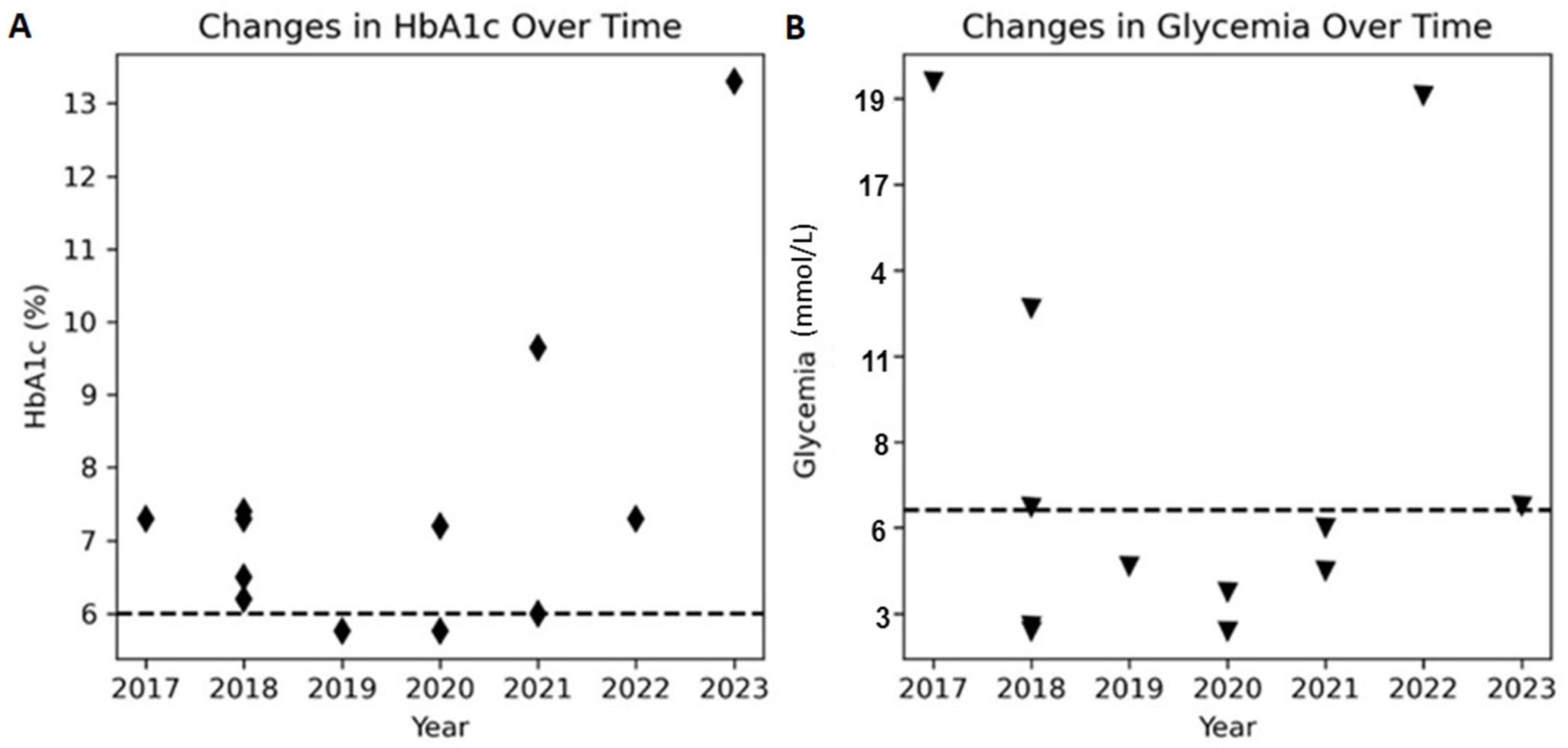
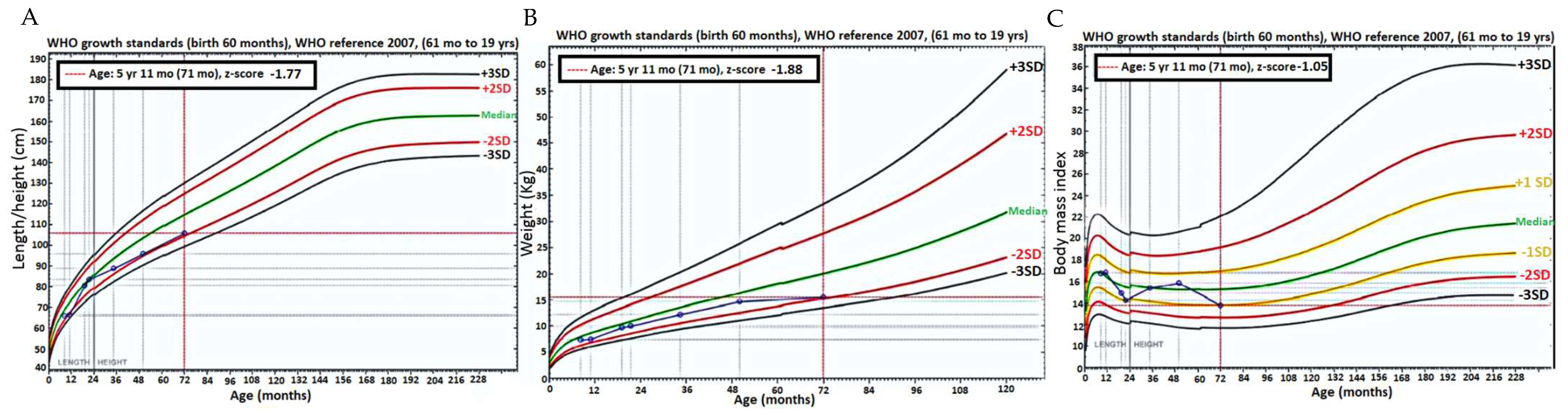
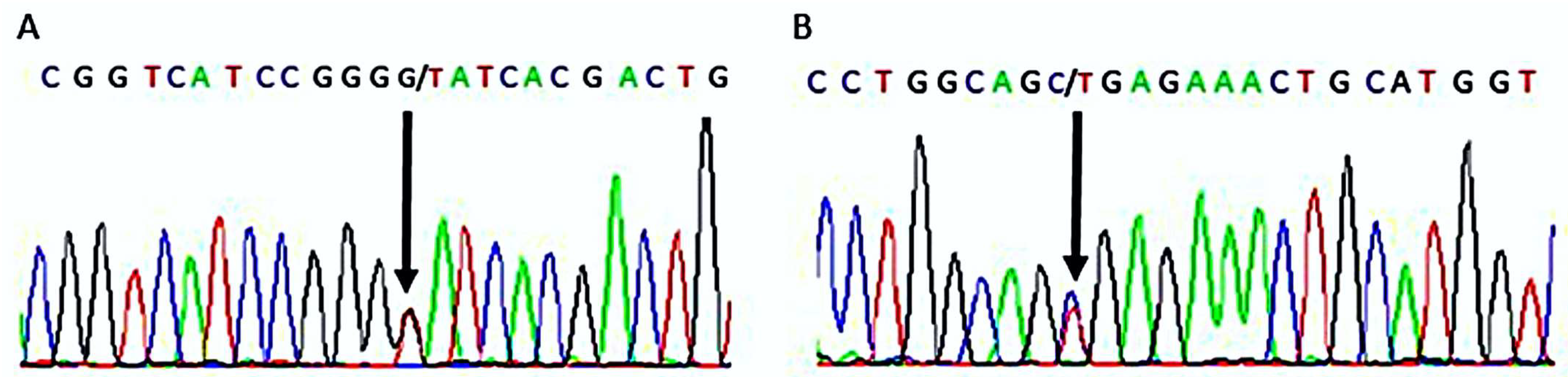
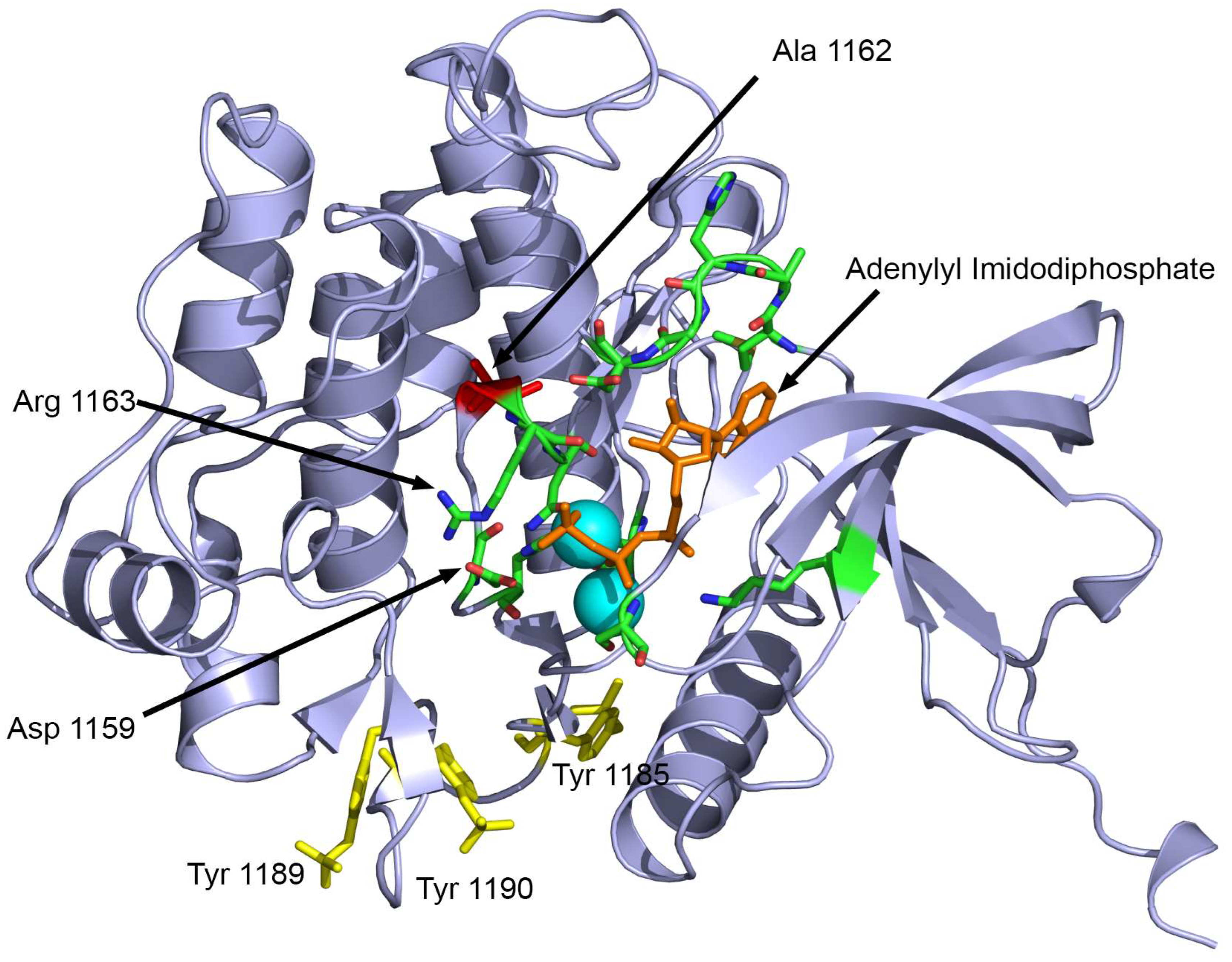
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Preparation and DNA Extraction
4.2. Whole-Exome Sequencing (WES) and Bioinformatic Analysis
4.3. Variant Validation
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hosoe, J.; Kadowaki, H.; Miya, F.; Aizu, K.; Kawamura, T.; Miyata, I.; Satomura, K.; Ito, T.; Hara, K.; Tanaka, M.; et al. Structural Basis and Genotype-Phenotype Correlations of INSR Mutations Causing Severe Insulin Resistance. Diabetes 2017, 66, 2713–2723. [Google Scholar] [CrossRef]
- Jiang, S.; Fang, Q.; Zhang, F.; Wan, H.; Zhang, R.; Wang, C.; Bao, Y.; Zhang, L.; Ma, X.; Lu, J.; et al. Functional characterization of insulin receptor gene mutations contributing to Rabson-Mendenhall syndrome—Phenotypic heterogeneity of insulin receptor gene mutations. Endocr. J. 2011, 58, 931–940. [Google Scholar] [CrossRef]
- Melvin, A.; O’Rahilly, S.; Savage, D.B. Genetic syndromes of severe insulin resistance. Curr. Opin. Genet. Dev. 2018, 50, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Laudes, M.; Barroso, I.; Luan, J.; Soos, M.A.; Yeo, G.; Meirhaeghe, A.; Logie, L.; Vidal-Puig, A.; Schafer, A.J.; Wareham, N.J.; et al. Genetic variants in human sterol regulatory element binding protein-1c in syndromes of severe insulin resistance and type 2 diabetes. Diabetes 2004, 53, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yu, F.; Ma, Z.; Lu, H.; Luo, J.; Sun, T.; Liu, Q.; Gan, S. INSR novel mutations identified in a Chinese family with severe INSR-related insulin resistance syndromes: A case report. Medicine 2022, 101, e32266. [Google Scholar] [CrossRef] [PubMed]
- Ben Abdelaziz, R.; Ben Chehida, A.; Azzouz, H.; Boudabbous, H.; Lascols, O.; Ben Turkia, H.; Tebib, N. A novel homozygous missense mutation in the insulin receptor gene results in an atypical presentation of Rabson-Mendenhall syndrome. Eur. J. Med. Genet. 2016, 59, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kadowaki, H.; Ando, A.; Quin, J.D.; MacCuish, A.C.; Yazaki, Y.; Akanuma, Y.; Kadowaki, T. Two aberrant splicings caused by mutations in the insulin receptor gene in cultured lymphocytes from a patient with Rabson-Mendenhall’s syndrome. J. Clin. Investig. 1998, 101, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Rabson, S.M.; Mendenhall, E.N. Familial hypertrophy of pineal body, hyperplasia of adrenal cortex and diabetes mellitus; report of 3 cases. Am. J. Clin. Pathol. 1956, 26, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Aftab, S.; Shaheen, T.; Asif, R.; Anjum, M.N.; Saeed, A.; Manzoor, J.; Cheema, H.A. Management challenges of Rabson Mendenhall syndrome in a resource limited country: A case report. J. Pediatr. Endocrinol. Metab. 2022, 35, 1429–1432. [Google Scholar] [CrossRef]
- Chrzanowska, J.; Skarul, J.; Zubkiewicz-Kucharska, A.; Borowiec, M.; Zmyslowska, A. Incomplete phenotypic presentation in a girl with rare Rabson-Mendenhall syndrome. Acta Diabetol. 2023, 60, 449–453. [Google Scholar] [CrossRef]
- Kumar, K.; Kohli, P.; Kumar, G. Rare Ocular Complication in a Patient with Rabson-Mendenhall Syndrome. Indian. J. Pediatr. 2021, 88, 192. [Google Scholar] [CrossRef]
- Kushi, R.; Hirota, Y.; Ogawa, W. Insulin resistance and exaggerated insulin sensitivity triggered by single-gene mutations in the insulin signaling pathway. Diabetol. Int. 2021, 12, 62–67. [Google Scholar] [CrossRef]
- Rojek, A.; Wikiera, B.; Noczynska, A.; Niedziela, M. Syndrome of Congenital Insulin Resistance Caused by a Novel INSR Gene Mutation. J. Clin. Res. Pediatr. Endocrinol. 2023, 15, 312–317. [Google Scholar] [CrossRef]
- Dagdeviren Cakir, A.; Saidov, S.; Turan, H.; Ceylaner, S.; Ozer, Y.; Kutlu, T.; Ercan, O.; Evliyaoglu, O. Two Novel Variants and One Previously Reported Variant in the Insulin Receptor Gene in Two Cases with Severe Insulin Resistance Syndrome. Mol. Syndromol. 2020, 11, 90–96. [Google Scholar] [CrossRef]
- Ogawa, W.; Araki, E.; Ishigaki, Y.; Hirota, Y.; Maegawa, H.; Yamauchi, T.; Yorifuji, T.; Katagiri, H. New classification and diagnostic criteria for insulin resistance syndrome. Endocr. J. 2022, 69, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.R.; Pendyala, G.S.; Shah, P.; Pustake, B.; Mopagar, V.; Padmawar, N. Severe insulin resistance syndrome—A rare case report and review of literature. Natl. J. Maxillofac. Surg. 2021, 12, 100–105. [Google Scholar] [CrossRef]
- Church, T.J.; Haines, S.T. Treatment Approach to Patients with Severe Insulin Resistance. Clin. Diabetes 2016, 34, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yu, J.; Yuan, X.; Wang, C.; Zhu, Z.; Zhang, A.; Gu, W. Clinical and Functional Characterization of Novel INSR Variants in Two Families With Severe Insulin Resistance Syndrome. Front. Endocrinol. 2021, 12, 606964. [Google Scholar] [CrossRef] [PubMed]
- Musso, C.; Cochran, E.; Moran, S.A.; Skarulis, M.C.; Oral, E.A.; Taylor, S.; Gorden, P. Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): A 30-year prospective. Medicine 2004, 83, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Okawa, M.C.; Cochran, E.; Lightbourne, M.; Brown, R.J. Long-Term Effects of Metreleptin in Rabson-Mendenhall Syndrome on Glycemia, Growth, and Kidney Function. J. Clin. Endocrinol. Metab. 2022, 107, e1032–e1046. [Google Scholar] [CrossRef] [PubMed]
- Berger, D.; Barroso, I.; Soos, M.; Yeo, G.; Schafer, A.J.; O’Rahilly, S.; Whitehead, J.P. Genetic variants of insulin receptor substrate-1 (IRS-1) in syndromes of severe insulin resistance. Functional analysis of Ala513Pro and Gly1158Glu IRS-1. Diabet. Med. 2002, 19, 804–809. [Google Scholar] [CrossRef]
- Semple, R.K.; Savage, D.B.; Cochran, E.K.; Gorden, P.; O’Rahilly, S. Genetic syndromes of severe insulin resistance. Endocr. Rev. 2011, 32, 498–514. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, S.S.; Ramaldes, L.A.; Gabbay, M.A.L.; Moises, R.C.S.; Dib, S.A. Use of a Sodium-Glucose Cotransporter 2 Inhibitor, Empagliflozin, in a Patient with Rabson-Mendenhall Syndrome. Horm. Res. Paediatr. 2021, 94, 313–316. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.-P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef] [PubMed]
- Schubach, M.; Maass, T.; Nazaretyan, L.; Röner, S.; Kircher, M. CADD v1.7: Using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res 2024, 52, D1143–D1154. [Google Scholar] [CrossRef]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Human Mutation 2009, 30, 1237–1244. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Moreira, R.O.; Zagury, R.L.; Nascimento, T.S.; Zagury, L. Multidrug therapy in a patient with Rabson-Mendenhall syndrome. Diabetologia 2010, 53, 2454–2455. [Google Scholar] [CrossRef]
- Cama, A.; de la Luz Sierra, M.; Quon, M.J.; Ottini, L.; Gorden, P.; Taylor, S.I. Substitution of glutamic acid for alanine 1135 in the putative “catalytic loop” of the tyrosine kinase domain of the human insulin receptor. A mutation that impairs proteolytic processing into subunits and inhibits receptor tyrosine kinase activity. J. Biol. Chem. 1993, 268, 8060–8069. [Google Scholar] [CrossRef] [PubMed]
- Aghababaie, A.S.; Ford-Adams, M.; Buchanan, C.R.; Arya, V.B.; Colclough, K.; Kapoor, R.R. A novel heterozygous mutation in the insulin receptor gene presenting with type A severe insulin resistance syndrome. J. Pediatr. Endocrinol. Metab. 2020, 33, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Gosavi, S.; Sangamesh, S.; Ananda Rao, A.; Patel, S.; Hodigere, V.C. Insulin, Insulin Everywhere: A Rare Case Report of Rabson-Mendenhall Syndrome. Cureus 2021, 13, e13126. [Google Scholar] [CrossRef] [PubMed]
- Moncada, V.Y.; Hedo, J.A.; Serrano-Rios, M.; Taylor, S.I. Insulin-Receptor Biosynthesis in Cultured Lymphocytes From an Insulin-Resistant Patient (Rabson-Mendenhall Syndrome): Evidence for Defect Before Insertion of Receptor Into Plasma Membrane. Diabetes 1986, 35, 802–807. [Google Scholar] [CrossRef]
- Kadowaki, T.; Kadowaki, H.; Accili, D.; Taylor, S.I. Substitution of lysine for asparagine at position 15 in the alpha-subunit of the human insulin receptor. A mutation that impairs transport of receptors to the cell surface and decreases the affinity of insulin binding. J. Biol. Chem. 1990, 265, 19143–19150. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.I.; Underhill, L.H.; Hedo, J.A.; Roth, J.; Rios, M.S.; Blizzard, R.M. Decreased Insulin Binding to Cultured Cells from a Patient with the Rabson-Mendenhall Syndrome: Dichotomy between Studies with Cultured Lymphocytes and Cultured Fibroblasts*. J. Clin. Endocrinol. Metab. 1983, 56, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Cochran, E.; Young, J.R.; Sebring, N.; DePaoli, A.; Oral, E.A.; Gorden, P. Efficacy of Recombinant Methionyl Human Leptin Therapy for the Extreme Insulin Resistance of the Rabson-Mendenhall Syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 1548–1554. [Google Scholar] [CrossRef]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Camats, N.; Fluck, C.E.; Audi, L. Oligogenic Origin of Differences of Sex Development in Humans. Int. J. Mol. Sci. 2020, 21, 1809. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Units | Values | Normal Range |
|---|---|---|---|
| Plasma glucose | mmol/dL | 17 | 3–6 |
| Insulin | pmol/L | 15,539 | 36–126 |
| HbA1c | % | 7.3 | 4.6-6 |
| LH | IU/L | 0.00 | <0.02–18.3 |
| FSH | mIU/L | 0.55 | 0–5.0 |
| Estradiol | pg/mL | <11 | 6–27 |
| Total Testosterone | nmol/L | 0.0007 | <0.3 |
| Androstenedione | nmol/L | 0.01 | 0.2–0.6 |
| 17-OHP | nmol/L | 0.1 | <3.03 |
| Cortisol in serum | nmol/L | 275.9 | (AM) 124–662 |
| Urea | mmol/L | 9.3 | 1.7–8.3 |
| Creatinine | μmol/L | 16.8 | 20–66 |
| Sodium | mmol/L | 138 | 133–145 |
| Potassium | mmol/L | 4.5 | 3.1–5.4 |
| Calcium | mmol/L | 1.31 | 1.1–1.3 |
| Tool | p.Gly111Val | p.Ala1162Val | Interpretation |
|---|---|---|---|
| SIFT | Disease (score 0) | Disease (score 0.001) | Benign (score > 0.05) Disease (score ≤ 0.05) |
| PROVEAN | Deleterious (score −7.6) | Deleterious (score −3.5) | Neutral (score > −2.5) Deleterious (score ≤ −2.5) |
| PolyPhen-2 | Probably damaging (score 1) | Probably damaging (score 0.99) | Probably damaging (score ≥ 0.8) Benign (score < 0.8) |
| PANTHER | Probably damaging (deleterious 0.85) | Probably damaging (deleterious 0.85) | Probably damaging (deleterious ≥ 0.5) Benign (deleterious < 0.5) |
| MutationTaster2021 | D | D | D (Disease) N (Neutral) |
| CADD | Deleterious (score 27.3) | Deleterious (score 29) | Deleterious (score ≥ 20) Benign (score < 20) |
| SNPs&GO | Disease (score 0.7) | Disease (score 0.65) | Disease (score > 0.05) Benign (score ≤ 0.05) |
| MutPred2 | Disease (score 0.95) | Disease (score 0.90) | Pathogenic (score ≥ 0.5) Benign (score < 0.5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas Velazquez, M.N.; Blanco, F.; Ayala-Lugo, A.; Franco, L.; Jolly, V.; Di Tore, D.; Martínez de Lapiscina, I.; Janner, M.; Flück, C.E.; Pandey, A.V. A Novel Mutation in the INSR Gene Causes Severe Insulin Resistance and Rabson–Mendenhall Syndrome in a Paraguayan Patient. Int. J. Mol. Sci. 2024, 25, 3143. https://doi.org/10.3390/ijms25063143
Rojas Velazquez MN, Blanco F, Ayala-Lugo A, Franco L, Jolly V, Di Tore D, Martínez de Lapiscina I, Janner M, Flück CE, Pandey AV. A Novel Mutation in the INSR Gene Causes Severe Insulin Resistance and Rabson–Mendenhall Syndrome in a Paraguayan Patient. International Journal of Molecular Sciences. 2024; 25(6):3143. https://doi.org/10.3390/ijms25063143
Chicago/Turabian StyleRojas Velazquez, Maria Natalia, Fabiola Blanco, Ana Ayala-Lugo, Lady Franco, Valerie Jolly, Denisse Di Tore, Idoia Martínez de Lapiscina, Marco Janner, Christa E. Flück, and Amit V. Pandey. 2024. "A Novel Mutation in the INSR Gene Causes Severe Insulin Resistance and Rabson–Mendenhall Syndrome in a Paraguayan Patient" International Journal of Molecular Sciences 25, no. 6: 3143. https://doi.org/10.3390/ijms25063143
APA StyleRojas Velazquez, M. N., Blanco, F., Ayala-Lugo, A., Franco, L., Jolly, V., Di Tore, D., Martínez de Lapiscina, I., Janner, M., Flück, C. E., & Pandey, A. V. (2024). A Novel Mutation in the INSR Gene Causes Severe Insulin Resistance and Rabson–Mendenhall Syndrome in a Paraguayan Patient. International Journal of Molecular Sciences, 25(6), 3143. https://doi.org/10.3390/ijms25063143