

Super-Enhancers and Their Parts: From Prediction Efforts to Pathognomonic Status
 , , and
, , and
Abstract
1. Introduction
2. Super-Enhancers: Historical Background, Characteristics, and Search Methods
2.1. Enhancer Clusters—SE Concept Development History
- Regulate cell-identity genes, including master transcription-factor (TF) genes (it is worth noting that SE clusters were found to not be associated with housekeeping genes);
- Have an order of magnitude higher concentration of TF, MC, and chromatin modification marks (H3K27ac and H3K4me1) compared to TEs;
- Have an order of magnitude greater length compared to TEs;
- Are sensitive to changes in the concentrations of MC and master TF;
- An enhancer cluster contains only those enhancers that are located no further than 12.5 kb from each other (a definition assumption made after mESCs analysis) [7].
2.2. SE–Promoter Interaction and SE-Mediated Gene-Activation Models
2.3. Hierarchy of SE Elements
- All SE constituents remain active (hypomethylated), and the expression of the target gene remains constant. These genes remain active in both naive and primed cells;
- All constituent enhancers acquire CpG methylation, resulting in the decommissioning of the SE and the cessation of gene expression associated with the SE. This scenario is typical for SEs that control genes related to maintaining a naive state;
- Some SE constituents acquire CpG methylation and become inactive, leading to a rearrangement of the SE and a reduction in the expression of the target gene. This allows for fine-tuning of gene expression;
- All or part of the SE constituents undergo CpG demethylation and become active, enhancing the expression of the target gene. These genes are typically associated with the primed state.
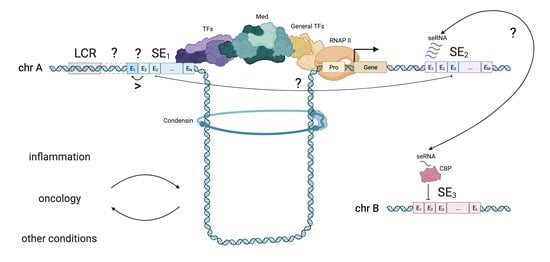
2.4. SE Elements Interaction and seRNA
2.5. SE Search Methods
2.6. SE Databases
2.7. SE Classification Attempts and Research Context
2.7.1. SE Classification
- Unique SEs—these SEs conform to the conventional definition of SEs and exhibit cell-type-specific gene regulatory functions;
- Nonunique SEs—this class of SEs is present in tissues that share a common function. For instance, the TJP3 (tight junction protein 3) gene, responsible for maintaining junctional integrity in intestinal cells of the digestive system organ tissues (such as the stomach, sigmoid colon, and small intestine), falls under this category;
- “Common” SEs—these SEs exhibit a strong association with genes that are universally highly expressed and are remarkably conserved throughout the human genome. Integrative analysis of 3D chromatin loops revealed that cell-type common SEs play a crucial role in the formation and rapid restoration of chromatin loops. Notably, loops enriched with common SEs displayed a 12-fold faster recovery rate compared to loops enriched with unique SEs [80].
2.7.2. LCR and SE Intersection
2.7.3. Evolutionary Conservation of SEs
3. Super-Enhancers and Diseases
3.1. Oncology
- chromosomal rearrangements leading to a convergence of SE with a specific oncogene;
- (super-)enhancer hijacking;
- SE focal amplifications;
- insertions, deletions, and other types of mutations in DNA that form binding sites for new TFs, leading to the formation of SE;
- insertions, deletions, and other types of mutations in the SE that disrupt TFs binding sites, leading to disruption of the SE function;
- deactivation of SE associated with some tumour-suppressor genes;
- disruption of TAD and sub-TAD boundaries;
- TFs increase in expression and overexpression, leading to pathological gene activation;
- viral oncogenes (Epstein–Barr and HPV viruses);
- some types of chemotherapy (e.g., cisplatin);
- other pathognomonic events.
3.1.1. Chromosomal Rearrangements and SE Dynamics
3.1.2. SNPs, Somatic Mutations, and SE Dynamics
3.1.3. Viral Infection-Mediated Changes and SE Dynamics
3.1.4. Global SE Alterations
3.1.5. Critical Proteins of SE Regulation Complex Overexpression
3.1.6. TFs Induce SE Activation and Regulate SE-Mediated Programs
3.1.7. CRCs and Autoregulatory Loops in TFs-Modulated SE Landscape
3.1.8. Sample Mechanism of TF-Mediated Pro-SE Looping: The Role of YY1
3.1.9. Copy Number Variation and Overexpression of TFs Leading to SEs Alteration
3.1.10. Spliced Isoforms of TFs Regulating SEs
3.1.11. Post-Translational Modifications Changing TFs Affinity
3.1.12. seRNA (eRNA)
3.2. Inflammation
3.3. Other Nosologies
4. Anti-SE Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Target | IC50 | Tumour Type | Reference(s) | |
|---|---|---|---|---|---|
| BET inhibitors | JQ1 | BD1 and BD2 of Brd2/Brd3/Brd4/Brdt | 77 nM/33 nM for BRD4 (1/2) | Various types | [16,84,183,222,223,242,243,244,245] |
| iBET151 | BD1 and BD2 of Brd2/Brd3/Brd4/Brdt | 0.79 μM for BRD4 | Diffuse large B-cell lymphoma, colorectal cancer, ovarian cancer | [16,224,225,226] | |
| OTX-015 | BD1 and BD2 of Brd2/Brd3/Brd4/Brdt | 0.449 µM | Diffuse large B-cell lymphoma, neuroblastoma | [16,228] | |
| BAY 1238097 | Strong binding of BD1, weaker binding of BD2 | <100 nM | Melanoma | [228] | |
| Volasertib | Dual kinase-bromodomain inhibitor | 300 nM/770 nM BD1/BD2 | Ovarian cancer | [226] | |
| BI 894999 | BD1 and BD2 | 5 nM/41 nM BD1/BD2 | Acute myeloid leukaemia | [246] | |
| ABBV-744 | Selective inhibitor of the BD2 domain | 4 nM | Prostate cancer | [229] | |
| CDK7 inhibitors | THZ1 | Cysteine residue located outside of the canonical kinase domain | <200 nM | Small-cell lung cancer, breast cancer, OSCC, ATL, NPC, ovarian cancer, GBM, osteosarcoma, cutaneous melanoma, hepatocellular carcinoma, chordoma, CML | [143,155,237,238,247,248,249,250,251,252,253,254,255] |
| THZ2 | Binding to CDK7 | 13.9 nM | TNBC, osteosarcoma | [237] | |
| SY-1365 | Covalent binding to CDK7 | 369 nM | Ovarian and breast cancer | [240] | |
| YKL-5-124 | Covalent binding to CDK7 | 8–60 nM | Neuroblastoma | [241] |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
| Cancer Type | Cancer Subtype(s) | Cell Line(s) with Shown Abnormal SEs | % of Cases with Discovered SE Alteration | Significant Gene(s) Associated with Abnormal SEs of Interest | Gene(s) Description (Why Authors Focus on This Gene/Gene Set) | TF(s) Associated with SE(s), Including Cell-Specific TF(s) | Estimated Molecular Pathogenesis of SEs | Affected Biological Pathways/Processes | Inhibitor(s) | Reference(s) |
|---|---|---|---|---|---|---|---|---|---|---|
| Skin tumours | ||||||||||
| Blastic plasmacytoid dendritic cell neoplasm (BPDCN) | BPCDN with t(6;8)(p21;q24) | In patients and the cell line, CAL-1 | 18.6% | RUNX2, MYC | MYC, widely known oncogene | RUNX2 | t(6;8)(p21;q24) translocation leads to the association of super-enhancer of RUNX2 and the MYC promoter | Cell proliferation, Ras signalling pathway, evasion of immunosurveillance | JQ1 | [122] |
| Cutaneous squamous cell carcinoma (cSCC) | — | Primary nonmetastatic (UT-SCC12A, UT-SCC91, UT-SCC105, UT-SCC111, UT-SCC118) and metastatic (UT-SCC7, UT-SCC59A, UT-SCC115) cSCC samples | Ranging from 42% to 75% of cases depending on the cell line (compared to 0% in normal cells) | BRD3OS (lncRNA-LINC00094), MMP1, MMP10, MMP13 | BRD3OS is transcribed from the opposite strand of BRD3 and is driven by a SE. The analysis of BRD3OS expression with RNA-ISH revealed a specific, mainly cytoplasmic signal for this lncRNA in cSCC cells. Significantly stronger BRD3OS expression is detected in metastatic cSCCs and is downregulated by the MEK1/ERK1/2 pathway | — | cSCC-associated LINC00094 SE-lncRNA | Cell growth, migration, invasion, metastasis, MEK1/ERK1/2 pathway | THZ1, JQ1 | [307] |
| Haematolymphoid tumours | ||||||||||
| Acute myeloid leukemia (AML) | Mixed-lineage leukaemia (MLL)-rearranged | NOMO-1, MV4-11, MOLM-13 cell lines, AML line carrying the AML1-ETO fusion protein (KASUMI-1) | — | MYC | — | Hematopoietic TFs (Erg, Lmo2, Pu.1, Cebpa and Cebpb) | Haematopoietic TFs increase in expression and overexpression, leading to pathological gene activation (parallelly increased ATPase activity of Brg1 subunit of SWI/SE complex, maintaining TF occupancy at SEs. SE hyperactivation | Cell proliferation | Potential: inhibitors of specific SWI/SNF subunits that have cancer maintenance function | [107] |
| AML | — | Primary peripheral blood samples, THP-1 cells | — | CAPG | CAPG is a cytoskeleton protein and contributes to AML progression through NF-κB pathway | — | — | NF-κB pathway | — | [308] |
| AML | AML with inv(3)/t(3;3) or 3q-AML | MUTZ-3, HNT-34, MOLM-1 cells | — | EVI1 | — | CEBPA, RUNX1 | Chromosomal rearrangements hijack GATA2 distal hematopoietic enhancer (G2DHE) and establish oncogenic SE, that requires PARP1 constitutive presence and involvement. PARP1 is shown to interact with RUNX1. Probably, PARP1 mediates chromatin remodelling/modulation | Cell proliferation | Talazoparib (not only inhibits catalytic activity but traps PARP1 on chromatin) | [309] |
| AML | AML with inv(3)/t(3;3) | MOLM-1, MUTZ-3 cells | — | EVI1 and more rarely PRDM homologs of the EVI1 gene (BLIMP1/PRDM1 or MEL1/PRDM16), GATA2 | AML with inv(3)/t(3;3) rearrangements is known to be related to EVI1 overexpression. The work aimed to test whether increased EVI1 transcription is caused by translocation of the enhancer | — | Chromosomal rearrangement (at 3q26.2 of enhancer element) leading to formation of SE regulating the EVI1 gene | Cell growth, proliferation | JQ1 | [117] |
| AML | — | NB4 cells | — | LYL1 (lymphoblastic leukemia 1) | LYL1 is a known TF and oncogene | — | — | Cell proliferation, p53 pathway, evasion of immunosurveillance | JQ1, GNE-987 degrader (proteolysis targeting chimaera) | [310] |
| AML | AML with t(3;8)(q26;q24) | Human myeloid cell line with t(3;8)(q26;q24), primary t(3;8) AML samples | — | EVI1 | — | GATA2, FLI1, ERG, RUNX1, LMO2, LYL1 | Chromosomal rearrangement t(3;8)(q26;q24) leads to translocation of MYC-SE (ARID1B, CDK6, THADA, or GATA2) to EVI1 locus. MYC-SE interacts with EVI1 promoter and upregulates EVI1 expression. Interaction between EVI1 and MYC-SE is possible due to CTCF binding site flanking EVI1 promoter | Cell proliferation | EVI1i to be developed | [108] |
| AML | AML with atypical 3q26-rearranged sequence (not harboring inv(3)/t(3;3)) | — | — | MECOM (EVI1) | — | RUNX1, LYL1, SCL, FLI1, ERG, LMO2, and GATA2 (key myeloid TFs) | Chromosomal rearrangements involving translocation of active SE regulating myeloid genes (THADA, CDK6, MYC, ARID1B, CD164, PROM1 (CD133), or FSCN1/EIF2AK1) to MECOM, which activates EVI1 expression, but not MS1-EVI1 (transcribed from the same gene but having a different promoter). In ~50% of cases of atypical AML underexpression of master TF GATA2 is witnessed, which might play a role in those rearrangements | — | BETi proposed to use but not tested | [116] |
| AML | — | MOLM-14 cell line | — | FOSL2, CDKN1B | — | IRF1, IRF8, CEBPA, and ETV6 (tumour-suppressor TF) | Suppression of SE activity by Mediator kinases (CDK8 (part of CDK8 module of Mediator complex) and its paralog CDK19) | AML cell proliferation | Cortistatin A (CA) (CDK8/CDK19i), I-BET151 (BETi) | [311] |
| AML | MLL-rearranged (MLL-r) and some non-MLL-r AML | OCI-AML3 AML cells, MLL-r leukemia lines (MOLM-13, MV4-11, OCI-AML2, and THP-1) | ~10% of adult AML cases, most childhood AML | MEF2D | — | HOXA9 | MLL rearrangements lead to SE formation and TF MEF2D overexpression | Cell renewal, CEBRE-centred-myeloid differentiation blockage | MEF2D inhibition, DOT1L inhibition (downregulation of HOXA9?) | [118] |
| AML | AML with t(12;22)(p13;q12) without MN1 fusion | — | t(12;22)(p13;q12) chromosomal rearrangement occurs in ~0.3% of myeloid neoplasms | MN1 | — | — | Chromosomal rearrangements involving translocation of active SE regulating ETV6 to MN1 (hypothesis) | — | Mediator kinase inhibitors proposed to be tested | [312] |
| AML | Paediatric AML | 11 primary AML samples, 7 primary T-ALL samples, 8 primary B-ALL samples, NB4, MV4–11, Kasumi-1, HL-60, THP-1 cells | — | LYL1, MPO, SPI1, ZFP36L2, HEMGN, ANKRD13D, RREB1, NACC1, ZEB2, SCYL1, ASNA1, TNRC18, GSE1, TRMT1, SLC39A13, FRMD8, PTMA, SPEN, PAF1 | — | SPI1, ZFP36L2, BRD4 | SEs associated with LYL1 are common to all three haematological diseases (AML, T-ALL, B-ALL). LYL1 and BRD4 function cooperatively with CEBPE and RUNX1 in AML; however, the expression of LYL1 might be regulated in both BRD4-dependent and BRD4–independent manners | Cell growth, survival, differentiation | GNE-987 (BRD4i) | [313] |
| AML | Acute erythroid leukemia | HEL, CMK11-5, UT7-EPO, K562 cells | — | GFI1 | GFI1 is a key regulator for neutrophil development | GFI1/GFI1B, CEBPA, TAL1, RUNX1, GATA1 | Sustainable repression of GFI1-SE by LSD1 leads to the absence of erythroid to myeloid switch. The SE is controlled by the coordination of TFs | Acute myeloid leukaemia, myeloid cell differentiation | LSD1 inhibitors: NCD38, NCD25 | [314] |
| Adult T-cell leukaemia (ATL) | Acute, lymphoma, chronic, smoldering, unknown ATL subtypes | 10 primary ATL samples, TL-Om1 cells | ~73% | TP73, TPRG1L, WRAP73, CCDC27, PIDD1, PERP, PHLDA3, FOSL2 | TP73 is highly activated under the control of a super-enhancer in ATL cells but not in normal T-cells or other haematological malignancies examined. Two distinct TP73 isoforms (TAp73 and Np73) are confirmed to be significantly higher in ATL cell lines than in T-ALL cell lines. Deletion of exons 2–3 accumulates at TP73 SE. The expression level of full-length TAp73 is associated with increased DNA damage and repair responses. TP73 positively regulates BATF3, JUNB, and FOSL2, which are critical transcription factors involved in ATL pathogenesis. An intragenic SE formation mechanism as a part of ATL clonal evolution is proposed | IRF4, NF-κB, BATF3, JUNB, FOSL2 | Intragenic SE formation of the TP73 and genetic deletion in this locus. Disruption of TAD and sub-TAD boundaries (chromatin looping). TFs increase in expression and overexpression, leading to pathological gene activation | Cell survival, proliferation, invasion, DNA damage response pathways | THZ1 | [156] |
| ATL | Acute and unfavourable chronic ATL subtypes | ST-1, KOB, KK-1 cells, 5 primary acute ATL samples and 1 primary sample with unfavourable chronic type of ATL (ATL-04) | 100% | IRF4, NOTCH1, DUSP22, AKAP1, KLHL8 | HBZ is encoded by the minus-strand of the HTLV-1 oncogenic provirus and induces BATF3. IRF4 and BATF3 activate TCR signaling and c-MYC. Studies suggest that IRF4 plays an important role in ATL survival and progression, including the activation of T-cell receptor (TCR) signalling. Mutations in IRF4 transcriptionally activate its downstream targets and are detected in ~10% of all ATL patients | IRF4 | SE hijacking. DNA mutations that form binding sites for new TFs, leading to the formation of SE. TFs increase in expression and overexpression, leading to pathological gene activation | Cell growth, survival (viability), proliferation, evasion of immunosurveillance | Alvocidib | [159] |
| T-cell acute lymphoblastic leukemia (T-ALL) | T-ALL with monoallelic overexpression of TAL1 lacking TAL1d or chromosomal translocations | Jurkat, MOLT-3 cells | 5.5% (146 paediatric tumours) | TAL1 | Oncogene, one of the core components | MYB, TAL1 complex (RUNX1, GATA3, TAL1, HEB, E2E) | Insertion introduces a new binding site for MYB TF, leading to the formation of SE. MYB recruits CBP and RUNX1, GATA-3, and TAL1 itself | Establishment of the T-ALL cell state | KRAB-dCas9 mediated repression of the E3 enhancer — | [128,315] |
| T-ALL | 5′TAL1 SE, TAL1-TCRB, TAL1-TCRD, SIL-TAL1 subtypes | 443 unselected primary samples from the GRAALL-2003, -2005 and the FRALLE 2000 trials (199 adult; 244 pediatric), T-ALL cell lines ATCC®, MOLT-4 cells | 5% of T-ALL have 5′TAL1 SE mutations | TAL1, TLX1, TLX3, CALM-AF10, MYB, LMO2 | TAL1 is a major transcription factor that is dysregulated in more than 50% of T-ALL. The SIL-TAL1 fusion transcript results from 90 kb of interstitial microdeletions fusing the 5′-end of the gene to the 3′-end of its neighbouring gene, STIL. Together with TAL1-TCRB and TAL1-TCRD mutations, SE formation is driven by creating de novo MYB transcription factor binding sites. MYB, in turn, recruits transcription co-activators and leads to TAL1 overexpression | TAL1, MYB | Intergenic mutations in DNA (upstream of the TAL1 promoter) that form binding sites for new TFs (MYB), leading to the formation of SE. Insertion introduces a new binding site for MYB TF, leading to SE formation | Cell survival, reprogramming, genomic instability, establishment of the T-ALL cell state | Mebendazole | [103] |
| T-ALL | — | Jurkat cells | — | TAL1, LMO2 | — | — | Disruption of TAD and sub-TAD boundaries: deletion or mutation of CTCF boundaries in insulated neighbourhoods leads to aberrant activation of oncogenes TAL1 and LMO2 by SEs | Establishment of the T-ALL cell state | — | [316] |
| T-ALL | — | Jurkat cells | ~5% of Jurkat cells | MYC | — | NOTCH1, RBPJ | Duplication of the MYC-SE | T-ALL cell growth, proliferation, metabolism | DBZ γ-secretase inhibitor suppresses NOTCH signalling | [106] |
| T-ALL | — | RPMI18402, Jurkat, MOLT-3 cells | — | CD47 | CD47, expressed on the tumour cell’s surface, serves as a ‘not eat me’ signal. Its binding to SIRPα, a receptor on immune cells, inhibits phagocytosis and thereby allows tumour cells to avoid immune surveillance | NFKB1 | Formation of aberrant SE, mechanisms is unknown | Evasion of immunosurveillance | JQ1 | [115] |
| B-cell lymphoma (BCL) | EBV-associated BCL | GM12878 lymphoblastoid cell line (LCL) derived from EBV-transformed B-cells | — | MYC, BCL2 | MYC heterodimerises with MAX to activate Cyclin D2 expression, promote cell cycle entry, and enable LCL DNA replication. BCL2 is known to inhibit proapoptotic proteins. SE-driven BCL2 overexpression can protect LCLs from MYC-induced apoptosis | STAT5, NFAT | Proposed model: EBNAs and cell transcription factor NF-kB activated by EBV protein LMP1 co-occupy enhancer sites, recruit cell TFs and nucleate EBV SE formation. In the previous study, the authors have shown that EBNA2 is crucial for MYC-SE formation, while its inactivation had little impact on BCL2. For NF-kB inactivation, the opposite was observed. In the following study, authors showed that eRNA is essential for enhancer–promoter looping in the MYC locus | Cell proliferation, survival, evasion of immunosurveillance | JQ1 | [185] |
| Diffuse large B-cell lymphoma (DLBCL) | — | Primary GCB and ABC DLBCL samples, HLY1, LY18, Karpas-422 (carry BCL6 SE mutations), LY10 (carry BCL2 SE mutations), RCK8, HLY1 (carry CXCR4 SE mutations) cell lines | ~92% of DLBCL samples carry at least 2 hypermutated active SEs. BCL6-SE is hypermutated in 59% of primary cases, BCL2-SE in ~33%, CXCR4-SE in ~19%) | BCL6, BCL2, CXCR4, PAX5, BCL7A, CIITA (specific for GSB subtype), RHEX (specific for ABC subtype) | — | B-cell specific TFs | SE-specific somatic hypermutation (SHM) induced by abnormal activity of activation-induced cytidine deaminase (AID). BCL6-SE mutation disrupts a BLIMP1 binding site. BLIMP1 is a master TF and a tumour suppressor. BCL2-SE/CXCR4-SE mutations disrupt NR3C1 binding site. Disruption of SE TF binding sites allows gene transcription to escape from the negative regulation of TF, which results in aberrant gene expression | Altered GC dynamics and prolonged retention of B-cells within the highly mutagenic environment of the DZ (dark zone) | Corticosteroid therapy to be studied | [317] |
| DLBCL | — | LY4 cells | — | CD47 | CD47, expressed on the tumour cell’s surface, serves as a ‘not eat me’ signal. Its binding to SIRPα, a receptor on immune cells, inhibits phagocytosis and thereby allows tumour cells to avoid immunosurveillance | — | Formation of aberrant SE, mechanisms is unknown | Evasion of immunosurveillance | — | [115] |
| Chronic lymphocytic leukaemia (CLL) | — | MEC1 cells | — | BMF | B cell lymphoma 2 (BCL2)-modifying factor (BMF) is BH3, the only proapoptotic member of the BCL2 protein family, which directly interacts with BCL2, thereby neutralizing its antiapoptotic activity | RELA (sub-unit of NF-kB protein complex) | SNP at 15q15.1, rs539846 C>A The alteration is conserved. RELA-binding motif leads to disruption of RELA binding, which results in reduction of BMF transcription | Negative regulation of apoptotic process, evasion of immunosurveillance | — | [12] |
| CLL | — | 23 primary blood samples | — | CXCR4, CD74, PAX5, CD5, KRAS, BCL2 | Genes playing role in CLL pathobiology and/or broadly across tumour types | PAX5 (dominates), FOXP1, RARA, ETS1, IRF2, IRF8 | Disrupted gene regulatory sites are observed in distal enhancers of the PAX5 locus in CLL | Cell proliferation, evasion of immunosurveillance, B-cell differentiation | JQ1 | [144] |
| Chronic myelogenous leukaemia (CML) | BCR-A BL positive CML | 3 primary CML CD34+ samples of relapsed CML after imatinib treatment, 1 CD34+CD38− primary sample of newly diagnosed CML | — | XBP1, E2F3, ETV6, SMAD3, CDK6 | XBP1 is vulnerable to low-concentration THZ1 treatment and ranked in the top 10% of actively expressed transcripts in RNA-seq. XBP1 is an oncogene, being a substrate for IRE1α, a sensor of unfolded protein response (UPR). IRE1α is activated in response to cell stress via autophosphorylation, then splices unspliced XBP1 RNA, thereby generating active XBP1 form (transcription factor) | — | Unknown | Cell survival, self-renewal capacity of leukaemia stem cells (LSCs), maintenance of LSCs function through IRE1-XBP1s pathway | THZ1, THZ1 + imatinib | [255] |
| CML | — | Primary blood samples, K562 cells | — | BUB1, CENPO, KIF2C, ORC1, RRM2 | These genes were chosen using WGCNA-based method. Expression of these genes is related to CML status and phase, positively regulated by super-enhancers and suppressed by JQ1 | SOX2, OCT4, Nanog | — | Megakaryocyte differentiation, hemostasis, cell cycle, chromosome organisation | JQ1 | [318] |
| Multiple myeloma (MM) | MM t(4;14)-positive subtype | 1 primary MM sample, LP-1, OPM2, H929, KMS28BM, KMS11 cells | 100% | HJURP, FGFR3 | HJURP is a centromeric protein that plays a central role in the incorporation and maintenance of the histone H3-like variant CENPA at centromeres. Overexpression of HJURP promotes cell growth and suppressed cell apoptosis in both t(4;14) and non-t(4;4) myeloma. SE-mediated epigenetic control is the primary driver for HJURP activation in t(4;14) MM and is important for the downstream functional effect of HJURP. NSD2 is important for the binding of BRD4 and the abundance of H3K27ac to the active parts of SE, the NSD2-BRD4 complex helps activate HJURP expression | NSD2 | Chromosomal rearrangements leading to convergence of SE with a specific oncogene (distal HJURP-SE interaction with HJURP promoter and consequent upregulation of the gene) | Cell growth, proliferation, survival, evasion of immunosurveillance | THZ1, JQ1 | [121] |
| Endocrine tumours | ||||||||||
| Thyroid cancer | Anaplastic thyroid carcinoma (ATC) | CAL-62 (ChIP-seq), CAL-62, 8505C, 8305C, C643 (THZ1 treatment) cells | — | PPP1R15A | Silencing of PPP1R15A via CRISPR–Cas9-mediated induces reduction in ATC cells’ colony-formation ability. PPP1R15A inhibition sensitises ATC cells to conventional chemotherapy. CDK7 expression is significantly higher in ATC samples compared to PTC samples | — | Probably, CDK7 overexpression | Cell growth, proliferation | THZ1 (CDK7 inhibitor), GBZ (PPP1R15A inhibitor) | [152] |
| Head and neck tumours | ||||||||||
| Squamous cell carcinoma (SCC) | Head and neck squamous cell carcinoma (HNSCC) | BICR-31 cells (cell line derived from a SCC of the tongue of a Caucasian male) | ~3% (SE amplification without KLF5 gene amplification) | KLF5 | — | — | Focal amplification of SEs | — | — | [105] |
| SCC | ESCC, HNSCC, LSCC subtypes | ESCC (KYSE70, (KYSE140, TE-5, TT), HNSCC (UMSCC1, 93UV147), LSCC (SK-MES-1, Calu-1, ChagoK1, H520, H2170, H226) cells | 100% of ESCC cells | lncRNA-CCAT1, TXNRD1 | CCAT1 is a novel SCC oncogenic lncRNA, its mRNA is upregulated in tumours compared with normal tissue in ESCC, HNSCC, LSCC. Mechanism: highly expressed CCAT1 recruits TP63 and SOX2 and forms with them proteins/RNA complex, localising at EGFR-SE and increasing its transcription, which activates MEK/ERK1/2 and PI3K/AKT signalling pathways | TP63, SOX2 | Genomic coamplification of TP63 and SOX2. TP63 and SOX2 cobind CCAT1-SE and promoter | Cell proliferation, survival through MEK/ERK1/2, PI3K/AKT pathways | — | [319] |
| SCC | Head and neck squamous cell carcinoma (HNSCC) | HN6 and SCC1 cell lines | — | MIR21 (miRNA, product MiR21-5p) | miR-21-5p is an oncogenic miRNA upregulated in many solid cancers, including HNSCC. In this study, miR-21-5p expression was shown to be significantly upregulated in HNSCC as compared to normal tissue (TCGA data) | FOSL1 | Dysregulation of FOSL1 | Cell proliferation, invasion | JQ1, I-BET151 | [320] |
| SCC | Head and neck squamous cell carcinoma (HNSCC) | UM-SCC1 cell line | — | SNAI2, FOSL1, CD44, EPHA2 | SNAI2 is a critical EMT transcriptional factor that maintains stemness and promotes invasive growth in HNSCC and multiple other cancers | FOSL1 (master regulator of HNSCC) | FOSL1 dysregulation: FOSL1 selectively associates with Mediator (MED1) to establish SEs | Cell migration, invasion, EMT, stemness | SR11302 (AP1 family inhibitor) | [162] |
| Nasopharyngeal carcinoma (NPC) | — | S26, S18, 6–10B, 5–8F cells | >80% | NDRG1, TRIB1, CASC8, KLHL38, ACTA1, EGFR, FOSL1, KLF5, ETS1 | hnRNPR is known to modulate RNA splicing and is directly involved in transcription. hnRNPR stabilises CCNB1 and CENPF mRNA to promote tumour metastasis. JUN-mediated seRNA-NPCM/hnRNPR can bind to the metastasis-promoting NDRG1 and TRIB1 loci and regulate transcription via R-loop formation. The dysregulated expression of the ACTA1 subunit is prone to form stress fibres and F-actin, which are associated with cytoskeletal stabilisation, cell survival, proliferation, and migration | Jun (c-Jun) together with c-Fos forms the AP-1 early TF. JUN gene is mapped to a chromosomal region involved in both translocations and deletions in human malignancies | Global enhancement of enhancer activity and specific SE activity, SE hijacking: the 3′-end of seRNA-NPCM hybridises with the SE region to form an R-loop, and the middle segment of seRNA-NPCM binds to hnRNPR at the promoter of distal and neighbouring genes (NDRG1 and TRIB1, accordingly). DNA translocations/deletions, forming binding sites for new TFs and leading to SE formation | Metastasis, NF-κB, EMT, TNFa pathways, cell motility, migration, proliferation | JQ1 | [182] |
| NPC | Basal-like (BC) and luminal-like (LC) NPC subtypes | Primary NPC samples, HK1, C666-1 (NPC), FaDu (hypopharyngeal carcinoma) cells | — | Associated with SE-HK1: TP63, ITGA6, CD44, WNT3, CAV2; Associated with SE-C666-1: FOXA1, CD74, CCL5, CCL20, CXCL13, CXCL17, TRAF3; BC-specific: ITGA6, WNT3, EGFR, CD44, IL-1β, CAV1, CAV2, KRT6A, KRT14 | NPC stems from malignant transformation of the pseudostratified ciliated columnar nasopharynx epithelium, which is composed of ciliated cells (CCs), nonciliated goblet secretory cells, and basal cells (BCs). BCs can self-renew and differentiate into luminal cells (LCs), including CCs and goblet cells. ΔNP63α is a prominent isoform of TP63 driven by SEs in NPC cells. ΔNP63α enhanced EGF-stimulated NF-κB activation in NPC cells by activating SE-mediated EGFR transcription. Depletion of ΔNP63α in NPC cells induces robust growth inhibition of NPC cells in vitro and in vivo. Levels of SE-associated transcripts are generally higher than those of TE-associated transcripts in HK1 and C666-1 cell lines | ΔNp63, TP63 | Genome-wide reorganisation/redistribution of the enhancer and SE landscape, due to the basal-to-luminal differentiation | Cell proliferation, tumourigenesis, evasion of immunosurveillance, basal-to-luminal gene expression signature switch, NF-κB pathway | JQ1 | [179] |
| NPC | — | EBV-positive cells C666-1, 2 untreated primary NPC samples, xenografts C15 (untreated NPC primary tumour) and C17, and C18 (NPC metastases) cells | — | ETV6 | ETV6 is a TF from the ETS family, known to be critical for oncogenesis. ETV6-SE is found in all samples and is not found in the control group in the study | NF-κB, IRF1, IRF2, ETS1, MYB, CTCT motifs uniquely enriched for NPC samples. YY1 and ZNF143 motifs are shared between cancer and control cells | Unknown. But NF-κB can be activated by EBV LMP2, which expression is consistently observed in NPC | Tumourigenesis | JQ1 | [136] |
| NPC | — | HK1cells | — | HAS3 | HAS3 overexpression promotes activation of the EGFR/AKT/ERK pathway | — | — | MAPK pathway, angiogenesis, cell proliferation, migration, evasion of immunosurveillance | JQ1 | [321] |
| Urinary and male genital tumours | ||||||||||
| Prostate cancer (ProCa) | Early-stage and late-stage subtypes | VCaP, LAPC4 cells | 100% | FOXA1, BRD4, Nkx3-1, GATA2, KLK2, KLK3, TMPRSS2, FKBP5, ACPP, ACP4, PMEPA1, IGF1R | Darolutamide inhibits androgen-induced changes at the AR cistrome. Darolutamide blocks enhancer activation and depletes AR and FOXA1 from enhancer and SE regions | — | Genome-wide reorganisation/redistribution of the enhancer and SE landscape, specific to the exact AR-dependent condition and cell type, disruption of TAD, and sub-TAD boundaries | Cell metabolism, growth, proliferation, metastasis, ERK pathway, PI3K/AKT/mTOR pathway | Darolutamide (AR antagonist) | [157] |
| ProCa | — | VCaP, LNCaP cells | — | MYC | Androgen receptor (AR) plays an important role in PCAT1-SE repression. Suppression of androgen activity increases MYC expression | AR, MYC, FOXA1, HOXB13 | — | DNA replication, cell cycle, homologous recombination, mismatch repair, p53 pathway, Wnt pathway, RNA transport, ribosome biogenesis | JQ1 | [150] |
| ProCa | ENZ-resistant castration-resistant prostate cancer (CRPC) | ENZ-resistant C4-2 cells | — | CHPT1 | CHPT1 catalyzes synthesis of PtdCho, a major component of cell membrane. Thus, CHPT1 overexpression increases PtdCho synthesis, thereby forming a less penetrating membrane, which may physically limit intake of toxic drugs into the cell | AR | TFs overexpression and seRNA, regulating BRD4 occupancy at SE via binding with BRD4 at the regions containing lysine-rich motifs. Formation of aberrant SE | Cell survival | JQ1, CPI-637 | [184] |
| ProCa | CRPC | 19 pairs of normal and tumour primary samples (2N, 3T specimens for ChIp-seq), C4-2B (metastatic CRPC), LAPC4, RWPE-1, LNCaP, VCaP, PC-3, 22Rv1, HEK293T cells | — | HOXB13, ACK1, FOLH1 | HOXB13 is a pioneer TF. ACK1 is a crucial regulator of CRPC growth, oncogene. FOLH1 is a ProCa diagnostic gene | HOXB13 (acK13-HOXB1) | HOXB13 acetylated by p300/CBP at lysine 13 (acK13-HOXB1) establishes tumour-specific SEs, and the following factors are likely to contribute to this: (1) acK13-HOXB significantly better binds chromatin remodelling proteins, bromodomain-containing proteins, and CTCF compared to unmodified HOXB13; (2) BRD9 specifically interacts only with acK13-HOXB13, not with HOXB13; (3) acK13-HOXB13 binding motif at SE region differs from unmodified protein | Tumourigenesis | (R)-9b, ACK1 inhibitor (shown on the prostate organoids) | [180] |
| Bladder cancer | — | UM-UC-3, T24 cells | — | LINC00162 | LINC00162 is an SE lncRNA, located on 21q22.3. It is encoded by sequence overlapping with SE and is found to be significantly upregulated with the highest fold change in bladder cancer cell line compared to normal cell line in the SE lncRNA microarray expression analysis. Suggested mechanism: LINC00162 binds THRAP3 and blocks its ability to positively regulate PTTG1IP expression, which leads to inhibition of PTTG1IP. PTTG1IP could inhibit the proliferation, promote cell apoptosis, and block G0/G1 phase | — | — | Cell proliferation, cell cycle | — | [190] |
| Central nervous system tumours | ||||||||||
| Medulloblastoma (MB) | Group 3 subtype (G3-MB) | Primary cell lines (D425, MB002, HD-MB03) | 100% | MYC, OTX2, CRX, ARL4D, AUTS2, BMF, IGF2BP3, KIF21B, KLHL29, LRP8, MARS1, PSMB5, SDK2, SSBP3, UPR | ARL4D is a member of the ARF protein family (of RAS superfamily of small GTPases). In cancer cells, ARF members are subverted for regulating proliferation, migration, and invasion. An OTX2-SE-ARL4D regulatory axis represents a subtype-specific tumour dependency and therapeutic target of G3-MB by contributing to maintaining cell cycle progression and inhibiting neural differentiation of tumour cells. BETi and CDK7i work synergistically on suppressing SE-driven core transcriptional regulatory network of G3-MB | OTX2, CRX | SE-driven core transcriptional regulatory network of G3-MB (SE-associated transcription or higher activation of ER stress and UPR). TFs increase in expression and overexpression, leading to pathological gene activation | Cell proliferation, cell cycle | THZ1, JQ1, Marizomib and their combinations | [171] |
| Neuroblastoma (NB) | NB with rs2168101 G>T | SKNSH and SHSY5Y neuroblastoma cells (G/G homozygous), Kelly (G/-, MYCN-amplified), BE2 (G/T, MYCN-amplified), primary patient samples | In the rs2168101-harbouring patients 28% of Europeans, 32% of Eastern Asians and almost no Africans carried protective T allele | LMO1 | LMO1 has been previously identified by the same research group as an oncogene in neuroblastoma. In 45 tumours: rs2168101 (G/G = 33, G/T = 12, T/T = 0) | GATA2/3 | SNP rs2168101 G>T Wild-type evolutionarily conserved G-allele is located at LMO1-SE. But interestingly, there is a more recent protective T-allele containing an SNP that disturbs GATA3 binding motif in the SE, which leads to decrease in LMO1 expression | Tumour formation (G-allele)/tumour susceptibility (T-allele) | — | [127] |
| NB | Lineage-committed adrenergic (ADRN) and mesenchymal (MES) NB subtypes | IMR32, SH-SY5Y, GIMEN, NBL-S, LAN-1, SKNBE-2, NGP, GICAN, SHEP cells | — | c-MYC, N-MYC, ALK, PHOX2A/B, NPM1, ODC1, CCNA2, TGM2, DKK3, FABP5, UFD1L, FASN, NCL; MES-associated: WWTR1 (TAZ), CREG1, PRRX1, SNAI2, MEOX2; ADRN-associated: PHOX2A, DLK1, GATA2, HAND1, NOTCH3, SNAI2, IMR32, NGF, NT3, NGFR | ISX-9 abrogates ALK and N-MYC functions in NB cells and induces NB subtype switching. ISX-9 affects histone acetylation and promotes SE weakening. ISX-9 reprogrammed NB cells from an ADRN into a MES-like (MES/GN) state which is incompatible with oncogenic growth | BORIS, NEUROD1, ATOH1, OLIG2, NEUROG2 | Genome-wide reorganisation/redistribution of the enhancer and SE landscape (ADRN-restricted SE activities regulating key ADRN oncogenes such as MYCN, ALK, PHOX2B might account for the MES vs. ADRN subtype specificity). SE hijacking. TFs increase in expression and overexpression, leading to pathological gene activation | Cell growth, proliferation, transdifferentiation | Isoxazole (ISX-9) | [175] |
| NB | — | SH-SY5Y (subclone with adrenergic (ADRN) phenotype) derived from SK-N-SH cells, 5 CD133-primary samples (2/5 had an isogenic pair of ADRN phenotype), SH-EP2 (subclone with undifferentiated mesenchymal (MES) phenotype) derived from SK-N-SH cells, 3 CD133+ primary samples (all with an isogenic pair of ADRN phenotype) | — | (1) DBH, CHGA, DLK1 (2) ASCL1, EYA1, GATA3, HAND1, SIX3 (ZNF536, PHOX2A, KLF13, SOX11, GATA2, KLF7, TFAP2B, ISL1, HEY1, DACH1, PHOX2B, PBX3, SATB1) (3) WNT5A, IGFBP2, FN1, IL13RA1 (4) MEOX1, MEOX2, SIX1, SIX4, SOX9, SMAD3 WWTR1 (PRRX1, CREG1, ELK4, DCAF6, ID1, MAML2, NOTCH2, CBFB, IFI16, ZNF217, EGR3, ZFP36L1 ABEP1) | (1) known markers of adrenergic differentiation (2) 18 ADRN-specific TFs. Although PHOX2B and PHOX2A are ADRN-specific, SEs associated with them are also found in some MES lines. (3) MES-specific genes (4) 20 MES-specific TFs PRRX1 reprogram super-enhancers and mRNA landscapes of ADRN cells toward a MES state. Thus, PRRX1 is probably the master regulator of MES program | GATA3, PRRX1 | Probably, GATA3, PRRX1 upregulation | ADRN phenotype establishment maintenance, MES phenotype establishment maintenance, phenotype reprogramming via EMT regulation, chemoresistance, relapse | — | [322] |
| NB | MYCN-amplified and nonamplified NB | 3 primary samples, Kelly, NGP, IMR-32, SH-SY5Y cells | — | MYCN, ALK, PHOX2B, HAND2 | MYCN-amplified cell lines (Kelly, NGP, IMR-32) were selectively targeted by THZ1. ALK is prooncogenic in NB. PHOX2B, HAND2 are master TFs that specify sympathetic neuronal cell identity | MYCN | Genomically amplified MYCN induces global transcription amplification | Tumourigenesis, cell proliferation | THZ1 | [165] |
| Glioma (GM) | Glioblastoma (GBM) | LN-18 and U251 cells, xenograft mouse models, primary GBM samples | — | TMEM44-AS1 | — | MYC | MED1, MYC overexpression observed | Glioma cell proliferation, migration, invasion, p38MAPK, IL-6, interferon pathways | Myci975 (MYCi, small-molecule Myc inhibitors) | [109] |
| Thoracic tumours | ||||||||||
| Lung adenocarcinoma (LUAD) | — | A549 cells | 100% | EphA2 | EphA2 is a member of the Eph receptor family. Its main binding ligand is ephrin A1, EphA2 acts through activating tyrosine phosphorylation of downstream substrate proteins and is degraded in normal cells after phosphorylation in contrast to cancer cells. EphA2-SE interacts with the EphA2 gene promoter and promotes transcription by recruiting FOSL2 and TCF7L2, resulting in abnormally high expression of EphA2, which activates signal pathways to promote tumour progression, which is confirmed by FOSL2 and TCF7L2 siRNA silencing analysis. CRISPR-based EphA2-SE deletion suppressed cell proliferation, migration, and invasion by decreasing the expression of EphA2 in vitro | TCF7L2, FOSL2 bind to EphA2-SE | Chromosomal rearrangements leading to convergence of SE (EphA2-SE) with a specific oncogene (EphA2). SE hijacking | Cell proliferation, invasion, migration, tumourigenesis, WNT/β-catenin pathway, PI3K/AKT pathway | Disruption of EphA2-SE, SY-5609 | [120] |
| LUAD | — | A549, NCI-H2009, NCI-H358 cells | ~17% co-occurring SE and MYC gene amplification ~2% SE amplification only | MYC | — | GATA3, FOXA1, NFE2L2, CEBPB | Focal amplification of SE | Anchorage-independent and clonogenic growth | — | [105] |
| LUAD | — | Primary samples, A549 cells | 43.66% | RAI14 | — | — | — | Cell proliferation | — | [323] |
| LUAD | — | Primary samples, A549, A427, PC-9, H1299, SW1573, H1975, H358 cells | — | LINC01977 | LINC01977 promotes malignancy via the canonical TGF-β/SMAD3 pathway | SMAD3 | TAM2 infiltration induces rich a TGF-β microenvironment. It makes SMAD3 bind the promoter and the SE of LINC01977, which upregulates LINC01977 expression | TGFb pathway, EMT | JQ1, SGC-CBP30 (small molecule, inhibitor of CBP/P300) | [189] |
| Small-cell lung cancer (SCLC) | SCLC-A, SCLC-P subtypes | Lu134A cells with TP53 and RB1 mutations, H2107, H128, H510, H69, H510, H211 cells | — | ASCL1, NKX2-1, INSM1, FOXA1, MIR375, MIR7-3, MIR200B, MIR429, miR-455-3p (suppressed in SCLC-P) | ASCL1 is a master transcription regulator; its inhibition leads to downregulation of TFS NKX2-1, INSM1, FOXA1. ASCL1 is crucial for neuroendocrine differentiation. miR-375 and miR-9-5p probably co-target YAP1, which defines SCL-Y subtype, and suppress it; NOTCH2 is a common target gene of miR-9-5p and miR-375, and this mechanism may contribute to suppression of Notch signalling and sustained expression of ASCL1. SE-associated miRNAs influence transcriptional networks, in particular targeting subtype-specific genes | ASCL1, POU2F3 | — | Cell-type identity regulation, intratumour heterogeneity formation | — | [164] |
| Lung cancer | — | Primary lung cancer-associated fibroblasts (CAF), adult NHLF fibroblasts | — | TBX4, TBX2, TBX5, SFRP1, ADM, THBS1, HOXA5, FOXL1, FOXP1, MEIS1, TGIF1 | TBX4 is the only TF associated with SE in NHLFs among multiple cell types and is unique to lung tissue. The TBX4-SE overlaps with a lung mesenchyme-specific enhancer, suggesting the retained activities of developmentally regulated enhancers in adult tissues. The loss of TBX4 is associated with the acquisition of the CAF phenotype by lung fibroblasts and TGF-β might be involved in this process | TBX4 | Mutations in DNA that form binding sites for new TFs (TBX4), leading to the formation of SE. TFs increase in expression and overexpression, leading to pathological gene activation | Cell proliferation, transdifferentiation, cellular contractility (collagen gel contraction) | — | [160] |
| Female genital tumours | ||||||||||
| Cervical cancer (CerCa) | — | HeLa cells | 100% | EphA2 | EphA2 is a member of the Eph receptor family. Its main binding ligand is ephrin A1, EphA2 acts through activating tyrosine phosphorylation of downstream substrate proteins and is degraded in normal cells after phosphorylation in contrast to cancer cells. EphA2-SE interacts with the EphA2 gene promoter and promotes transcription by recruiting FOSL2 and TCF7L2, resulting in abnormally high expression of EphA2, which activates signal pathways to promote tumour progression, which is confirmed by FOSL2 and TCF7L2 siRNA silencing analysis. CRISPR-based EphA2-SE deletion suppressed cell proliferation, migration, and invasion by decreasing the expression of EphA2 in vitro | TCF7L2, FOSL2 bind to EphA2-SE | Chromosomal rearrangements leading to convergence of SE (EphA2-SE) with a specific oncogene (EphA2). SE hijacking | Cell proliferation, invasion, migration, tumourigenesis, WNT/β-catenin pathway, PI3K/AKT pathway | Disruption of EphA2-SE, SY-5609 | [120] |
| CerCa | HPV-related CerCa | HeLa, S12, Ca Ski, SCC-047, SCC-090, Si-Ha cells | — | Cis-genes (CASC8 and HMGA2), trans-genes (PLXNB2, CGAS) | PLXNB2 and CASC8 levels are declined in 3/3 HeLa ecDNA-reduced monoclones, and CGAS and HMGA2 levels are declined in 2/2 S12 ecDNA-reduced monoclones | TP53 | HPV integration generates HPV breakpoint-induced cellular SE (BP-cSEs), further local genomic amplification and TAD remodelling. BP-cSEs cis- and trans-regulate the targeted gene expression, including ecDNA form | TP53 pathway | — | [129] |
| CerCa | HPV-related CerCa | W12 subclones, C-33A, C-411, CasKi, HeLa, ME-180, SiHa cells | — | E6/E7 cellular genes (e.g., FOSL2, neighbouring with integration site) | E6/E7 viral oncogenes downregulation in HPV cancer cells is known to result in reduced proliferation and senescence. FOSL2 expression is weakly reduced after the disruption of BRD4 binding to SE | — | HPV16 integration, subsequent SE hijacking and focal amplification Mechanism: HPV16 integrates into transcriptional active region of a host genome (chr 2 p23.2) and hijacks adjacent enhancer element, then viral genome and flanking cellular DNA sequences co-amplify, which leads to viral-cellular de novo SE formation | Cell proliferation increased genomic instability at the integration locus | iBET762, JQ1 | [130,131] |
| Uterine corpus endometrial carcinoma (UCEC) | — | Ishikawa cells | ~10% co-occurring SEs and MYC gene amplification, ~4% SEs amplification only | MYC | — | — | Focal amplification of SE | — | — | [105] |
| Ovarian cancer (OvCa) | High-grade serous ovarian cancer (HGSOC) | OVCAR-3 cells | ~11% of patients from TCGA dataset had BRD4 copy-number amplification | RAE1 (SE60), EPHA2, MAB21L3 (SE14) | In this study, SE14 and SE60 are determined as prominent SEs among top 86 SEs, via CRISPRi and CRISPR-deletion assays combined with RNA sequencing. SEs direct target genes are defined using Hi-C | SOX4, ATF4, YY1 (SE60), ELF3, KLF, and JUN (SE14) | BRD4 and SE copy-number amplification | Cell proliferation, migration, ERRB3 pathway | — | [111] |
| OvCa | Cisplatin-resistant high-grade serous ovarian cancer (HGSOC), epithelial ovarian cancer (EOC) subtypes | OVCAR3, A2780 CP70 (cisplatin-resistant) cells, orthotopic xenograft mouse model | — | ALDH1A1 | ALDH1A1 is important in epithelial ovarian cancer (EOC) due to its crucial role in maintenance of cells with stem-like features. Its knockdown has been shown to increase EOC cell sensitivity to chemotherapy | — | Cisplatin induces SE activation (concluded based on elevated seRNA expression after cisplatin treatment) and possible BRD4 overexpression/amplification, seRNA contributes to SE-derived transcription | Stemness | Cisplatin+JQ1 (synergistic effect) | [183] |
| OvCa | Cisplatin-resistant high-grade serous ovarian cancer (HGSOC) | OVCAR4, OVCAR3, OV81, SKOV3 (cisplatin-resistant) cells, xenograft mouse models | — | SOX9 | SOX9 plays role in establishment and maintenance of chemoresistance | SOX9 | Cisplatin-induced SE formation and/or activation | Cell migration, chemosensitivity, immunosurveillance | JQ1, JQ1+cisplatin (synergetic effect) | [324] |
| OvCa | Clear-cell ovarian cancer (CCOC), endometrioid ovarian cancer (EnOC), high-grade serous ovarian cancer (HGSOC), mucinous ovarian cancer (MOC) subtypes | 20 primary epithelial ovarian cancer (EOC) samples, 5 samples of each histotype (ChiP-seq), OVCA429, CaOV3, UWB1.289 cells | — | lncRNA-UCA1 | UCA1 was identified as a driver of EOC development. Suggested mechanism, based on experiments results: UCA1 binding to AMOTp130 facilitates AMOTp130-YAP interaction, which promotes YAP and its target genes activation via YAP dephosphorylation and YAP nuclear translocation. Strategy for prioritizing and mechanistic analysis of lncRNAs is suggested in the study | — | Unknown. Acquired in EOC | Cell proliferation, survival through Hippo-YAP signalling regulation | JQ1 | [139] |
| OvCa | — | A2780 cells (not treated), A2780 cells (treated with cisplatin) | — | ISL1, PAX2, PRRX1, ZIC1, ATF5 (downregulated SE) GATA2, NKX2.5, SNAI2, MZF1, EGR1 (upregulated SE) | ISL1 is known to be involved in cell survival and mesenchymal transition. ISL1 is identified as the major cell-type-specific SE regulator (according to the authors, ISL1 is expressed in 75% of ovarian cancers). Mechanism: repeated cisplatin treatment downregulates ISL1 expression, and thereby induces SEs redistribution. It may lead to the acquisition of new phenotypic features, which serve as an adaptation mechanism in cells undergoing near-to-death experience | ISL1 | Cisplatin treatment leading to ISL1 downregulation | Drug-resistance associated with acquisition of new phenotypic features | — | [308] |
| Soft tissue and bone tumours | ||||||||||
| Osteosarcoma (OS) | — | Osteosarcoma primary samples, 143B, SJSA1, ZOS, ZOSM cells | 100% | LIF, SOX2, OCT4, NANOG, CD133, NOTCH1, HES1, KRT19, HEY1, HEYL | LIF is a cytokine that affects cell growth by inhibiting differentiation and is critical for the maintenance of osteosarcoma stem cell-like properties. UTX binds to the LIF promoter and modulates SE signals. LIF inhibition reduces SE and enhancer signals of the NOTCH1 signalling pathway | UTX | Chromosomal rearrangements leading to convergence of SE (LIF-SE, stem cell related) with a specific oncogene (LIF). SE hijacking | Cell invasion, metastasis, EMT, JAK/STAT3, NOTCH1 pathways | GSK-J4 (UTXi) | [168] |
| OS | — | U2OS, 143B cell lines (ChIP-seq), SJSA-1, U2-OS, HOS, G-292, MNNG/HOS, 143B and MG-63 (other experiments), metastatic and nonmetastasic osteosarcoma primary samples | — | CDK6, TGFB2 | Critical oncogenes in osteosarcoma | MYC | MYC is aberrantly expressed in osteosarcoma, especially in metastasis, and drives the strong expression of SEs-associated oncogenes | Cell proliferation. migration, invasion, metastasis | THZ1 | [155] |
| OS | — | SJSA-1, U2-OS cells | — | SRSF3, FOXK1 (1) MYC, TCF7L2, RUNX2, MDM2 (2) | These genes are downregulated in both cell lines after THZ2 treatment and are in 10% of genes more sensitive to THZ2 in each cell line (1) or in at least one of cell lines (2). SRSF3, MYC, TCF7L2, RUNX2, MDM2 have been shown to be involved in malignant potential in osteosarcoma | — | CDK7 is overexpressed in osteosarcoma tissues compared to normal tissues in paired specimens from 35 patients | Cell proliferation, migration, cell development (GO for all SE-associated genes) | THZ2 (specific CDK7 inhibitor) | [151] |
| OS | — | Surgical specimens of osteosarcoma, human osteosarcoma cell lines (143B, SJSA1) | — | Max-like protein X (MLX) | The chosen gene plays a role in proliferation, determination, and differentiation and is overexpressed in patient samples. SE-driven MLX is overexpressed in osteosarcoma, positively regulates SLC7A11 (transporter for cysteine and glutamate), and prevents ferroptosis | — | — | Oxidative phosphorylation, cell redox homeostasis, cell proliferation, migration, lipid metabolism, cysteine metabolism, ferroptosis, response to reactive oxygen | — | [325] |
| Ewing sarcoma | — | A673, SKNMC, TC32, TC71, EW8, TCC446, EWS502 cells, 3 primary samples from https://doi.org/10.1016/j.ccell.2014.10.004, https://doi.org/10.18632/oncotarget.4903 | >90% | MEIS1, EWS-FLI1, APCDD1, CCND1, NKX2–2, BCL11B, MYC, IGF2BP1, STEAP1, SOX5, CDC25A, LRP6, LEF1, NKD1 | Fusion gene EWS-FLI1 orchestrates the transcriptional dysregulation in Ewing sarcoma. MEIS1 is a homeodomain TF and an oncogenic factor in Ewing sarcoma. APCDD1 is a transmembrane glycoprotein that is associated with the Wnt/β-catenin signalling pathway. MEIS1 and EWS-FLI1 transcriptionally activate APCDD1 in Ewing sarcoma | MEIS1, EWS-FLI1 | Chromosomal rearrangements leading to convergence of SE (APCDD1-SE) with a specific oncogene (APCDD1). SE hijacking: MEIS1-SE, MENKX2–2-SE, APCDD1-SE, CCND-SE. TFs increase in expression and overexpression, leading to pathological gene activation | Cell proliferation, evasion of immunosurveillance | THZ1 | [124] |
| Ewing sarcoma | — | TC32, TC71 cells | — | Cyclin D1 (CCND1), caveolin 1 (CAV1), glycogen synthase kinase 3 alpha (GSK-3α), microtubule-associated protein tau (MAPT) | A combination of SE profiling, near-whole genome shRNA-based and small-molecule drug sensitivity screening is done for identification of cyclin D1 and CDK4 as Ewing sarcoma-selective dependencies | EWS/FLI1 | A somatic chromosomal translocation leads to the formation of fusion protein EWS/FLI1. This oncogenic TF forms an aberrant transcriptional profile | Cell proliferation, cell cycle, p53 pathway | GSK-3 inhibitors (SB216763 and CHIR-99021), PD0332991 (CDK4/6 inhibitor) | [123] |
| Rhabdomyosarcoma (RMS) | Fusion-positive (FP-RMS), fusion-positive alveolar (FP-ARMS), RMS with mutations in receptor tyrosine kinase/RAS pathways (FN-RMS) | RH4, RH5, SCMC cells, 3 FP-RMS and 5 FN-RMS primary samples (GSE83726) | 100% of cells, >60% of tumours | PAX3-FOXO1, MYOD1, MYCN, MYOG, IGF1R/IGF2, FGFR4, ALK, HDAC5, MEST | The genes encode the primary driver of RMS program, master TFs, receptor tyrosine kinases, histone deacetylase, and enhancer-bound coactivator, respectively. MEST and IGF2 are involved in mesoderm development | PAX3-FOXO1 (excluding MYOG), MYOD1, MYCN, MYOG | t(2;13)(q35;q14) chromosomal translocation forms the fusion gene, the product of which, PAX3-FOXO1, is a pioneer factor establishing de novo SEs via opening chromatin, recruiting BRD4 and other MTFs to SEs. Further study showed that SEs activity depends on subunit of nucleosome remodelling and histone deacetylase (NuRD) complex, CHD4, which enhances DNA accessibility for PAX3-FOXO1 and HDAC2, and consequently, BRD4 binding to SE regions | Specification of RMS cell identity, cell maintenance in myoblastic state | JQ1 | [119,167] |
| Breast tumours | ||||||||||
| Breast cancer (BrCa) | — | MCF7 cells | 100% | EphA2 | EphA2 is a member of the Eph receptor family. Its main binding ligand is ephrin A1, EphA2 acts through activating tyrosine phosphorylation of downstream substrate proteins and is degraded in normal cells after phosphorylation in contrast to cancer cells. EphA2-SE interacts with the EphA2 gene promoter and promotes transcription by recruiting FOSL2 and TCF7L2, resulting in abnormally high expression of EphA2, which activates signal pathways to promote tumour progression, which is confirmed by FOSL2 and TCF7L2 siRNA silencing analysis. CRISPR-based EphA2-SE deletion suppressed cell proliferation, migration, and invasion by decreasing the expression of EphA2 in vitro | TCF7L2, FOSL2 bind to EphA2-SE | Chromosomal rearrangements lead to convergence of SE (EphA2-SE) with a specific oncogene (EphA2). SE hijacking | Cell proliferation, invasion, migration, tumourigenesis, WNT/β-catenin pathway, PI3K/AKT pathway | Disruption of EphA2-SE, SY-5609 | [120] |
| BrCa | Estrogen receptor positive and progesterone receptor positive subtype (ER+PR+), human epidermal growth factor positive (HER2+ER−, PR−) subtypes | MCF7 and HCC1954 cell lines; 1 ER+PR+ patient-derived xenograft sample | - | CD47 | CD47, expressed on the tumour cell’s surface, serves as a ‘not eat me’ signal. Its binding to SIRPα, a receptor on immune cells, and inhibits phagocytosis and thereby allows tumour cells to avoid immune surveillance | NFKB1 (binds to constituent enhancer E5), PPARα | Formation of aberrant SE, mechanisms are to be discovered. TNF inflammatory pathway leads to activation of NFKB1 translocation to the nucleus | Escape of immune surveillance | BR inhibitors (JQ1, I-BET151, PFI-1): JQ1 is less toxic among them; infliximab (trademark, Remicade) blocks TNF pathway; CD47 antibody combined with infiximab shows best results | [115] |
| BrCa | ER+PR+, HER2+, TN BrCa subtypes | MCF7 (ER+PR+), HCC1954 (HER2+), BT549, MDAMB231, Hs578T (TN) cells | >80% | NSMCE2, MAL2 | NSMCE2 encodes a member of the E3 ubiquitin-related modifier (SUMO) ligase family, which is part of the SMC 5/6 complex. It is essential for DNA DSB repair through HR and chromosomal segregation during cell division. NSMCE2-reduced expression has been shown in studies to improve the efficacy of chemotherapy drugs. MAL2 is a transmembrane protein that is essential for transcytosis, its overexpression reduces cancer cells’ antigen presentation but may accumulate HER2 accumulation | NF1, a tumour-suppressor gene that has been reported to be associated with an increase in breast cancer risk when lost (via in silico simulation) | Large tandem duplications in breast cancers (e.g., copy number gains of the NSMCE2 and MAL2 loci) lead to SE formation. Deactivation of some SE-associated tumour suppressor genes | Cell-cycle deregulation (G1-S transition defects), tumourigenesis, HER2 pathway | JQ1, I-BET151 | [104] |
| BrCa | — | T47D/A1-2 cells | 100% | DDIT4 | Glucocorticoid (dexametazone, Dex) treatment alters the SE landscape in breast cancer cells. A Dex-specific SE encompasses DDIT4 (a GR target gene that is implicated in a variety of cancers and exhibits a highly heterogeneous Dex response) and four GR binding sites. Each binding site is uniquely required to promote or suppress DDIT4 expression. Dex treatment triggers a loop-switching mechanism to induce DDIT4 transcription | STAT1/3, SREBF1/2, SP1/3, E2F1, E2F2 | Genome-wide reorganisation/redistribution of the enhancer and SE landscape, disruption of TAD and sub-TAD boundaries (chromatin looping). Modulation of the frequency and amplitude of transcriptional bursts | Cell proliferation, tumourigenesis | Dex | [149] |
| BrCa | Stem-like and non-stem-like BrCa subtypes | BPLER (stem-like breast cancer-cell model), HMLER (matched nonstem tumour cell (nsTC)-like cell model), SUM159PT, SUM149PT, MCF7, MDA-MB-157 cells | >50% | c-MYC, CD44, CDKN1B, SLUG, VDR, SMAD3, VEGFA, XBP1, RB1, EZH2, EP300 c-JUN, HOXA2, HOXA10, CD44, BCL-XL, HDAC7, DHRS2, CLU, CDKN1A, BHLHE40 | HDAC1/3/7 inhibition by MS-275 reduces H3K27ac and represses SE-associated gene transcription at stem-cell transcription factor genes. Long-term HDAC1/3 double knockdown maintains HDAC7 downregulation in stem-like breast cancer cells. A subset of stem-cell TF genes may be differentially regulated by HDAC1/3/7 in stem-like BrCa cells | — | Genome-wide reorganisation/redistribution of the enhancer and SE landscape, specific to the cell-type and disruption of TAD and sub-TAD boundaries. | — | MS-275 (entinostat), HDAC1-, HDAC3-, and HDAC7-specific siRNAs | [112] |
| BrCa | Estrogen receptor positive and progesterone receptor positive subtype (ER+PR+), human epidermal growth factor positive (HER2+ER-, PR-) subtypes | MCF7, HCC1954 cells, 1 ER+PR+ patient-derived xenograft sample | — | CD47 | CD47, expressed on the tumour cells surface, serves as a “do not eat me” signal. Its binding to SIRPα, a receptor on immune cells, inhibits phagocytosis and thereby allows tumour cells to avoid immunosurveillance | NFKB1 (binds to constituent enhancer E5), PPARα | Aberrant SE formation, mechanisms are unknown. TNF inflammatory pathway leads to activation of NFKB1 translocation to the nucleus | Evasion of immunosurveillance | BR inhibitors: JQ1, I-BET151, PFI-1 JQ1 is less toxic among them, CD47 antibody combined with infliximab (blocks TNF pathway) showed best results | [115] |
| BrCa | Radioresistant BrCa | HB-2, MCF10A (modified to overexpress ENC1) cells, constructed radio-resistant BC cells (MDAMB-231/RaR, BT549/RaR), metastatic mice model, primary samples | — | ENC1 | ENC1 overexpression promotes Hippo-YAP1/TAZ pathway through:
| TCF4 | TCF4 TF overexpression leads to aberrant activity of ENC1-SE | Radioresistance, recurrence, metastasis promotion via Hippo-YAP1/TAZ pathway | — | [326] |
| BrCa | Basal-like, triple-negative (ER-/PR-/HER2-) BrCa subtype (TNBC) | BT549, HCC1937, MDA-MB436, MDA-MB-231, SUM149, SUM159, MDA-MB468, HCC1395, SUM1315 | 60% | FOXC1, MET, ANLN | FOXC1 and MET are known oncogenes highly expressed in TNBC and associated with worse clinical outcomes. ANLN is a novel TNBC-specific gene. FOXC1 exhibits significantly higher expression in TNBC samples compared to non-TNBC samples (Her2, LumA, LumB BC subtypes and normal breast tissue) in the METABRIC cohort. Recently, ANLN was shown to regulate breast cancer cell migration and stemness | HIF1A, FOXC1, SP1, YY1 (FOXC1 SE), SP3, YY1, RBPJ, PBX1, TFE3, ZNF148, NR2C1, NFYB, ESRRA | Unknown (however, FOXC1-SE focal amplification in genomic analysis was not found) | Tumourigenesis, cell migration, invasion (MET), metastasis (FOXC1), stemness (ANLN) | JQ1 | [102] |
| BrCa | — | MCF7 cells | — | GNA13, PDPK1 WNT4, LRP5 (downregulation) | These genes are known to be involved in drug resistance. For GNA13 and PDPK1 occupancy by BRD4/LSD1/NuRD complex is examined and confirmed | — | BRD4 reorganises chromatin and facilitates recruitment of complex LSD1/NuRD to SEs, BRD4/LSD1/NuRD complex occupies SEs and represses SE-associated genes through SE activity restriction | Chemoresistance (evasion of immunosurveillance), PI3K-AKT, WNT, Hippo pathways | — | [154] |
| BrCa | — | MCF10A cells (MCF10A-AT1, MCF10A-DCIS, MCF10A-CA1), patient samples | — | RARRES1, MIR200B (enhancer, not identified as SE) | RP11-379F4.4, RP11-465B22 are cis-acting SE-lncRNAs, i.e., lncRNAs transcribed from SE and functioning at the loci they are being transcribed. RP11-379F4.4 downregulates RARRES1, which suppresses invasion, respectively, and thereby probably contributes to the progression of ductal carcinoma in situ (DCIS) lesion to invasive ductal carcinoma (IDC). RP11-465B22 upregulates miR-200b expression, which is known to inhibit proliferation, metastasis, and angiogenesis. Thus, increased expression of these SE-lncRNAs could be used as markers of disease progression | — | Unknown | Cell invasion (RP11-379F4.4) | — | [137] |
| BrCa | — | MDA-MB-23, BT549 cells | — | ENC1 | ENC1 overexpression affects breast cancer BrCa recurrence and radio resistance, promoting through the Hippo pathway:
| TCF4 | Increased TCF4 expression results in increased SE activity | Recurrence and radio resistance via Hippo pathway | — | [326] |
| BrCa | TNBC BrCa | 10 primary samples, HCC38 and other cells (23 cell lines in total) | — | BAMBI, EGFR, MYC | BMP and activin membrane-bound inhibitor (BAMBI) is a transmembrane protein, TGFβ pseudo-receptor. SEs are found in all TNBC primary samples | — | Acquired in BrCa | Cell growth, proliferation | — | [101] |
| BrCa | — | HMEC, SUM149, SUM159, MDA-MB-231 (ChIP-seq), MDA-MB-231, BT549 (CRISPR-Cas9), HMEC (Hi-C) cells | — | RCAN1.4, isoform of RCAN1 (RCAN) | Clinical relevance of RCAN1.4 and RUNX3 in BrCa is accessed via in silico analysis and immunohistochemical staining. RCAN1 is found to be one of the most downregulated genes at chr 21q22 in BrCa compared to adjacent normal tissues (from TCCG cohort). Significant RCAN1 decline is observed in all molecular subtypes (LumA, LumB, Her2, TNBC). RCAN1.4 blocks calcineurin-NFATc1 signalling (CaN/NFATc1 signalling), precisely, it regulates the nuclear localisation of NFATc1 by inhibiting calcineurin activity | RUNX3 | Over 90% of RCAN1.4 expressions are SE-driven and ensured by activity of RUNX3, binding RCAN1.4 promoter and SE-region. RUNX3 downregulation, frequently observed in BrCa, probably leads to SE disruption and consequently to decreased RCAN1.4 expression | Tumour suppression | JQ1 | [327] |
| BrCa | ERα-positive BrCa | RET MCF7 (oestrogen-treated), T47D, ZR-75-1 cells | 30% | RET | RET is known to be important in BrCa. In this study, RET is defined as an ERSE-associated gene, found among the top genes most induced by oestrogen. Furthermore, RET was able to rescue the defects in ERSE-driven gene transcription during BRD4 knockdown. Suggested mechanism: RET activates RET/RAS/RAF/MEK/ERK/p90RSK/ERα phosphorylation cascade, and thereby forms positive feedback loop (BRD4/ERα–RET–ERα), activating ERα and promoting transcription of ERα target genes including RET itself | ERα | BRD4 is crucial for transcriptional activation of ERα-SE-associated genes and functions as a master regulator in. BRD4 was significantly induced on ER-SEs under oestrogen treatment | Cell growth, proliferation, migration, invasion, cell cycle | Cotreatment with JQ1 and BLU-667 (RET inhibitor) | [166] |
| BrCa | BRCA1mut/+ BrCa | Primary samples tissues, BRCA1mut/+ MCF10A, BRCA1(185delAG)/+ MCF10A cells (the most common pathogenic mutation) | — | TNFAIP3, SOD2 | Both genes are associated with BRCA1 in various human cell lines | GATA family (GATA2, GATA3, and GATA4) for SEs missing in BRCA1mut/+ | BRCA1 plays a role in chromatin reorganisation, mutations in BRCA1 lead to SE structure disruption | NF-kB pathway, epithelial cell differentiation, DNA stability, inflammatory response, stress response | — | [328] |
| BrCa | — | MCF7/TamR cells derived from MCF7 continuously treated with tamoxifen more than 1 year | — | ATP1A1−AS1 | ATP1A1−AS1 is among SE-associated lncRNAs which potentially play a role in tamoxifen resistance | FOXA1 | Downregulation of SE-associated lncRNA ATP1A1−AS1 leads to downregulation of ATP1A1, which can cause tamoxifen resistance | Wnt, Rap1, Hippo, AMPK, mTOR pathways, regulation of cytoskeleton organisation, focal adhesion, protein digestion and absorption, hormone bioprocessing | — | [329] |
| BrCa | HPV-related BrCa | CaSki, SiHa, HeLa cells | — | EGFR, c-MET | — | — | HPV16 E6 destabilises the histone demethylase KDM5C | — | — | [132] |
| BrCa | HER2+, TNBC BrCa | HER2+: HCC-1954 cells TNBC: HCC-1937, SUM-159, MDA-MB-231, MDA-MB-436 cells | — | CD274, CD273 | CD274 and CD273 are genes encoding PD-L1 and PD-L2, respectively. Immune checkpoints PD-1/PD-L1 (or PD-L2) pair have proven to be the major targets in multiple cancer types | — | Unknown. SE, located between CD274 and CD273 genes, is supposed to be activated through signal-pathway alteration rather than genetic mutations. | Evasion of immunosurveillance due to coexpression of PD-L1 and PD-L2 in cancer cells | JQ-1, I-BET-762 | [330] |
| BrCa | Basal-like BrCa (BLBC) | HCC1806, HCC1937 cells | — | KLF5 (Krüpple-like factor 5) | KLF5 is overexpressed in BLBC and is known to promote breast cancer cell proliferation and survival | — | Overexpression of SE-regulated KLF5 | Cell cycle, cell proliferation, migration | JQ1, THZ1, compound 870 (BRD4 inhibitor) | [331] |
| Digestive system tumours | ||||||||||
| Colorectal carcinoma (CRC) | CRC with rs6854845 G>T SNP | FHC, HCT-116, SW-480 cells with SNP rs685484 introduced by CRISPR–cas9 | — | CXCLs, EPGN, EREG | — | — | rs6854845 mutation affects SE 3D structure | Inflammatory response, cell proliferation | — | [332] |
| CRC | — | HCT-116, SW-1116 cells, 41 primary tumour and adjacent normal tissue samples from patients (TCGA cohort) | — | MYC, RNF43, GPRC5A | Oncogenes | FOXQ1, HNF4A, PPARG show regulatory effect on RNF43 and GPRC5A | TFs upregulated in cancer may change local CpG methylation levels via binding to DNA and establish new SE at corresponding loci, thereby driving oncogenes overexpression | Cell proliferation, survival, tumourigenesis | JQ1 | [333] |
| CRC | — | HCT-116 cells | — | MYC | MYC is a known oncogene, in which abnormal activity drives a rapid cell cycle. β-catenin mediates AHCTF1 recruitment to TCF4-binding site within SE and thus mediates SE anchoring to the nuclear pore through AHCTF1. In its turn, SE recruits transcriptionally active MYC allele to the nuclear pore, and that facilitates export of nuclear MYC transcripts to cytoplasm and helps them avoid rapid nuclear degradation. Increased MYC transcripts export rate results in post-transcriptional dysregulation of MYC expression | — | WNT signalling establishes oncogenic SE | Cell proliferation | BC21 treatment inhibits WNT signalling | [334] |
| CRC | — | HCT-116 cells | 100% | EphA2 | EphA2 is a member of the Eph receptor family. Its main binding ligand is ephrin A1, EphA2 acts through activating tyrosine phosphorylation of downstream substrate proteins and is degraded in normal cells after phosphorylation in contrast to cancer cells. EphA2-SE interacts with the EphA2 gene promoter and promotes transcription by recruiting FOSL2 and TCF7L2, resulting in abnormally high expression of EphA2, which activates signal pathways to promote tumour progression, which is confirmed by FOSL2 and TCF7L2 siRNA silencing analysis. CRISPR-based EphA2-SE deletion suppressed cell proliferation, migration, and invasion by decreasing the expression of EphA2 in vitro | TCF7L2, FOSL2 bind to EphA2-SE | Chromosomal rearrangements lead to convergence of SE (EphA2-SE) with a specific oncogene (EphA2). SE hijacking | Cell proliferation, invasion, migration, tumourigenesis, WNT/β-catenin pathway, PI3K/AKT pathway | Disruption of EphA2-SE, SY-5609 | [120] |
| CRC | — | 33 pairs of CRC and peritumoural samples, HCT-116, HT-29, LoVo | 100% | ACC005592.2 (SPRY4-AS1), OLFM4, MLEC, DSCAML1, HAS1 | SPRY4-AS1 has 6 transcripts, which represent distinct annotated isoforms of a single lncRNA gene. Knockdown of AC005592.2 inhibits CRC cell proliferation, invasion, and migration and induces apoptosis, whereas overexpression has the opposite effect on cancer cells. CRC progression can be attenuated by blocking WNT/β-catenin signalling via OLFM4 (a secreted glycoprotein), negative regulation | — | CRC-associated ACC005592.2 SE-lncRNA | Cell proliferation, invasion, migration, evasion of immunosurveillance | SPRY4-AS1-SE siRNAs | [192] |
| CRC | — | HCT-116, DLD1 cells | 100% | lncRNA-LINC00857 | HSF1 can stimulate acetyltransferase P300-mediated SE activity to facilitate LINC00857 expression, contributing to SLC1A5/ASCT2-mediated glutamine transport (through the so-called LINC00857–ANXA11 axis). Other enhancer-mediated lncRNAs are closely associated with HSF1 in CRC | HSF1, a conserved TF protecting cells from cellular stress responses, may act as a scaffold for other transcriptional regulators | HSF1-associated SE-lncRNA. TFs increase in expression and recruitment, stimulating SE activity | Cell metabolism, proliferation, migration, reprogramming | HSF-siRNAs | [191] |
| CRC | — | HCT116, RKO, HT29, SW620 SW480, SW48, HCT15 cells | — | HOXB8, MYC | HOXB8 is the most significantly upregulated HOXB gene in CRC and it is consistently overexpressed in all the pathological subtypes of colorectal tumours. HOXB8 with BACH1 co-occupies TSS of the BACH1 gene and activates BACH1 transcription, which leads to the activation of metastasis-modulating genes (HMOX1, FTH1, PSAP, SOCS2) regulated by BACH1 and HOXB8 as cofactors within the same complex | MYC | Unknown. Supposed mechanism: MYC activation due to the gain of SE promotes initiation of other transcriptional cascades driven by MYC binding SEs | Cell migration, invasion | JQ1, iBET-762 | [163] |
| CRC | All four consensus molecular subtypes (CMS) [The consensus molecular subtypes of colorectalcancer|Nature Medicine] | 15 primary samples (3 CMS1, 4 CMS2, 3 CMS3, 4 CMS4, 2 with unknown molecular subtype) CRC organoids, cytokine-stimulated HT-29 cells, xenograft models | 80% | PDZK1IP1 | PDZK1IP1 is suggested to increase pentose phosphate pathway activity and thus enhance cancer cells’ glucose absorption and reductive capacity, which enables tumour growth under oxidative stress | STAT3, NF-κB | Inflammation cytokines and tumour microenvironment are supposed to reprogramme epigenetic landscape of CRC at CTCF sites, often flanking SEs | Cell proliferation, tumourigenesis | — | [208] |
| CRC | — | HT29, LoVo, HCT15, HCT116, SW620, COLO205 cells, 72 paired primary normal colon and CRC samples | — | ETS2 | ETS2 is a TF, which expression has been shown to be significantly upregulated in primary CRC tissues. It is speculated that SE-addicted ETS2 upregulation promotes transcription of ETS2, downstream genes that manage inflammation, and thus enhances predisposition to inflammatory bowel disease (IBD), CRC-predisposed disease, and CRC emergence | MECOM (MDS1 and EVI1 complex locus) | SNP rs2836754 T/C The risk SNP for IBD located at noncoding region, where ETS2-SE was predicted. MECOM prefers binding at T allele over C, thus, genetic variant probably underlies disease-specific ETS2-SE activation | Chronic inflammation (hypothesis) | — | [125] |
| CRC | — | LoVo, caco-2, HT29, SW620, SW480, HCT116, RKO cell lines; 38 primary tumour samples | — | IL-20RA | Relationship between IL20RA and CRC remains unclear. In the study, IL-20RA is significantly increased in tumour tissues in comparison with normal tissues. Based on DEGs found via RNA-seq in LoVo cells with and without IL-20RA knockdown, authors suggested that IL-20RA can regulate MAPK pathway | — | — | Cell proliferation, migration, invasion, EMT, evasion of immunosurveillance (probably through IL-6-Jak-STAT3 positive and immune response negative regulation); angiogenesis | JQ1, iBET-151 | [225] |
| CRC | — | Primary samples, HT-29, HCT-116, SW480, LoVo, FHC (immortalised colon epithelial cell line) cells | 71.4% | RP11-569A11.1 (SE-LncRNA) | Downregulation of SE-LncRNA, RP11-569A11.1, leads to tumour progression because it regulates IFIT2, a tumour suppressor | — | Downregulation of SE-LncRNA, RP11-569A11.1 | IFN, TNFa, NF-kB pathways, DNA methylation, cell proliferation, migration, evasion of immunosurveillance | — | [186] |
| CRC (liver metastasis) | — | ChIP-seq: SW620, HCT116 cells with and without metastatic capabilities respectively. Western blotting: HCT116, HCT8, SW620, COLO205 cells, MC38 sublines with different metastatic competence, 4 primary samples | — | CCDC137, BCL2L1 | BCL2L1 is a known pro-survival gene. BCL21 product, Bcl-XL, is reduced after SR-483 treatment, while other apoptotic proteins are not. CCDC137—predicted SE-associated oncogene, whose function in CRC remained unknown at the time of the study. It is shown to be crucial for the maintenance of cancer stem cells’ (CSCs) traits. SEs regulation of these genes was confirmed using CRISPR | — | Unknown, probably, overexpression of CDK12, elongation regulator and crucial component of SE machinery. SEs formation | Metastasis, cell proliferation, migration, invasion, survival, stemness | SR-4835, selective inhibitor of CDK12 | [153] |
| CRC (liver metastasis) | — | Primary samples, KM12SM, V410, V457, V576, V784, V855, V866 | 64% of primary samples | Integrins, S100A family, cadherins, filamins, collagens, HAS, laminins, TGF-beta family, Wnt family, targets of Wnt pathway | — | HNF-1α (a key regulator), AP1, ETS, FOX, and KLF families | HNF1-alpha upregulates transcription-changing SE landscapes and must be a key regulator in liver metastasis | Wnt pathway, TGF-beta pathway, EMT | — | [335] |
| Gastric (stomach) adenocarcinoma (STAD) | — | 11 primary samples, AGS, MKN45 cells | — | TM4SF1-AS1 (representative lncRNA) | SE-associated lncRNA TM4SF1-AS1 suppresses T-cell-mediated immune killing | — | — | Immunosurveillance, cancer microRNAs, cellular senescence, protein folding in endoplasmic reticulum, cell cycle | — | [188] |
| STAD | EBV, CIN (chromosomal instability), GS (genomically stable) STAD subtypes Based on histology:
| OCUM-1, SNU16, FU97, KATOIII, MKN7, NCC-59, RERF-GC-1B, YCC-21, YCC-22, YCC-3, YCC-7 cells, 19 primary (1 EBV, 10 GS, 8 GIN) samples | 53% | CLDN4 locus, ELF3 locus, CCAT1, CDH17, SMURF1, LINC00299 | CLDN4 locus genes are associated with SEs and are found to be gained when compared to normal tissue in multiple patients. CLDN4 expression is associated with PC progression and prognosis. ELF3 is predicted as a cancer gene in a number of malignant neoplasms. It is shown to be downregulated after CRISPR–Cas9 inhibition of enhancer elements within predicted SE in the study. CCAT1, CDH17 are known to be overexpressed in STAD. SMURF1, LINC00299 are suggested to be novel genes associated with STAD | CDX2 and HNF4a co-occupy SE | Unknown. But CDX2 and HNF4a co-occupancy is required for SEs maintenance in STAD | Cell invasion, angiogenesis, evasion of immunosurveillance | — | [138] |
| Pancreatic ductal adenocarcinoma (PDAC) | — | Panc-1 cells | 100% | EphA2 | EphA2 is a member of the Eph receptor family. Its main binding ligand is ephrin A1; EphA2 acts through activating tyrosine phosphorylation of downstream substrate proteins and is degraded in normal cells after phosphorylation in contrast to cancer cells. EphA2-SE interacts with the EphA2 gene promoter and promotes transcription by recruiting FOSL2 and TCF7L2, resulting in abnormally high expression of EphA2, which activates signal pathways to promote tumour progression, which is confirmed by FOSL2 and TCF7L2 siRNA silencing analysis. CRISPR-based EphA2-SE deletion suppressed cell proliferation, migration, and invasion by decreasing the expression of EphA2 in vitro | TCF7L2, FOSL2 bind to EphA2-SE | Chromosomal rearrangements lead to convergence of SE (EphA2-SE) with a specific oncogene (EphA2). SE hijacking | Cell proliferation, cell invasion, cell migration, tumourigenesis, WNT/β-catenin pathway, PI3K/AKT pathway | Disruption of EphA2-SE, SY-5609 | [120] |
| PDAC | Adenosquamous carcinoma of the pancreas (ASCP) | Primary ASCP samples in the recruiting clinical trial https://classic.clinicaltrials.gov/ct2/show/NCT04896073 accessed on 10 September 2023 | — | c-MYC | Minnelide is a water-soluble prodrug (14–0-phosponooymethyl triptolide disodium salt) of the Chinese medicinal herb triptolide. Inhibition by Minnelide of the XPB subunit of the TFIIH transcription complex leads to disruption of the SE activity | c-MYC, XPB, CDK7, TFIIH, BRD4 | Disruption of TAD and sub-TAD boundaries (chromatin looping) | Cell proliferation, transdifferentiation | Minnelide™, triptolide | [110,142] |
| PDAC | — | MIA PaCa-2, PANC-1, P4057, PSN1, AsPC1, Capan-1, BxPC-3, PA-TU8902, PA-TU8988S, PANC-1, CFPAC-1 cells, CAF primary samples (CW5, CAF08, CW1, B010A, B010C, B009B, BF1) | — | POLR2E, PARK7, MYC, COL1A1, COL1A2, TGFBI | Genes related to transcription and extracellular matrix. PARK7 and MYC have both been reported previously to be associated with PDAC. COL1A1, COL1A2, TGFBI genes have been shown to be abundant in advanced-stage PDAC | — | — | Transcription regulation, apoptosis and immune function, extracellular matrix organisation, angiogenesis, and hypoxia (for all SE-associated genes) | Triptolide (TPL). TPL inhibits XPB subunits of the TFIIH complex | [142] |
| PDAC | — | PANC-1, CFPAC-1 cells | — | TGFBR2 | This work investigates the role of SEs in the TGF-β signalling regulation. from TGF-β family ligands and receptors are examined in this study. TGFRB2, one of TGF-β receptors, activates TGFRB1 through phosphorylation, which phosphorylates Smad2 and Smad3 | — | — | Cell migration, EMT promotion via TGF-β pathway | JQ1 | [336] |
| Gastrointestinal stromal tumours (GIST) | Succinate dehydrogenase deficient (SDH-deficient) GIST | — | — | FGF3/4, KIT | — | — | Hyper-methylation of allele-specific CTCF sites caused by SDH lesion leads to CTCF occlusion and disruption of insulated neighbourhood boundaries, followed by aberrant interaction between oncogene FGF3/FGF4, promoters and ANO1-SE | Driving tumour proliferation through RTK pathway | BGJ-398, a potent and selective inhibitor of FGFR1-4 and sunitinib (potent activity against KIT) | [337] |
| Oesophageal squamous cell carcinoma (ESCC) | — | KYSE70, KYSE140, KYSE150, KYSE450, KYSE510, TE3, TE7, TT cells | 100% | MFI2-AS1, HOTAIR, XIST, SNHG5, SNHG15, SNHG7, SNHG1, SNHG14, SNHG6, SNHG12, SNHG10, LINC00094, LINC00338, LINC00205, ANAPC10-DLEU2 | KLF5 is highly expressed in multiple cancer types. KLF5 is also able to occupy the lncRNA-RP1 promoter to enhance RP1 expression, which plays an oncogenic role in breast cancer | TCF3, KLF5 might cooperatively regulate expression of LINC00094 through activation of its SE and promoter | Competing endogenous RNAs (ceRNAs), including ce-lncRNAs, chromosomal rearrangements leading to convergence of SE with specific oncogenes. TFs increase in expression and overexpression, leading to pathological gene activation | Cell growth, survival (clonogenicity), proliferation, migration | THZ1 | [196] |
| ESCC | — | TE7, TE5, TT, KYSE510, KYSE140, cells | 60% | lncRNA-LINC01503 | qRT-PCR of 113 ESCC and matched nontumour oesophageal tissues reveal significant LINC01503 overexpression in cancer, which correlates with shorter overall survival and disease-free survival. LINC01503 silencing via siRNA demonstrates the requirement of this lncRNA for ESCC cell proliferation in this study. LINC01503 interacts with EBP-1 and interrupts EBP-1 interaction with the p85 subunit of PI3K, also LINC01503 reduces ERK2 dephosphorylation by blocking its interaction with DUSP6. Thereby, LINC01503 regulates PI3K/Akt and ERK/MAPK pathways | ΔNp63 | TP63 genomic amplification TP63, previously known to be amplified in ESCC, establishes and maintains SEs activity | Cell proliferation, migration, invasion through activation of Akt and MAPK pathways | — | [178] |
| ESCC | — | KYSE510, TE-7 cells | — | RUNX1, YAP1, DNAJB1, PAK4 | These genes are shown to be important for proliferation and to be significantly decreased after THZ1 treatment in both cell lines. Also, SREBF2 is shown to be an SE-associated gene, important for growth but only in KYSE510. PAK4 is known to be involved in cancer progression by activating RAF/MEK/ERK and PI3K/AKT pathways | — | — | Cell proliferation | THZ1, KPT-9274 (small-molecule inhibitor if SE-associated gene PAK4) | [248] |
| ESCC | — | Primary samples, KYSE180, TE6 cells | >30% of ESCC | KRT14, FAT2, PTHLH | — | ΔNp63 | BRDT is uniquely expressed in a group of ESCC. BRDT occupies some ΔNp63-dependent super-enhancers to modulate cancerogenic gene expression. | Cell migration | MZ1 degrader (proteolysis targeting chimaera) | [338] |
| ESCC | — | 8 ESCC cell lines, TE5, TT, KYSE140 cells | Strong expression of ALDH3A1 protein in ESCC tumours (54.5%) | TP63, SOX2, KLF5, ALDH3A1 | TP63, SOX2 and KLF5 are the master TFs, which form, drive, and maintain an ESCC-specific core regulatory circuitry program. Other 19 TP63/SOX2/KLF5-interacting TFs might play roles in transcription repression (DR1, HIC2, CUX1), chromatin modification and remodelling (ARID2, ZZZ3, HMGA1, HMGA2), and responding to specific stimuli (STAT1, STAT3). ALDH3A1 is defined as a novel and crucial target of core regulatory circuitry, also its expression was higher in ESCC compared to EAC samples and wasn’t detected in nonmalignant oesophagus epithelium. Authors suggest that ALDH3A1, isoenzyme of aldehyde dehydrogenase superfamily, may be biologically relevant in development of alcohol-associated ESCC | TP63, SOX2, and KLF5 | TP63, SOX2, and KLF5 trio-bind SE regions, enhance its accessibility, and cooperatively regulate SE-associated gene transcription | Cell viability, clonogenic capacity, cell growth, cell proliferation | ARV-771 and romidepsin combination (inhibited BET and HDAC respectively) | [169] |
| Hepatocellular carcinoma (HCC) | NAFLD-HCC | 12 primary liver tumour and paired nontumour samples, LO2, HepG2 cells | 100% | SIRT7, STK25, HIF1A, DNMT3B, EED, HDAC10 | The concomitant SE activation of SIRT7 with the PRC1/2 genes such as CBX3, CBX4, CBX8, and EED in the primary NAFLD-associated HCCs suggests their concerted epigenetic action for gene regulation of lipid, amino acid, and carbohydrate metabolism, in addition to oxidative stress and immune regulators. SIRT7 may cooperate with EZH2, the enzymatic subunit of PRC2, for gene silencing, and SIRT7-SE facilitates gene transcription by enhancing co-occupancy of C/EBP and BRD4. The study also discovered frequent SE activation of NAD-dependent SIRTs | PRC1/2, EGR2, IRF8, SOCS3, ZBTB16 | Somatically gained SIRT7-SE, mutations in DNA that form binding sites for new TFs, leading to the formation of SE. Genome-wide reorganisation/redistribution of the enhancer and SE landscape. Deactivation of SE associated with tumour suppressor gene | Cell metabolism, growth, cancer phenotypes, angiogenesis, evasion of immunosurveillance | JQ1 | [194] |
| HCC | — | HepG2, SK-Hep-1, HCC cells | — | LINC01004 | High expression of E2F1 leads to high expression of LINC01004, which contributes to tumour development. LINC01004 is among SE-associated lncRNAs which are significantly negatively correlated with HCC patients’ prognosis | E2F1 | Overexpression of E2F1 leads to high expression of LINC01004, which contributes to tumour development | Cell proliferation, migration | JQ1 | [187] |
| HCC | — | HepG2 cells | — | HCCL5 (uncharacterised cytoplasmic lncRNA) | HCCL5 is a novel uncharacterised cytoplasmic lncRNA. HCCL5 accelerates EMT phenotype in HCC cells through EMT master transcription factors Snail, Slug, ZEB1, and Twist1 expression upregulation | ZEB1 (also interacts with HCCL5 promoter) | — | G1–S transition, EMT, cell growth, migration, invasion, metastasis, evasion of immunosurveillance | — | [339] |
| HCC | — | TCGA-LIHC data | — | RTKN2, HS3ST5, SQSTM1, ETV4, ACSL6 | SQSTM1 is a multifunctional protein that binds ubiquitin and regulates activation of the nuclear factor kappa-B (NF-kB) signalling pathway. ETV4 belongs to the PEA3 subfamily of the ETS transcription factor, a cancer-promoting transcription factor. HS3ST5 is a member of the heparan sulphate 3-O-sulfotransferase family. RTKN2, a novel identified Rho-GTPase effector protein, is a new Rho-GTP enzyme that regulates many cell processes, including cell survival and cycle progression. 3 genes in the 4-gene signature (RTKN2, HS3ST5, SQSTM1, ETV4) have been previously associated with HCC | — | — | Cell growth, proliferation, migration, survival, cell cycle, evasion of immunosurveillance | — | [158] |
| HCC | — | Huh7, HepG2 cells, 50 primary samples (from TCGA) | — | SPHK1, YAP1, CCND1, E2F2, EGFR, MYC | SPHK1 mediates phosphorylation of SP1, which is crucial for sphingolipid metabolism and adjusts prosurvival functions. In this study, SPHK1 is observed to be significantly upregulated in HCC compared to corresponding normal tissues, and its upregulation exhibited significant correlation with histologic grade poor, overall survival, and disease-free survival. Blockade of SPHK1-SE regions via CRISPRi reduced SPHK1 expression in HepG2 cells | — | Trans-acting factors within the SE complex, namely CDK7, BRD4, EP300, and MED showed significant overexpression in HCC, which could serve as an additional booster of SE-driven oncogene transcription | Cell proliferation, angiogenesis, motility, cell death, cell cycle, DNA repair, MAPK, Wnt, NF-κB pathways | THZ1 inhibits CDK7, CBP30, JQ1, OTX015 target EP300 and BRD4 | [143] |
| HCC | — | PLC-PRF-5 cells | — | QKI | QK1 is known to be associated with human cancer development and progression. QKI mediates alternative splicing and circRNAs formation, in this study it is shown to cause circ-0008150 and circ-0007821 formation, which adsorb miR-615-5p, targeted vimentin, and miR-381-3p, targeted Zeb1 respectively. QKI overexpression also promoted expression of other EMT-related TFs: Twist and Snail families. YY1 is highly expressed in primary HCC tumours compared to normal liver tissues | YY1 | YY1–p65–p300 complex increases QKI expression: YY1 binds to SE and promoter of QK1, while p65 binds to the QK1 promoter and p300 serves as mediator stabilising the complex | EMT, cell proliferation, migration, angiogenesis, cellular response to VEGF stimulus, NIK/NF-kB pathway | Hyperoside | [176] |
| HCC | — | HepG2, Huh7 cells (ChIP-seq), HepG2, Huh7, SK-Hep1, SNU-449 cells (AJUBA function) | — | AJUBA | AJUBA, defined as a SE-associated gene in this study, is expressed at a relatively high level in HCC cells among cells of other cancer types. The SE is confirmed using CRISPR–Cas9. Mechanism of EMT promotion: AJUBA (Ajuba LIM protein) recruits and interacts with TRAF6, which enhances the phosphorylation of Akt, thereby increasing Akt activity toward GSK-3β | TCF4, Wnt signalling pathway member | Unknown | Cell migration, invasion, EMT promotion via Akt/GSK-3β/Snail pathway | — | [325] |
References
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone Modifications at Human Enhancers Reflect Global Cell-Type-Specific Gene Expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Bulger, M.; Groudine, M. Functional and Mechanistic Diversity of Distal Transcription Enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional Regulatory Elements in the Human Genome. Annu. Rev. Genom. Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef] [PubMed]
- The FANTOM Consortium; Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; et al. An Atlas of Active Enhancers across Human Cell Types and Tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E.M. Transcription Factors: From Enhancer Binding to Developmental Control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Bruter, A.V.; Rodionova, M.D.; Varlamova, E.A.; Shtil, A.A. Super-Enhancers in the Regulation of Gene Transcription: General Aspects and Antitumor Targets. Acta Naturae 2021, 13, 4–15. [Google Scholar] [CrossRef]
- Wang, X.; Cairns, M.J.; Yan, J. Super-Enhancers in Transcriptional Regulation and Genome Organization. Nucleic Acids Res. 2019, 47, 11481–11496. [Google Scholar] [CrossRef]
- Lowrey, P.L.; Takahashi, J.S. Mammalian Circadian Biology: Elucidating Genome-Wide Levels of Temporal Organization. Annu. Rev. Genom. Hum. Genet. 2004, 5, 407–441. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, Y. Enhancer and Super-enhancer: Positive Regulators in Gene Transcription. Anim. Models Exp. Med. 2018, 1, 169–179. [Google Scholar] [CrossRef]
- Stower, H. Super Enhancers. Nat. Rev. Genet. 2013, 14, 367. [Google Scholar] [CrossRef]
- Tuan, D.; Solomon, W.; Li, Q.; London, I.M. The “Beta-like-Globin” Gene Domain in Human Erythroid Cells. Proc. Natl. Acad. Sci. USA 1985, 82, 6384–6388. [Google Scholar] [CrossRef]
- Li, Q.; Peterson, K.R.; Fang, X.; Stamatoyannopoulos, G. Locus Control Regions. Blood 2002, 100, 3077–3086. [Google Scholar] [CrossRef]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; Van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and Cohesin Connect Gene Expression and Chromatin Architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.M.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and Characterization of Super-Enhancer-Associated Dependencies in Diffuse Large B Cell Lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Young, R.A.; Sharp, P.A. Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell 2017, 168, 1000–1014.e15. [Google Scholar] [CrossRef]
- Pérez-Rico, Y.A.; Boeva, V.; Mallory, A.C.; Bitetti, A.; Majello, S.; Barillot, E.; Shkumatava, A. Comparative Analyses of Super-Enhancers Reveal Conserved Elements in Vertebrate Genomes. Genome Res. 2017, 27, 259–268. [Google Scholar] [CrossRef]
- Goh, K.Y.; Inoue, T. A Large Transcribed Enhancer Region Regulates C. elegans Bed-3 and the Development of Egg Laying Muscles. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2018, 1861, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, M.; Bishop, J.; Teng, Y.; Cao, Y.; Beall, B.D.; Li, S.; Liu, T.; Fang, Q.; Fang, C.; et al. Identification and Functional Validation of Super-Enhancers in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2022, 119, e2215328119. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chen, D.; Schumacher, J.; Durantini, D.; Engelhorn, J.; Chen, M.; Carles, C.C.; Kaufmann, K. Dynamic Control of Enhancer Activity Drives Stage-Specific Gene Expression during Flower Morphogenesis. Nat. Commun. 2019, 10, 1705. [Google Scholar] [CrossRef] [PubMed]
- Kainth, A.S.; Chowdhary, S.; Pincus, D.; Gross, D.S. Primordial Super-Enhancers: Heat Shock-Induced Chromatin Organization in Yeast. Trends Cell Biol. 2021, 31, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.C.J.; Stitzel, M.L.; Taylor, D.L.; Orozco, J.M.; Erdos, M.R.; Akiyama, J.A.; Van Bueren, K.L.; Chines, P.S.; Narisu, N.; NISC Comparative Sequencing Program; et al. Chromatin Stretch Enhancer States Drive Cell-Specific Gene Regulation and Harbor Human Disease Risk Variants. Proc. Natl. Acad. Sci. USA 2013, 110, 17921–17926. [Google Scholar] [CrossRef]
- Khan, A.; Mathelier, A.; Zhang, X. Super-Enhancers Are Transcriptionally More Active and Cell Type-Specific than Stretch Enhancers. Epigenetics 2018, 13, 910–922. [Google Scholar] [CrossRef] [PubMed]
- Niemitz, E. Super Stretchy Enhancers. Nat. Genet. 2013, 45, 1417. [Google Scholar] [CrossRef]
- Jia, Q.; Chen, S.; Tan, Y.; Li, Y.; Tang, F. Oncogenic Super-Enhancer Formation in Tumorigenesis and Its Molecular Mechanisms. Exp. Mol. Med. 2020, 52, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Rajderkar, S.; Barozzi, I.; Zhu, Y.; Hu, R.; Zhang, Y.; Li, B.; Alcaina Caro, A.; Fukuda-Yuzawa, Y.; Kelman, G.; Akeza, A.; et al. Topologically Associating Domain Boundaries Are Required for Normal Genome Function. Commun. Biol. 2023, 6, 435. [Google Scholar] [CrossRef]
- Gong, Y.; Lazaris, C.; Sakellaropoulos, T.; Lozano, A.; Kambadur, P.; Ntziachristos, P.; Aifantis, I.; Tsirigos, A. Stratification of TAD Boundaries Reveals Preferential Insulation of Super-Enhancers by Strong Boundaries. Nat. Commun. 2018, 9, 542. [Google Scholar] [CrossRef]
- Cubeñas-Potts, C.; Corces, V.G. Topologically Associating Domains: An Invariant Framework or a Dynamic Scaffold? Nucleus 2015, 6, 430–434. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, T.I.; Zhao, K.; et al. Control of Cell Identity Genes Occurs in Insulated Neighborhoods in Mammalian Chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef]
- Zeng, X.; Lee, H.K.; Wang, C.; Achikeh, P.; Liu, C.; Hennighausen, L. The Interdependence of Mammary-Specific Super-Enhancers and Their Native Promoters Facilitates Gene Activation during Pregnancy. Exp. Mol. Med. 2020, 52, 682–690. [Google Scholar] [CrossRef]
- Vos, E.S.M.; Valdes-Quezada, C.; Huang, Y.; Allahyar, A.; Verstegen, M.J.A.M.; Felder, A.-K.; Van Der Vegt, F.; Uijttewaal, E.C.H.; Krijger, P.H.L.; De Laat, W. Interplay between CTCF Boundaries and a Super Enhancer Controls Cohesin Extrusion Trajectories and Gene Expression. Mol. Cell 2021, 81, 3082–3095.e6. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, S.D.; Davidson, S.; Shchuka, V.M.; Singh, G.; Malek-Gilani, N.; Langroudi, L.; Martchenko, A.; So, V.; Macpherson, N.N.; Mitchell, J.A. Enhancers and Super-Enhancers Have an Equivalent Regulatory Role in Embryonic Stem Cells through Regulation of Single or Multiple Genes. Genome Res. 2017, 27, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, M.; Backer, S.; Auradé, F.; Wong, M.M.-K.; Wurmser, M.; Pierre, R.; Langa, F.; Do Cruzeiro, M.; Schmitt, A.; Concordet, J.-P.; et al. A Fast Myosin Super Enhancer Dictates Muscle Fiber Phenotype through Competitive Interactions with Myosin Genes. Nat. Commun. 2022, 13, 1039. [Google Scholar] [CrossRef] [PubMed]
- Man, J.C.K.; Van Duijvenboden, K.; Krijger, P.H.L.; Hooijkaas, I.B.; Van Der Made, I.; De Gier-de Vries, C.; Wakker, V.; Creemers, E.E.; De Laat, W.; Boukens, B.J.; et al. Genetic Dissection of a Super Enhancer Controlling the Nppa-Nppb Cluster in the Heart. Circ. Res. 2021, 128, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Oudelaar, A.M.; Harrold, C.L.; Hanssen, L.L.P.; Telenius, J.M.; Higgs, D.R.; Hughes, J.R. A Revised Model for Promoter Competition Based on Multi-Way Chromatin Interactions at the α-Globin Locus. Nat. Commun. 2019, 10, 5412. [Google Scholar] [CrossRef] [PubMed]
- Wijgerde, M.; Grosveld, F.; Fraser, P. Transcription Complex Stability and Chromatin Dynamics in Vivo. Nature 1995, 377, 209–213. [Google Scholar] [CrossRef]
- Bartman, C.R.; Hsu, S.C.; Hsiung, C.C.-S.; Raj, A.; Blobel, G.A. Enhancer Regulation of Transcriptional Bursting Parameters Revealed by Forced Chromatin Looping. Mol. Cell 2016, 62, 237–247. [Google Scholar] [CrossRef]
- Allahyar, A.; Vermeulen, C.; Bouwman, B.A.M.; Krijger, P.H.L.; Verstegen, M.J.A.M.; Geeven, G.; Van Kranenburg, M.; Pieterse, M.; Straver, R.; Haarhuis, J.H.I.; et al. Enhancer Hubs and Loop Collisions Identified from Single-Allele Topologies. Nat. Genet. 2018, 50, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Braikia, F.-Z.; Oudinet, C.; Dauba, A.; Khamlichi, A.A. Two Modes of Cis-Activation of Switch Transcription by the IgH Superenhancer. Proc. Natl. Acad. Sci. USA 2019, 116, 14708–14713. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chng, W.-J.; Zhou, J. Super-Enhancers: Critical Roles and Therapeutic Targets in Hematologic Malignancies. J. Hematol. Oncol. 2019, 12, 77. [Google Scholar] [CrossRef]
- Tang, F.; Yang, Z.; Tan, Y.; Li, Y. Super-Enhancer Function and Its Application in Cancer Targeted Therapy. Npj Precis. Onc. 2020, 4, 2. [Google Scholar] [CrossRef]
- Huang, J.; Li, K.; Cai, W.; Liu, X.; Zhang, Y.; Orkin, S.H.; Xu, J.; Yuan, G.-C. Dissecting Super-Enhancer Hierarchy Based on Chromatin Interactions. Nat. Commun. 2018, 9, 943. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.; Hughes, J.R.; Babbs, C.; Davies, J.O.J.; Graham, B.J.; Hanssen, L.L.P.; Kassouf, M.T.; Oudelaar, A.M.; Sharpe, J.A.; Suciu, M.C.; et al. Genetic Dissection of the α-Globin Super-Enhancer In Vivo. Nat. Genet. 2016, 48, 895–903. [Google Scholar] [CrossRef]
- Honnell, V.; Norrie, J.L.; Patel, A.G.; Ramirez, C.; Zhang, J.; Lai, Y.-H.; Wan, S.; Dyer, M.A. Identification of a Modular Super-Enhancer in Murine Retinal Development. Nat. Commun. 2022, 13, 253. [Google Scholar] [CrossRef]
- Blayney, J.W.; Francis, H.; Rampasekova, A.; Camellato, B.; Mitchell, L.; Stolper, R.; Cornell, L.; Babbs, C.; Boeke, J.D.; Higgs, D.R.; et al. Super-Enhancers Include Classical Enhancers and Facilitators to Fully Activate Gene Expression. Cell 2023, 186, 5826–5839.e18. [Google Scholar] [CrossRef]
- Bell, E.; Curry, E.W.; Megchelenbrink, W.; Jouneau, L.; Brochard, V.; Tomaz, R.A.; Mau, K.H.T.; Atlasi, Y.; De Souza, R.A.; Marks, H.; et al. Dynamic CpG Methylation Delineates Subregions within Super-Enhancers Selectively Decommissioned at the Exit from Naive Pluripotency. Nat. Commun. 2020, 11, 1112. [Google Scholar] [CrossRef]
- Dukler, N.; Gulko, B.; Huang, Y.-F.; Siepel, A. Is a Super-Enhancer Greater than the Sum of Its Parts? Nat. Genet. 2017, 49, 2–3. [Google Scholar] [CrossRef]
- Shin, H.Y.; Willi, M.; Yoo, K.H.; Zeng, X.; Wang, C.; Metser, G.; Hennighausen, L. Hierarchy within the Mammary STAT5-Driven Wap Super-Enhancer. Nat. Genet. 2016, 48, 904–911. [Google Scholar] [CrossRef]
- Giles, K.E.; Woolnough, J.L.; Atwood, B. ncRNA Function in Chromatin Organization. In Epigenetic Gene Expression and Regulation; Elsevier: Amsterdam, The Netherlands, 2015; pp. 117–148. ISBN 978-0-12-799958-6. [Google Scholar]
- Bose, D.A.; Donahue, G.; Reinberg, D.; Shiekhattar, R.; Bonasio, R.; Berger, S.L. RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 2017, 168, 135–149.e22. [Google Scholar] [CrossRef] [PubMed]
- Gorbovytska, V.; Kim, S.-K.; Kuybu, F.; Götze, M.; Um, D.; Kang, K.; Pittroff, A.; Brennecke, T.; Schneider, L.-M.; Leitner, A.; et al. Enhancer RNAs Stimulate Pol II Pause Release by Harnessing Multivalent Interactions to NELF. Nat. Commun. 2022, 13, 2429. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as Non-Coding RNA Transcription Units: Recent Insights and Future Perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef]
- Sigova, A.A.; Abraham, B.J.; Ji, X.; Molinie, B.; Hannett, N.M.; Guo, Y.E.; Jangi, M.; Giallourakis, C.C.; Sharp, P.A.; Young, R.A. Transcription Factor Trapping by RNA in Gene Regulatory Elements. Science 2015, 350, 978–981. [Google Scholar] [CrossRef]
- Rahnamoun, H.; Lee, J.; Sun, Z.; Lu, H.; Ramsey, K.M.; Komives, E.A.; Lauberth, S.M. RNAs Interact with BRD4 to Promote Enhanced Chromatin Engagement and Transcription Activation. Nat. Struct. Mol. Biol. 2018, 25, 687–697. [Google Scholar] [CrossRef]
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs Associate with Mediator to Enhance Chromatin Architecture and Transcription. Nature 2013, 494, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-L.; Fei, T.; Chen, Y.; Li, T.; Gao, Y.; Wang, X.; Sun, T.; Sweeney, C.J.; Lee, G.-S.M.; Chen, S.; et al. Enhancer RNAs Participate in Androgen Receptor-Driven Looping That Selectively Enhances Gene Activation. Proc. Natl. Acad. Sci. USA 2014, 111, 7319–7324. [Google Scholar] [CrossRef]
- Li, W.; Notani, D.; Ma, Q.; Tanasa, B.; Nunez, E.; Chen, A.Y.; Merkurjev, D.; Zhang, J.; Ohgi, K.; Song, X.; et al. Functional Roles of Enhancer RNAs for Oestrogen-Dependent Transcriptional Activation. Nature 2013, 498, 516–520. [Google Scholar] [CrossRef]
- Tan, Y.; Li, Y.; Tang, F. Oncogenic seRNA Functional Activation: A Novel Mechanism of Tumorigenesis. Mol. Cancer 2020, 19, 74. [Google Scholar] [CrossRef]
- Wu, M.; Shen, J. From Super-Enhancer Non-Coding RNA to Immune Checkpoint: Frameworks to Functions. Front. Oncol. 2019, 9, 1307. [Google Scholar] [CrossRef]
- Choi, J.; Lysakovskaia, K.; Stik, G.; Demel, C.; Söding, J.; Tian, T.V.; Graf, T.; Cramer, P. Evidence for Additive and Synergistic Action of Mammalian Enhancers during Cell Fate Determination. eLife 2021, 10, e65381. [Google Scholar] [CrossRef]
- Pefanis, E.; Wang, J.; Rothschild, G.; Lim, J.; Kazadi, D.; Sun, J.; Federation, A.; Chao, J.; Elliott, O.; Liu, Z.-P.; et al. RNA Exosome-Regulated Long Non-Coding RNA Transcription Controls Super-Enhancer Activity. Cell 2015, 161, 774–789. [Google Scholar] [CrossRef]
- Ishov, A.M.; Gurumurthy, A.; Bungert, J. Coordination of Transcription, Processing, and Export of Highly Expressed RNAs by Distinct Biomolecular Condensates. Emerg. Top. Life Sci. 2020, 4, 281–291. [Google Scholar] [CrossRef]
- Gurumurthy, A.; Shen, Y.; Gunn, E.M.; Bungert, J. Phase Separation and Transcription Regulation: Are Super-Enhancers and Locus Control Regions Primary Sites of Transcription Complex Assembly? BioEssays 2019, 41, 1800164. [Google Scholar] [CrossRef]
- Gurumurthy, A.; Yu, D.T.; Stees, J.R.; Chamales, P.; Gavrilova, E.; Wassel, P.; Li, L.; Stribling, D.; Chen, J.; Brackett, M.; et al. Super-Enhancer Mediated Regulation of Adult β-Globin Gene Expression: The Role of eRNA and Integrator. Nucleic Acids Res. 2021, 49, 1383–1396. [Google Scholar] [CrossRef]
- Kvon, E.Z.; Waymack, R.; Gad, M.; Wunderlich, Z. Enhancer Redundancy in Development and Disease. Nat. Rev. Genet. 2021, 22, 324–336. [Google Scholar] [CrossRef]
- Frankel, N.; Davis, G.K.; Vargas, D.; Wang, S.; Payre, F.; Stern, D.L. Phenotypic Robustness Conferred by Apparently Redundant Transcriptional Enhancers. Nature 2010, 466, 490–493. [Google Scholar] [CrossRef]
- Osterwalder, M.; Barozzi, I.; Tissières, V.; Fukuda-Yuzawa, Y.; Mannion, B.J.; Afzal, S.Y.; Lee, E.A.; Zhu, Y.; Plajzer-Frick, I.; Pickle, C.S.; et al. Enhancer Redundancy Provides Phenotypic Robustness in Mammalian Development. Nature 2018, 554, 239–243. [Google Scholar] [CrossRef]
- Tobias, I.C.; Abatti, L.E.; Moorthy, S.D.; Mullany, S.; Taylor, T.; Khader, N.; Filice, M.A.; Mitchell, J.A. Transcriptional Enhancers: From Prediction to Functional Assessment on a Genome-Wide Scale. Genome 2021, 64, 426–448. [Google Scholar] [CrossRef]
- Quang, D.X.; Erdos, M.R.; Parker, S.C.J.; Collins, F.S. Motif Signatures in Stretch Enhancers Are Enriched for Disease-Associated Genetic Variants. Epigenetics Chromatin 2015, 8, 23. [Google Scholar] [CrossRef]
- Zehnder, T.; Benner, P.; Vingron, M. Predicting Enhancers in Mammalian Genomes Using Supervised Hidden Markov Models. BMC Bioinformatics 2019, 20, 157. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zhang, X. Integrative Modeling Reveals Key Chromatin and Sequence Signatures Predicting Super-Enhancers. Sci. Rep. 2019, 9, 2877. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Yue, W.; Zhu, Z.; Wu, X.; Yu, S.; Shen, Q.; Pan, Q.; Xu, W.; Zhang, R.; et al. Super-Enhancers Conserved within Placental Mammals Maintain Stem Cell Pluripotency. Proc. Natl. Acad. Sci. USA 2022, 119, e2204716119. [Google Scholar] [CrossRef]
- Lin, C.Y.; Orlando, D.A.; Abraham, B.J. Rose: Rank Ordering Of Super-Enhancers. 2013. Available online: http://younglab.wi.mit.edu/super_enhancer_code.html (accessed on 1 September 2023).
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Gomez, R.L.; Woods, L.M.; Ramachandran, R.; Abou Tayoun, A.N.; Philpott, A.; Ali, F.R. Super-Enhancer Associated Core Regulatory Circuits Mediate Susceptibility to Retinoic Acid in Neuroblastoma Cells. Front. Cell Dev. Biol. 2022, 10, 943924. [Google Scholar] [CrossRef]
- Ryu, J.; Kim, H.; Yang, D.; Lee, A.J.; Jung, I. A New Class of Constitutively Active Super-Enhancers Is Associated with Fast Recovery of 3D Chromatin Loops. BMC Bioinform. 2019, 20, 127. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, B.; Shaw, T.I.; Fridley, B.L.; Duckett, D.R.; Tan, A.C.; Teng, M. Summarizing Internal Dynamics Boosts Differential Analysis and Functional Interpretation of Super Enhancers. Nucleic Acids Res. 2022, 50, 3115–3127. [Google Scholar] [CrossRef]
- Koutsi, M.A.; Pouliou, M.; Champezou, L.; Vatsellas, G.; Giannopoulou, A.-I.; Piperi, C.; Agelopoulos, M. Typical Enhancers, Super-Enhancers, and Cancers. Cancers 2022, 14, 4375. [Google Scholar] [CrossRef]
- Hamdan, F.H.; Johnsen, S.A. Super Enhancers—New Analyses and Perspectives on the Low Hanging Fruit. Transcription 2018, 9, 123–130. [Google Scholar] [CrossRef]
- Jia, R.; Gao, Y.; Guo, S.; Li, S.; Zhou, L.; Gou, C.; Huang, Y.; Fan, M.; Chen, Y. Super Enhancer Profiles Identify Key Cell Identity Genes During Differentiation From Embryonic Stem Cells to Trophoblast Stem Cells Super Enhencers in Trophoblast Differentiation. Front. Genet. 2021, 12, 762529. [Google Scholar] [CrossRef]
- Kai, Y.; Li, B.E.; Zhu, M.; Li, G.Y.; Chen, F.; Han, Y.; Cha, H.J.; Orkin, S.H.; Cai, W.; Huang, J.; et al. Mapping the Evolving Landscape of Super-Enhancers during Cell Differentiation. Genome Biol. 2021, 22, 269. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.U.; Pi, D.; Yao, S.; Nawaz, A.; Ali, F.; Ali, S. piEnPred: A Bi-Layered Discriminative Model for Enhancers and Their Subtypes via Novel Cascade Multi-Level Subset Feature Selection Algorithm. Front. Comput. Sci. 2021, 15, 156904. [Google Scholar] [CrossRef]
- Perez-Rathke, A.; Sun, Q.; Wang, B.; Boeva, V.; Shao, Z.; Liang, J. CHROMATIX: Computing the Functional Landscape of Many-Body Chromatin Interactions in Transcriptionally Active Loci from Deconvolved Single Cells. Genome Biol. 2020, 21, 13. [Google Scholar] [CrossRef]
- Pfeifer, B. Bayesian Post-Hoc Regularization of Random Forests. arXiv 2023, arXiv:2306.03702. [Google Scholar]
- Yuwen, L.; Chen, S.; Yuan, X. G2Basy: A Framework to Improve the RNN Language Model and Ease Overfitting Problem. PLoS ONE 2021, 16, e0249820. [Google Scholar] [CrossRef]
- Fiorentini, N.; Pellegrini, D.; Losa, M. Overfitting Prevention in Accident Prediction Models: Bayesian Regularization of Artificial Neural Networks. Transp. Res. Rec. J. Transp. Res. Board 2023, 2677, 1455–1470. [Google Scholar] [CrossRef]
- Wu, B.; Liu, Z.; Yuan, Z.; Sun, G.; Wu, C. Reducing Overfitting in Deep Convolutional Neural Networks Using Redundancy Regularizer. In Artificial Neural Networks and Machine Learning—ICANN 2017; Lintas, A., Rovetta, S., Verschure, P.F.M.J., Villa, A.E.P., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2017; Volume 10614, pp. 49–55. ISBN 978-3-319-68611-0. [Google Scholar]
- Han, H.; Jiang, X. Overcome Support Vector Machine Diagnosis Overfitting. Cancer Inform. 2014, 13s1, CIN.S13875. [Google Scholar] [CrossRef]
- Chung, H.-R. Computational Methods for Epigenomic Analysis. In Computational Epigenetics and Diseases; Elsevier: Amsterdam, The Netherlands, 2019; pp. 11–22. ISBN 978-0-12-814513-5. [Google Scholar]
- Yang, B.; Peng, Y.; Leung, H.C.-M.; Yiu, S.-M.; Chen, J.-C.; Chin, F.Y.-L. Unsupervised Binning of Environmental Genomic Fragments Based on an Error Robust Selection of L-Mers. BMC Bioinform. 2010, 11, S5. [Google Scholar] [CrossRef]
- Khan, A.; Zhang, X. dbSUPER: A Database of Super-Enhancers in Mouse and Human Genome. Nucleic Acids Res. 2016, 44, D164–D171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, C.; Zhao, J.; Zhang, Y.; Zhao, X.; Feng, C.; Zhang, G.; Zhu, J.; Wang, F.; Qian, F.; et al. SEdb 2.0: A Comprehensive Super-Enhancer Database of Human and Mouse. Nucleic Acids Res. 2023, 51, D280–D290. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, D.; Gu, Y.; Wang, C.; Zhang, M.; Lin, X.; Xing, J.; Wang, H.; Zhang, Y. SEA Version 3.0: A Comprehensive Extension and Update of the Super-Enhancer Archive. Nucleic Acids Res. 2019, 48, D198–D203. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, S.; Shang, S.; Zhang, B.; Li, S.; Wang, X.; Wang, F.; Su, J.; Wu, Q.; Liu, H.; et al. SEA: A Super-Enhancer Archive. Nucleic Acids Res. 2016, 44, D172–D179. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.-C.; Li, X.-C.; Guo, J.-C.; Zhao, J.-M.; Li, Y.-Y.; Tang, Z.-D.; Zhou, L.-W.; Zhang, J.; Bai, X.-F.; Jiang, Y.; et al. SEanalysis: A Web Tool for Super-Enhancer Associated Regulatory Analysis. Nucleic Acids Res. 2019, 47, W248–W255. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.-C.; Zhou, L.-W.; Li, Y.-Y.; Yu, Z.-M.; Li, L.-D.; Wang, Y.-Z.; Xu, M.-C.; Wang, Q.-Y.; Li, C.-Q. SEanalysis 2.0: A Comprehensive Super-Enhancer Regulatory Network Analysis Tool for Human and Mouse. Nucleic Acids Res. 2023, 51, W520–W527. [Google Scholar] [CrossRef]
- Raisner, R.; Bainer, R.; Haverty, P.M.; Benedetti, K.L.; Gascoigne, K.E. Super-Enhancer Acquisition Drives Oncogene Expression in Triple Negative Breast Cancer. PLoS ONE 2020, 15, e0235343. [Google Scholar] [CrossRef]
- Huang, H.; Hu, J.; Maryam, A.; Huang, Q.; Zhang, Y.; Ramakrishnan, S.; Li, J.; Ma, H.; Ma, V.W.S.; Cheuk, W.; et al. Defining Super-Enhancer Landscape in Triple-Negative Breast Cancer by Multiomic Profiling. Nat. Commun. 2021, 12, 2242. [Google Scholar] [CrossRef]
- Smith, C.; Touzart, A.; Simonin, M.; Tran-Quang, C.; Hypolite, G.; Latiri, M.; Andrieu, G.P.; Balducci, E.; Dourthe, M.-É.; Goyal, A.; et al. Harnessing the MYB-Dependent TAL1 5’super-Enhancer for Targeted Therapy in T-ALL. Mol. Cancer 2023, 22, 12. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, C.; Oh, J.; Choudhery, Z.; Shi, W.; Valdes, G.; Betancur, P. NSMCE2, a Novel Super-Enhancer-Regulated Gene, Is Linked to Poor Prognosis and Therapy Resistance in Breast Cancer. BMC Cancer 2022, 22, 1056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of Focally Amplified Lineage-Specific Super-Enhancers in Human Epithelial Cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-Driven MYC Enhancer Promotes T Cell Development, Transformation and Acute Lymphoblastic Leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.-S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in Acute Leukemia Maintenance and Enhancer-Mediated Myc Regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef]
- Ottema, S.; Mulet-Lazaro, R.; Erpelinck-Verschueren, C.; Van Herk, S.; Havermans, M.; Arricibita Varea, A.; Vermeulen, M.; Beverloo, H.B.; Gröschel, S.; Haferlach, T.; et al. The Leukemic Oncogene EVI1 Hijacks a MYC Super-Enhancer by CTCF-Facilitated Loops. Nat. Commun. 2021, 12, 5679. [Google Scholar] [CrossRef]
- Bian, E.; Chen, X.; Cheng, L.; Cheng, M.; Chen, Z.; Yue, X.; Zhang, Z.; Chen, J.; Sun, L.; Huang, K.; et al. Super-Enhancer-Associated TMEM44-AS1 Aggravated Glioma Progression by Forming a Positive Feedback Loop with Myc. J. Exp. Clin. Cancer Res. 2021, 40, 337. [Google Scholar] [CrossRef]
- Skorupan, N.; Ahmad, M.I.; Steinberg, S.M.; Trepel, J.B.; Cridebring, D.; Han, H.; Von Hoff, D.D.; Alewine, C. A Phase II Trial of the Super-Enhancer Inhibitor MinnelideTM in Advanced Refractory Adenosquamous Carcinoma of the Pancreas. Future Oncol. 2022, 18, 2475–2481. [Google Scholar] [CrossRef]
- Kelly, M.R.; Wisniewska, K.; Regner, M.J.; Lewis, M.W.; Perreault, A.A.; Davis, E.S.; Phanstiel, D.H.; Parker, J.S.; Franco, H.L. A Multi-Omic Dissection of Super-Enhancer Driven Oncogenic Gene Expression Programs in Ovarian Cancer. Nat. Commun. 2022, 13, 4247. [Google Scholar] [CrossRef]
- Caslini, C.; Hong, S.; Ban, Y.J.; Chen, X.S.; Ince, T.A. HDAC7 Regulates Histone 3 Lysine 27 Acetylation and Transcriptional Activity at Super-Enhancer-Associated Genes in Breast Cancer Stem Cells. Oncogene 2019, 38, 6599–6614. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef]
- Saint-André, V.; Federation, A.J.; Lin, C.Y.; Abraham, B.J.; Reddy, J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Models of Human Core Transcriptional Regulatory Circuitries. Genome Res. 2016, 26, 385–396. [Google Scholar] [CrossRef]
- Betancur, P.A.; Abraham, B.J.; Yiu, Y.Y.; Willingham, S.B.; Khameneh, F.; Zarnegar, M.; Kuo, A.H.; McKenna, K.; Kojima, Y.; Leeper, N.J.; et al. A CD47-Associated Super-Enhancer Links pro-Inflammatory Signalling to CD47 Upregulation in Breast Cancer. Nat. Commun. 2017, 8, 14802. [Google Scholar] [CrossRef]
- Ottema, S.; Mulet-Lazaro, R.; Beverloo, H.B.; Erpelinck, C.; Van Herk, S.; Van Der Helm, R.; Havermans, M.; Grob, T.; Valk, P.J.M.; Bindels, E.; et al. Atypical 3q26/MECOM Rearrangements Genocopy Inv(3)/t(3;3) in Acute Myeloid Leukemia. Blood 2020, 136, 224–234. [Google Scholar] [CrossRef]
- Gröschel, S.; Sanders, M.A.; Hoogenboezem, R.; de Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; van der Velden, V.H.J.; Havermans, M.; Avellino, R.; van Lom, K.; et al. A Single Oncogenic Enhancer Rearrangement Causes Concomitant EVI1 and GATA2 Deregulation in Leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, P.; Galbo, P.M.; Zhou, X.; Aryal, S.; Qiu, S.; Zhang, H.; Zhou, Y.; Li, C.; Zheng, D.; et al. Transcription Factor MEF2D Is Required for the Maintenance of MLL-Rearranged Acute Myeloid Leukemia. Blood Adv. 2021, 5, 4727–4740. [Google Scholar] [CrossRef]
- Marques, J.G.; Gryder, B.E.; Pavlovic, B.; Chung, Y.; Ngo, Q.A.; Frommelt, F.; Gstaiger, M.; Song, Y.; Benischke, K.; Laubscher, D.; et al. NuRD Subunit CHD4 Regulates Super-Enhancer Accessibility in Rhabdomyosarcoma and Represents a General Tumor Dependency. eLife 2020, 9, e54993. [Google Scholar] [CrossRef]
- Cui, S.; Wu, Q.; Liu, M.; Su, M.; Liu, S.; Shao, L.; Han, X.; He, H. EphA2 Super-Enhancer Promotes Tumor Progression by Recruiting FOSL2 and TCF7L2 to Activate the Target Gene EphA2. Cell Death Dis. 2021, 12, 264. [Google Scholar] [CrossRef]
- Jia, Y.; Zhou, J.; Tan, T.K.; Chung, T.-H.; Chen, Y.; Chooi, J.-Y.; Sanda, T.; Fullwood, M.J.; Xiong, S.; Toh, S.H.M.; et al. Super Enhancer-Mediated Upregulation of HJURP Promotes Growth and Survival of t(4;14)-Positive Multiple Myeloma. Cancer Res. 2022, 82, 406–418. [Google Scholar] [CrossRef]
- Kubota, S.; Tokunaga, K.; Umezu, T.; Yokomizo-Nakano, T.; Sun, Y.; Oshima, M.; Tan, K.T.; Yang, H.; Kanai, A.; Iwanaga, E.; et al. Lineage-Specific RUNX2 Super-Enhancer Activates MYC and Promotes the Development of Blastic Plasmacytoid Dendritic Cell Neoplasm. Nat. Commun. 2019, 10, 1653. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Vallurupalli, M.; Chen, L.; Crompton, B.; Cowley, G.; Vazquez, F.; Weir, B.A.; Tsherniak, A.; Parasuraman, S.; Kim, S.; et al. Functional, Chemical Genomic, and Super-Enhancer Screening Identify Sensitivity to Cyclin D1/CDK4 Pathway Inhibition in Ewing Sarcoma. Oncotarget 2015, 6, 30178–30193. [Google Scholar] [CrossRef]
- Lin, L.; Huang, M.; Shi, X.; Mayakonda, A.; Hu, K.; Jiang, Y.-Y.; Guo, X.; Chen, L.; Pang, B.; Doan, N.; et al. Super-Enhancer-Associated MEIS1 Promotes Transcriptional Dysregulation in Ewing Sarcoma in Co-Operation with EWS-FLI1. Nucleic Acids Res. 2019, 47, 1255–1267. [Google Scholar] [CrossRef]
- Chen, Y.; Ying, Y.; Wang, M.; Ma, C.; Jia, M.; Shi, L.; Wang, S.; Zheng, X.; Chen, W.; Shu, X. A Distal Super-Enhancer Activates Oncogenic ETS2 via Recruiting MECOM in Inflammatory Bowel Disease and Colorectal Cancer. Cell Death Dis. 2023, 14, 8. [Google Scholar] [CrossRef]
- Kandaswamy, R.; Sava, G.P.; Speedy, H.E.; Beà, S.; Martín-Subero, J.I.; Studd, J.B.; Migliorini, G.; Law, P.J.; Puente, X.S.; Martín-García, D.; et al. Genetic Predisposition to Chronic Lymphocytic Leukemia Is Mediated by a BMF Super-Enhancer Polymorphism. Cell Rep. 2016, 16, 2061–2067. [Google Scholar] [CrossRef]
- Oldridge, D.A.; Wood, A.C.; Weichert-Leahey, N.; Crimmins, I.; Sussman, R.; Winter, C.; McDaniel, L.D.; Diamond, M.; Hart, L.S.; Zhu, S.; et al. Genetic Predisposition to Neuroblastoma Mediated by a LMO1 Super-Enhancer Polymorphism. Nature 2015, 528, 418–421. [Google Scholar] [CrossRef]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. An Oncogenic Super-Enhancer Formed through Somatic Mutation of a Noncoding Intergenic Element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef]
- Tian, R.; Huang, Z.; Li, L.; Yuan, J.; Zhang, Q.; Meng, L.; Lang, B.; Hong, Y.; Zhong, C.; Tian, X.; et al. HPV Integration Generates a Cellular Super-Enhancer Which Functions as ecDNA to Regulate Genome-Wide Transcription. Nucleic Acids Res. 2023, 51, 4237–4251. [Google Scholar] [CrossRef]
- Dooley, K.E.; Warburton, A.; McBride, A.A. Tandemly Integrated HPV16 Can Form a Brd4-Dependent Super-Enhancer-Like Element That Drives Transcription of Viral Oncogenes. mBio 2016, 7, e01446-16. [Google Scholar] [CrossRef] [PubMed]
- Warburton, A.; Redmond, C.J.; Dooley, K.E.; Fu, H.; Gillison, M.L.; Akagi, K.; Symer, D.E.; Aladjem, M.I.; McBride, A.A. HPV Integration Hijacks and Multimerizes a Cellular Enhancer to Generate a Viral-Cellular Super-Enhancer That Drives High Viral Oncogene Expression. PLoS Genet. 2018, 14, e1007179. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Loo, J.X.; Shi, X.; Xiong, W.; Guo, Y.; Ke, H.; Yang, M.; Jiang, Y.; Xia, S.; Zhao, M.; et al. E6 Protein Expressed by High-Risk HPV Activates Super-Enhancers of the EGFR and c-MET Oncogenes by Destabilizing the Histone Demethylase KDM5C. Cancer Res. 2018, 78, 1418–1430. [Google Scholar] [CrossRef]
- Shen, H.; Xu, W.; Guo, R.; Rong, B.; Gu, L.; Wang, Z.; He, C.; Zheng, L.; Hu, X.; Hu, Z.; et al. Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell 2016, 165, 331–342. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, C.-Y.; Gao, C.; Chen, J.-D.; Chen, J.-J.; Wang, Z.; Ju, L.-G.; Tang, S.-B.; Yao, J.; Li, F.; et al. Regulation of KDM5C Stability and Enhancer Reprogramming in Breast Cancer. Cell Death Dis. 2022, 13, 843. [Google Scholar] [CrossRef]
- Zhou, H.; Schmidt, S.C.S.; Jiang, S.; Willox, B.; Bernhardt, K.; Liang, J.; Johannsen, E.C.; Kharchenko, P.; Gewurz, B.E.; Kieff, E.; et al. Epstein-Barr Virus Oncoprotein Super-Enhancers Control B Cell Growth. Cell Host Microbe 2015, 17, 205–216. [Google Scholar] [CrossRef]
- Ke, L.; Zhou, H.; Wang, C.; Xiong, G.; Xiang, Y.; Ling, Y.; Khabir, A.; Tsao, G.S.; Zeng, Y.; Zeng, M.; et al. Nasopharyngeal Carcinoma Super-Enhancer–Driven ETV6 Correlates with Prognosis. Proc. Natl. Acad. Sci. USA 2017, 114, 9683–9688. [Google Scholar] [CrossRef]
- Ropri, A.S.; DeVaux, R.S.; Eng, J.; Chittur, S.V.; Herschkowitz, J.I. Cis-Acting Super-Enhancer lncRNAs as Biomarkers to Early-Stage Breast Cancer. Breast Cancer Res. 2021, 23, 101. [Google Scholar] [CrossRef]
- Ooi, W.F.; Xing, M.; Xu, C.; Yao, X.; Ramlee, M.K.; Lim, M.C.; Cao, F.; Lim, K.; Babu, D.; Poon, L.-F.; et al. Epigenomic Profiling of Primary Gastric Adenocarcinoma Reveals Super-Enhancer Heterogeneity. Nat. Commun. 2016, 7, 12983. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Spindler, T.J.; De Souza Fonseca, M.A.; Corona, R.I.; Seo, J.-H.; Dezem, F.S.; Li, L.; Lee, J.M.; Long, H.W.; Sellers, T.A.; et al. Super-Enhancer-Associated LncRNA UCA1 Interacts Directly with AMOT to Activate YAP Target Genes in Epithelial Ovarian Cancer. iScience 2019, 17, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Lawrenson, K.; Freedman, M.L. NCBI GEO: Epigenomic and Transcriptomic Analyses of Epithelial Ovarian Cancer. GSE121103 (2020). Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121103 (accessed on 1 September 2023).
- Li, Q.-L.; Lin, X.; Yu, Y.-L.; Chen, L.; Hu, Q.-X.; Chen, M.; Cao, N.; Zhao, C.; Wang, C.-Y.; Huang, C.-W.; et al. Genome-Wide Profiling in Colorectal Cancer Identifies PHF19 and TBC1D16 as Oncogenic Super Enhancers. Nat. Commun. 2021, 12, 6407. [Google Scholar] [CrossRef] [PubMed]
- Noel, P.; Hussein, S.; Ng, S.; Antal, C.E.; Lin, W.; Rodela, E.; Delgado, P.; Naveed, S.; Downes, M.; Lin, Y.; et al. Triptolide Targets Super-Enhancer Networks in Pancreatic Cancer Cells and Cancer-Associated Fibroblasts. Oncogenesis 2020, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Tsang, F.H.; Law, C.; Tang, T.C.; Cheng, C.L.; Chin, D.W.; Tam, W.V.; Wei, L.; Wong, C.C.; Ng, I.O.; Wong, C. Aberrant Super-Enhancer Landscape in Human Hepatocellular Carcinoma. Hepatology 2019, 69, 2502–2517. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.J.; Federation, A.J.; Schwartz, L.S.; Kasar, S.; Klitgaard, J.L.; Lenci, R.; Li, Q.; Lawlor, M.; Fernandes, S.M.; Souza, A.; et al. Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia. Cancer Cell 2018, 34, 982–995.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, D.; Tang, D.; Huang, W. Abnormal Activations of Super-Enhancers Enhance the Carcinogenicity in Lung Adenocarcinoma. Cancer Manag. Res. 2020, 12, 8509–8518. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Flam, E.L.; Danilova, L.; Kelley, D.Z.; Stavrovskaya, E.; Guo, T.; Considine, M.; Qian, J.; Califano, J.A.; Favorov, A.; Fertig, E.J.; et al. Differentially Methylated Super-Enhancers Regulate Target Gene Expression in Human Cancer. Sci. Rep. 2019, 9, 15034. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Yang, F.; Mackintosh, C.; Jayani, R.S.; Oh, S.; Jin, C.; Nair, S.J.; Merkurjev, D.; Ma, W.; Allen, S.; et al. Super-Enhancer Redistribution as a Mechanism of Broad Gene Dysregulation in Repeatedly Drug-Treated Cancer Cells. Cell Rep. 2020, 31, 107532. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.A.; Trotter, K.W.; Day, C.R.; Ward, J.M.; Inoue, K.; Rodriguez, J.; Archer, T.K. Multimodal Regulatory Elements within a Hormone-Specific Super Enhancer Control a Heterogeneous Transcriptional Response. Molecular Cell 2022, 82, 803–815.e5. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wu, Y.; Nouri, M.; Spisak, S.; Russo, J.W.; Sowalsky, A.G.; Pomerantz, M.M.; Wei, Z.; Korthauer, K.; Seo, J.-H.; et al. Androgen Receptor and MYC Equilibration Centralizes on Developmental Super-Enhancer. Nat. Commun. 2021, 12, 7308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, W.; Zou, C.; Zhao, Z.; Lai, Y.; Shi, Z.; Xie, X.; Huang, G.; Wang, Y.; Zhang, X.; et al. Targeting Super-Enhancer–Associated Oncogenes in Osteosarcoma with THZ2, a Covalent CDK7 Inhibitor. Clin. Cancer Res. 2020, 26, 2681–2692. [Google Scholar] [CrossRef]
- Cao, X.; Dang, L.; Zheng, X.; Lu, Y.; Lu, Y.; Ji, R.; Zhang, T.; Ruan, X.; Zhi, J.; Hou, X.; et al. Targeting Super-Enhancer-Driven Oncogenic Transcription by CDK7 Inhibition in Anaplastic Thyroid Carcinoma. Thyroid 2019, 29, 809–823. [Google Scholar] [CrossRef]
- Dai, W.; Wu, J.; Peng, X.; Hou, W.; Huang, H.; Cheng, Q.; Liu, Z.; Luyten, W.; Schoofs, L.; Zhou, J.; et al. CDK12 Orchestrates Super-enhancer-associated CCDC137 Transcription to Direct Hepatic Metastasis in Colorectal Cancer. Clin. Transl. Med. 2022, 12, e1087. [Google Scholar] [CrossRef]
- Liu, B.; Liu, X.; Han, L.; Chen, X.; Wu, X.; Wu, J.; Yan, D.; Wang, Y.; Liu, S.; Shan, L.; et al. BRD4-Directed Super-Enhancer Organization of Transcription Repression Programs Links to Chemotherapeutic Efficacy in Breast Cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2109133119. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Z.; Huang, Z.; Chen, D.-C.; Zhu, X.-X.; Wang, Y.-Z.; Yan, Y.-W.; Tang, S.; Madhavan, S.; Ni, W.; et al. Super Enhancer Inhibitors Suppress MYC Driven Transcriptional Amplification and Tumor Progression in Osteosarcoma. Bone Res. 2018, 6, 11. [Google Scholar] [CrossRef]
- Ong, J.Z.L.; Yokomori, R.; Wong, R.W.J.; Tan, T.K.; Ueda, R.; Ishida, T.; Iida, S.; Sanda, T. Requirement for TP73 and Genetic Alterations Originating from Its Intragenic Super-Enhancer in Adult T-Cell Leukemia. Leukemia 2022, 36, 2293–2305. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, S.J.; Nevedomskaya, E.; Lesche, R.; Newman, R.; Mumberg, D.; Haendler, B. Darolutamide Antagonizes Androgen Signaling by Blocking Enhancer and Super-enhancer Activation. Mol. Oncol. 2020, 14, 2022–2039. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhou, Z.; Long, M.; Lin, Q.; Qiu, M.; Chen, P.; Huang, Q.; Qiu, J.; Jiang, Y.; Wen, Q.; et al. A Novel Signature Constructed by Super-Enhancer-Related Genes for the Prediction of Prognosis in Hepatocellular Carcinoma and Associated with Immune Infiltration. Front. Oncol. 2023, 13, 1043203. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Ando, K.; Imaizumi, Y.; Mishima, H.; Kinoshita, A.; Kobayashi, Y.; Kitanosono, H.; Kato, T.; Sawayama, Y.; Sato, S.; et al. Alvocidib Inhibits IRF4 Expression via Super-enhancer Suppression and Adult T-cell Leukemia/Lymphoma Cell Growth. Cancer Sci. 2022, 113, 4092–4103. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Miyashita, N.; Mikami, Y.; Noguchi, S.; Yamauchi, Y.; Suzukawa, M.; Fukami, T.; Ohta, K.; Asano, Y.; Sato, S.; et al. TBX4 Is Involved in the Super-Enhancer-Driven Transcriptional Programs Underlying Features Specific to Lung Fibroblasts. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 314, L177–L191. [Google Scholar] [CrossRef]
- Sanda, T.; Lawton, L.N.; Barrasa, M.I.; Fan, Z.P.; Kohlhammer, H.; Gutierrez, A.; Ma, W.; Tatarek, J.; Ahn, Y.; Kelliher, M.A.; et al. Core Transcriptional Regulatory Circuit Controlled by the TAL1 Complex in Human T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 209–221. [Google Scholar] [CrossRef]
- Zhang, M.; Hoyle, R.G.; Ma, Z.; Sun, B.; Cai, W.; Cai, H.; Xie, N.; Zhang, Y.; Hou, J.; Liu, X.; et al. FOSL1 Promotes Metastasis of Head and Neck Squamous Cell Carcinoma through Super-Enhancer-Driven Transcription Program. Mol. Ther. 2021, 29, 2583–2600. [Google Scholar] [CrossRef]
- Ying, Y.; Wang, Y.; Huang, X.; Sun, Y.; Zhang, J.; Li, M.; Zeng, J.; Wang, M.; Xiao, W.; Zhong, L.; et al. Oncogenic HOXB8 Is Driven by MYC-Regulated Super-Enhancer and Potentiates Colorectal Cancer Invasiveness via BACH1. Oncogene 2020, 39, 1004–1017. [Google Scholar] [CrossRef]
- Miyakawa, K.; Miyashita, N.; Horie, M.; Terasaki, Y.; Tanaka, H.; Urushiyama, H.; Fukuda, K.; Okabe, Y.; Ishii, T.; Kuwahara, N.; et al. ASCL1 Regulates Super-enhancer-associated miRNAs to Define Molecular Subtypes of Small Cell Lung Cancer. Cancer Sci. 2022, 113, 3932–3946. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef]
- Zheng, Z.; Xia, L.; Hu, G.; Liu, J.; Hu, Y.; Chen, Y.; Peng, J.; Zhang, W.; Liu, W. Super-Enhancer-Controlled Positive Feedback Loop BRD4/ERα–RET–ERα Promotes ERα-Positive Breast Cancer. Nucleic Acids Res. 2022, 50, 10230–10248. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Yohe, M.E.; Chou, H.-C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3–FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; He, Y.; He, J.; Wang, L.; Liu, Z.; Yang, J.; Gao, Z.; Lu, G.; Zou, C.; Zhao, W. Epigenetic Profiling Identifies LIF as a Super-Enhancer-Controlled Regulator of Stem Cell–like Properties in Osteosarcoma. Mol. Cancer Res. 2020, 18, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-Y.; Jiang, Y.; Li, C.-Q.; Zhang, Y.; Dakle, P.; Kaur, H.; Deng, J.-W.; Lin, R.Y.-T.; Han, L.; Xie, J.-J.; et al. TP63, SOX2, and KLF5 Establish a Core Regulatory Circuitry That Controls Epigenetic and Transcription Patterns in Esophageal Squamous Cell Carcinoma Cell Lines. Gastroenterology 2020, 159, 1311–1327.e19. [Google Scholar] [CrossRef]
- Zhang, T.; Song, X.; Zhang, Z.; Mao, Q.; Xia, W.; Xu, L.; Jiang, F.; Dong, G. Aberrant Super-Enhancer Landscape Reveals Core Transcriptional Regulatory Circuitry in Lung Adenocarcinoma. Oncogenesis 2020, 9, 92. [Google Scholar] [CrossRef]
- Li, M.; Han, Y.; Wang, C.; Kang, W.; Jiang, W.; Zhang, L.; Tang, Y. Dissecting Super-Enhancer Driven Transcriptional Dependencies Reveals Novel Therapeutic Strategies and Targets for Group 3 Subtype Medulloblastoma. J. Exp. Clin. Cancer Res. 2022, 41, 311. [Google Scholar] [CrossRef]
- Yoshino, S.; Suzuki, H.I. The Molecular Understanding of Super-Enhancer Dysregulation in Cancer. Nagoya J. Med. Sci. 2022, 84, 216. [Google Scholar]
- Perez, M.W.; Sias-Garcia, O.; Daramola, A.; Wei, H.; Terrell, M.; Rashid, R.; Park, W.D.; Duong, K.; Horton, T.M.; Li, F.; et al. Defining the Transcriptional Control of Pediatric AML Highlights RARA as a Superenhancer-Regulated Druggable Dependency. Blood Adv. 2021, 5, 4864–4876. [Google Scholar] [CrossRef]
- Thirant, C.; Ignacimouttou, C.; Lopez, C.K.; Diop, M.; Le Mouël, L.; Thiollier, C.; Siret, A.; Dessen, P.; Aid, Z.; Rivière, J.; et al. ETO2-GLIS2 Hijacks Transcriptional Complexes to Drive Cellular Identity and Self-Renewal in Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell 2017, 31, 452–465. [Google Scholar] [CrossRef]
- Koeniger, A.; Polo, P.; Brichkina, A.; Finkernagel, F.; Visekruna, A.; Nist, A.; Stiewe, T.; Daude, M.; Diederich, W.E.; Gress, T.M.; et al. Tumor-Suppressive Disruption of Cancer Subtype-Associated Super Enhancer Circuits by Small Molecule Treatment. NAR Cancer 2023, 5, zcad007. [Google Scholar] [CrossRef]
- Han, J.; Meng, J.; Chen, S.; Wang, X.; Yin, S.; Zhang, Q.; Liu, H.; Qin, R.; Li, Z.; Zhong, W.; et al. YY1 Complex Promotes Quaking Expression via Super-Enhancer Binding during EMT of Hepatocellular Carcinoma. Cancer Res. 2019, 79, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.-J.; Jiang, Y.-Y.; Jiang, Y.; Li, C.-Q.; Lim, M.-C.; An, O.; Mayakonda, A.; Ding, L.-W.; Long, L.; Sun, C.; et al. Super-Enhancer-Driven Long Non-Coding RNA LINC01503, Regulated by TP63, Is Over-Expressed and Oncogenic in Squamous Cell Carcinoma. Gastroenterology 2018, 154, 2137–2151.e1. [Google Scholar] [CrossRef]
- Cai, J.; Chen, S.; Yi, M.; Tan, Y.; Peng, Q.; Ban, Y.; Yang, J.; Li, X.; Zeng, Z.; Xiong, W.; et al. ΔNp63α Is a Super Enhancer-Enriched Master Factor Controlling the Basal-to-Luminal Differentiation Transcriptional Program and Gene Regulatory Networks in Nasopharyngeal Carcinoma. Carcinogenesis 2020, 41, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Yang, W.; Renganathan, A.; Weimholt, C.; Angappulige, D.H.; Nguyen, T.; Sprung, R.W.; Andriole, G.L.; Kim, E.H.; Mahajan, N.P.; et al. Acetylated HOXB13 Regulated Super Enhancer Genes Define Therapeutic Vulnerabilities of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2022, 28, 4131–4145. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Huang, Q.; Ren, H.; Yang, M. The Mechanism and Function of Super Enhancer RNA. Genesis 2021, 59, e23422. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Tan, Y.; Li, Y.; Wu, Y.; Wang, J.; Tang, F. JUN-Induced Super-Enhancer RNA Forms R-Loop to Promote Nasopharyngeal Carcinoma Metastasis. Cell Death Dis. 2023, 14, 459. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef]
- Wen, S.; He, Y.; Wang, L.; Zhang, J.; Quan, C.; Niu, Y.; Huang, H. Aberrant Activation of Super Enhancer and Choline Metabolism Drive Antiandrogen Therapy Resistance in Prostate Cancer. Oncogene 2020, 39, 6556–6571. [Google Scholar] [CrossRef]
- Liang, J.; Zhou, H.; Gerdt, C.; Tan, M.; Colson, T.; Kaye, K.M.; Kieff, E.; Zhao, B. Epstein–Barr Virus Super-Enhancer eRNAs Are Essential for MYC Oncogene Expression and Lymphoblast Proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 14121–14126. [Google Scholar] [CrossRef]
- Chen, H.; Zheng, J.; Yan, L.; Zhou, X.; Jiang, P.; Yan, F. Super-enhancer–Associated Long Noncoding RNA RP11-569A11.1 Inhibited Cell Progression and Metastasis by Regulating IFIT2 in Colorectal Cancer. Clin. Lab. Anal. 2021, 35, e23780. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Wang, Y.; Zhao, X.; Su, T. E2F1 Combined with LINC01004 Super-Enhancer to Promote Hepatocellular Carcinoma Cell Proliferation and Metastasis. Clin. Epigenetics 2023, 15, 17. [Google Scholar] [CrossRef]
- Peng, L.; Peng, J.-Y.; Cai, D.-K.; Qiu, Y.-T.; Lan, Q.-S.; Luo, J.; Yang, B.; Xie, H.-T.; Du, Z.-P.; Yuan, X.-Q.; et al. Immune Infiltration and Clinical Outcome of Super-Enhancer-Associated lncRNAs in Stomach Adenocarcinoma. Front. Oncol. 2022, 12, 780493. [Google Scholar] [CrossRef]
- Zhang, T.; Xia, W.; Song, X.; Mao, Q.; Huang, X.; Chen, B.; Liang, Y.; Wang, H.; Chen, Y.; Yu, X.; et al. Super-Enhancer Hijacking LINC01977 Promotes Malignancy of Early-Stage Lung Adenocarcinoma Addicted to the Canonical TGF-β/SMAD3 Pathway. J. Hematol. Oncol. 2022, 15, 114. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, R.; Wu, S.; Shen, L.; Ke, M.; Ouyang, Y.; Lin, M.; Lyu, Y.; Sun, B.; Zheng, Z.; et al. Super-Enhancer LncRNA LINC00162 Promotes Progression of Bladder Cancer. iScience 2020, 23, 101857. [Google Scholar] [CrossRef]
- Shen, Q.; Wang, R.; Liu, X.; Song, P.; Zheng, M.; Ren, X.; Ma, J.; Lu, Z.; Li, J. HSF1 Stimulates Glutamine Transport by Super-Enhancer-Driven lncRNA LINC00857 in Colorectal Cancer. Cancers 2022, 14, 3855. [Google Scholar] [CrossRef]
- Yan, L.; Chen, H.; Tang, L.; Jiang, P.; Yan, F. Super-Enhancer-Associated Long Noncoding RNA AC005592.2 Promotes Tumor Progression by Regulating OLFM4 in Colorectal Cancer. BMC Cancer 2021, 21, 187. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Shi, C.; Hong, W.; Li, P.; Zhou, X.; Fu, W.; Lin, L.; Zhang, J. Super-Enhancer-Driven lncRNA-DAW Promotes Liver Cancer Cell Proliferation through Activation of Wnt/β-Catenin Pathway. Mol. Ther.-Nucleic Acids 2021, 26, 1351–1363. [Google Scholar] [CrossRef]
- Wu, F.; Xu, L.; Tu, Y.; Cheung, O.K.W.; Szeto, L.L.M.; Mok, M.T.S.; Yang, W.; Kang, W.; Cao, Q.; Lai, P.B.S.; et al. Sirtuin 7 Super-Enhancer Drives Epigenomic Reprogramming in Hepatocarcinogenesis. Cancer Lett. 2022, 525, 115–130. [Google Scholar] [CrossRef]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.-A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B. A High-Resolution Map of the Three-Dimensional Chromatin Interactome in Human Cells. Nature 2013, 503, 290–294. [Google Scholar] [CrossRef]
- Wang, Q.; Peng, L.; Chen, Y.; Liao, L.; Chen, J.; Li, M.; Li, Y.; Qian, F.; Zhang, Y.; Wang, F.; et al. Characterization of Super-enhancer-associated Functional lncRNAs Acting as ceRNAs in ESCC. Mol. Oncol. 2020, 14, 2203–2230. [Google Scholar] [CrossRef]
- Natoli, G. Control of NF- B-Dependent Transcriptional Responses by Chromatin Organization. Cold Spring Harb. Perspect. Biol. 2009, 1, a000224. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the Enhancer Landscape during Macrophage Activation Is Coupled to Enhancer Transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.J.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.H.; Kung, A.L.; et al. NF-κB Directs Dynamic Super Enhancer Formation in Inflammation and Atherogenesis. Mol. Cell 2014, 56, 219–231. [Google Scholar] [CrossRef]
- Schmidt, S.F.; Larsen, B.D.; Loft, A.; Nielsen, R.; Madsen, J.G.S.; Mandrup, S. Acute TNF-Induced Repression of Cell Identity Genes Is Mediated by NFκB-Directed Redistribution of Cofactors from Super-Enhancers. Genome Res. 2015, 25, 1281–1294. [Google Scholar] [CrossRef]
- Higashijima, Y.; Matsui, Y.; Shimamura, T.; Nakaki, R.; Nagai, N.; Tsutsumi, S.; Abe, Y.; Link, V.M.; Osaka, M.; Yoshida, M.; et al. Coordinated Demethylation of H3K9 and H3K27 Is Required for Rapid Inflammatory Responses of Endothelial Cells. EMBO J. 2020, 39, e103949. [Google Scholar] [CrossRef]
- Xiao, X.; Fan, Y.; Li, J.; Zhang, X.; Lou, X.; Dou, Y.; Shi, X.; Lan, P.; Xiao, Y.; Minze, L.; et al. Guidance of Super-Enhancers in Regulation of IL-9 Induction and Airway Inflammation. J. Exp. Med. 2018, 215, 559–574. [Google Scholar] [CrossRef]
- Peeters, J.G.C.; Vervoort, S.J.; Tan, S.C.; Mijnheer, G.; de Roock, S.; Vastert, S.J.; Nieuwenhuis, E.E.S.; van Wijk, F.; Prakken, B.J.; Creyghton, M.P.; et al. Inhibition of Super-Enhancer Activity in Autoinflammatory Site-Derived T Cells Reduces Disease-Associated Gene Expression. Cell Rep. 2015, 12, 1986–1996. [Google Scholar] [CrossRef]
- Vahedi, G.; Kanno, Y.; Furumoto, Y.; Jiang, K.; Parker, S.C.J.; Erdos, M.R.; Davis, S.R.; Roychoudhuri, R.; Restifo, N.P.; Gadina, M.; et al. Super-Enhancers Delineate Disease-Associated Regulatory Nodes in T Cells. Nature 2015, 520, 558–562. [Google Scholar] [CrossRef]
- Baillie, J.K.; Arner, E.; Daub, C.; De Hoon, M.; Itoh, M.; Kawaji, H.; Lassmann, T.; Carninci, P.; Forrest, A.R.R.; Hayashizaki, Y.; et al. Analysis of the Human Monocyte-Derived Macrophage Transcriptome and Response to Lipopolysaccharide Provides New Insights into Genetic Aetiology of Inflammatory Bowel Disease. PLoS Genet. 2017, 13, e1006641. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. TERT Promoter Mutations and Telomerase Reactivation in Urothelial Cancer. Science 2015, 347, 1006–1010. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, B.; LaFave, L.M.; Earl, A.S.; Chiang, Z.; Hu, Y.; Ding, J.; Brack, A.; Kartha, V.K.; Tay, T.; et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 2020, 183, 1103–1116.e20. [Google Scholar] [CrossRef]
- Zhou, R.W.; Xu, J.; Martin, T.C.; Zachem, A.L.; He, J.; Ozturk, S.; Demircioglu, D.; Bansal, A.; Trotta, A.P.; Giotti, B.; et al. A Local Tumor Microenvironment Acquired Super-Enhancer Induces an Oncogenic Driver in Colorectal Carcinoma. Nat. Commun. 2022, 13, 6041. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Nannenberg, E.; Van Der Werf, C. Cardiogenetics, 25 Years a Growing Subspecialism. Neth. Heart J. 2020, 28, 39–43. [Google Scholar] [CrossRef]
- Cashman, T.J.; Trivedi, C.M. Super Enhancers: Enhancing Human Cardiogenesis. Circ. Res. 2020, 127, 1156–1158. [Google Scholar] [CrossRef]
- VanOudenhove, J.; Yankee, T.N.; Wilderman, A.; Cotney, J. Epigenomic and Transcriptomic Dynamics during Human Heart Organogenesis. Circ. Res. 2020, 127, e184–e209. [Google Scholar] [CrossRef]
- Ounzain, S.; Pedrazzini, T. Super-Enhancer Lncs to Cardiovascular Development and Disease. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 1953–1960. [Google Scholar] [CrossRef]
- Gong, J.; Qiu, C.; Huang, D.; Zhang, Y.; Yu, S.; Zeng, C. Integrative Functional Analysis of Super Enhancer SNPs for Coronary Artery Disease. J. Hum. Genet. 2018, 63, 627–638. [Google Scholar] [CrossRef]
- Guo, N.; Zhang, N.; Yan, L.; Cao, X.; Wang, J.; Wang, Y. Correlation between Genetic Polymorphisms within the MAPK1/HIF-1/HO-1 Signaling Pathway and Risk or Prognosis of Perimenopausal Coronary Artery Disease. Clin. Cardiol. 2017, 40, 597–604. [Google Scholar] [CrossRef]
- Sun, W.; Yao, S.; Tang, J.; Liu, S.; Chen, J.; Deng, D.; Zeng, C. Integrative Analysis of Super Enhancer SNPs for Type 2 Diabetes. PLoS ONE 2018, 13, e0192105. [Google Scholar] [CrossRef] [PubMed]
- Nott, A.; Holtman, I.R.; Coufal, N.G.; Schlachetzki, J.C.M.; Yu, M.; Hu, R.; Han, C.Z.; Pena, M.; Xiao, J.; Wu, Y.; et al. Brain Cell Type–Specific Enhancer–Promoter Interactome Maps and Disease—Risk Association. Science 2019, 366, 1134–1139. [Google Scholar] [CrossRef]
- GERAD Consortium; Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; Van Cauwenberghe, C.; Kolen, K.V.; Geller, F.; Sottejeau, Y.; Harold, D.; et al. Increased Expression of BIN1 Mediates Alzheimer Genetic Risk by Modulating Tau Pathology. Mol. Psychiatry 2013, 18, 1225–1234. [Google Scholar] [CrossRef]
- Zhang, Y.; Day, K.; Absher, D.M. STAT3-Mediated Allelic Imbalance of Novel Genetic Variant Rs1047643 and B-Cell-Specific Super-Enhancer in Association with Systemic Lupus Erythematosus. eLife 2022, 11, e72837. [Google Scholar] [CrossRef]
- Yamagata, K.; Nakayamada, S.; Tanaka, Y. Critical Roles of Super-Enhancers in the Pathogenesis of Autoimmune Diseases. Inflamm. Regen. 2020, 40, 16. [Google Scholar] [CrossRef]
- Alcalà-Vida, R.; Awada, A.; Boutillier, A.-L.; Merienne, K. Epigenetic Mechanisms Underlying Enhancer Modulation of Neuronal Identity, Neuronal Activity and Neurodegeneration. Neurobiol. Dis. 2021, 147, 105155. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Murakami, S.; Li, R.; Nagari, A.; Chae, M.; Camacho, C.V.; Kraus, W.L. Distinct Roles for BET Family Members in Estrogen Receptor α Enhancer Function and Gene Regulation in Breast Cancer Cells. Mol. Cancer Res. 2019, 17, 2356–2368. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Del Gaudio, N.; Migliaccio, A.; Altucci, L. Epigenetic Drugs against Cancer: An Evolving Landscape. Arch. Toxicol. 2014, 88, 1651–1668. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Yang, X.; Lin, J.; Cao, Z.; Lu, C.; Yang, Z.; Zheng, M.; Pan, R.; Cai, W. Super-Enhancer Induced IL-20RA Promotes Proliferation/Metastasis and Immune Evasion in Colorectal Cancer. Front. Oncol. 2021, 11, 724655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, P.; Jing, Y.; Yan, Y.; Cai, M.-C.; Zhang, M.; Zhang, S.; Peng, H.; Ji, Z.-L.; Di, W.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy in Ovarian Cancer by Downregulating FoxM1. Theranostics 2016, 6, 219–230. [Google Scholar] [CrossRef]
- Crosby, E.J.; Acharya, C.R.; Haddad, A.-F.; Rabiola, C.A.; Lei, G.; Wei, J.-P.; Yang, X.-Y.; Wang, T.; Liu, C.-X.; Wagner, K.U.; et al. Stimulation of Oncogene-Specific Tumor-Infiltrating T Cells through Combined Vaccine and αPD-1 Enable Sustained Antitumor Responses against Established HER2 Breast Cancer. Clin. Cancer Res. 2020, 26, 4670–4681. [Google Scholar] [CrossRef]
- Gelato, K.A.; Schöckel, L.; Klingbeil, O.; Rückert, T.; Lesche, R.; Toedling, J.; Kalfon, E.; Héroult, M.; Lejeune, P.; Mönning, U.; et al. Super-Enhancers Define a Proliferative PGC-1α-Expressing Melanoma Subgroup Sensitive to BET Inhibition. Oncogene 2018, 37, 512–521. [Google Scholar] [CrossRef]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective Inhibition of the BD2 Bromodomain of BET Proteins in Prostate Cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-Cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef]
- Bakshi, S.; McKee, C.; Walker, K.; Brown, C.; Chaudhry, G.R. Toxicity of JQ1 in Neuronal Derivatives of Human Umbilical Cord Mesenchymal Stem Cells. Oncotarget 2018, 9, 33853–33864. [Google Scholar] [CrossRef]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain Inhibitor OTX015 in Patients with Acute Leukaemia: A Dose-Escalation, Phase 1 Study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain Inhibitor OTX015 in Patients with Lymphoma or Multiple Myeloma: A Dose-Escalation, Open-Label, Pharmacokinetic, Phase 1 Study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Lewin, J.; Soria, J.-C.; Stathis, A.; Delord, J.-P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients with Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Herbschleb, K.; Massard, C.; Woodcock, V.; Soria, J.-C.; Walter, A.O.; Ewerton, F.; Poelman, M.; Benson, N.; Ocker, M.; et al. First-in-Human Phase I Study of the Bromodomain and Extraterminal Motif Inhibitor BAY 1238097: Emerging Pharmacokinetic/Pharmacodynamic Relationship and Early Termination Due to Unexpected Toxicity. Eur. J. Cancer 2019, 109, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Aftimos, P.G.; Bechter, O.; Awada, A.; Jungels, C.; Dumez, H.; Huyvaert, N.; Costermans, J.; Lee, C.; Meeus, M.-A.; Burkard, U.; et al. Phase I First-in-Man Trial of a Novel Bromodomain and Extra-Terminal Domain (BET) Inhibitor (BI 894999) in Patients (Pts) with Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 2504. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting Transcription Regulation in Cancer with a Covalent CDK7 Inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Li, B.; Wang, B.; Zhu, C.; Tang, D.; Pang, J.; Zhao, J.; Sun, C.; Qiu, M.; Qian, Z. Cyclin-dependent Kinase 7 Inhibitor THZ1 in Cancer Therapy. Chronic Dis. Transl. Med. 2019, 5, 155–169. [Google Scholar] [CrossRef]
- Hu, S.; Marineau, J.J.; Rajagopal, N.; Hamman, K.B.; Choi, Y.J.; Schmidt, D.R.; Ke, N.; Johannessen, L.; Bradley, M.J.; Orlando, D.A.; et al. Discovery and Characterization of SY-1365, a Selective, Covalent Inhibitor of CDK7. Cancer Res. 2019, 79, 3479–3491. [Google Scholar] [CrossRef]
- Gao, Y.; Volegova, M.; Nasholm, N.; Das, S.; Kwiatkowski, N.; Abraham, B.J.; Zhang, T.; Gray, N.S.; Gustafson, C.; Krajewska, M.; et al. Synergistic Anti-Tumor Effect of Combining Selective CDK7 and BRD4 Inhibition in Neuroblastoma. Front. Oncol. 2022, 11, 773186. [Google Scholar] [CrossRef]
- Tolani, B.; Gopalakrishnan, R.; Punj, V.; Matta, H.; Chaudhary, P.M. Targeting Myc in KSHV-Associated Primary Effusion Lymphoma with BET Bromodomain Inhibitors. Oncogene 2014, 33, 2928–2937. [Google Scholar] [CrossRef]
- Sengupta, D.; Kannan, A.; Kern, M.; Moreno, M.A.; Vural, E.; Stack, B.; Suen, J.Y.; Tackett, A.J.; Gao, L. Disruption of BRD4 at H3K27Ac-Enriched Enhancer Region Correlates with Decreased c-Myc Expression in Merkel Cell Carcinoma. Epigenetics 2015, 10, 460–466. [Google Scholar] [CrossRef]
- Tögel, L.; Nightingale, R.; Chueh, A.C.; Jayachandran, A.; Tran, H.; Phesse, T.; Wu, R.; Sieber, O.M.; Arango, D.; Dhillon, A.S.; et al. Dual Targeting of Bromodomain and Extraterminal Domain Proteins, and WNT or MAPK Signaling, Inhibits c-MYC Expression and Proliferation of Colorectal Cancer Cells. Mol. Cancer Ther. 2016, 15, 1217–1226. [Google Scholar] [CrossRef]
- Sin-Chan, P.; Mumal, I.; Suwal, T.; Ho, B.; Fan, X.; Singh, I.; Du, Y.; Lu, M.; Patel, N.; Torchia, J.; et al. A C19MC-LIN28A-MYCN Oncogenic Circuit Driven by Hijacked Super-Enhancers Is a Distinct Therapeutic Vulnerability in ETMRs: A Lethal Brain Tumor. Cancer Cell 2019, 36, 51–67.e7. [Google Scholar] [CrossRef]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The Novel BET Bromodomain Inhibitor BI 894999 Represses Super-Enhancer-Associated Transcription and Synergizes with CDK9 Inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-Y.; Lin, D.-C.; Mayakonda, A.; Hazawa, M.; Ding, L.-W.; Chien, W.-W.; Xu, L.; Chen, Y.; Xiao, J.-F.; Senapedis, W.; et al. Targeting Super-Enhancer-Associated Oncogenes in Oesophageal Squamous Cell Carcinoma. Gut 2017, 66, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.W.J.; Ngoc, P.C.T.; Leong, W.Z.; Yam, A.W.Y.; Zhang, T.; Asamitsu, K.; Iida, S.; Okamoto, T.; Ueda, R.; Gray, N.S.; et al. Enhancer Profiling Identifies Critical Cancer Genes and Characterizes Cell Identity in Adult T-Cell Leukemia. Blood 2017, 130, 2326–2338. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Peng, H.; Wang, X.; Yin, X.; Ma, P.; Jing, Y.; Cai, M.-C.; Liu, J.; Zhang, M.; Zhang, S.; et al. Preclinical Efficacy and Molecular Mechanism of Targeting CDK7-Dependent Transcriptional Addiction in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 1739–1750. [Google Scholar] [CrossRef]
- Meng, W.; Wang, J.; Wang, B.; Liu, F.; Li, M.; Zhao, Y.; Zhang, C.; Li, Q.; Chen, J.; Zhang, L.; et al. CDK7 Inhibition Is a Novel Therapeutic Strategy against GBM Both In Vitro and In Vivo. Cancer Manag. Res. 2018, 10, 5747–5758. [Google Scholar] [CrossRef]
- Eliades, P.; Abraham, B.J.; Ji, Z.; Miller, D.M.; Christensen, C.L.; Kwiatkowski, N.; Kumar, R.; Njauw, C.N.; Taylor, M.; Miao, B.; et al. High MITF Expression Is Associated with Super-Enhancers and Suppressed by CDK7 Inhibition in Melanoma. J. Investig. Dermatol. 2018, 138, 1582–1590. [Google Scholar] [CrossRef]
- Sharifnia, T.; Wawer, M.J.; Chen, T.; Huang, Q.-Y.; Weir, B.A.; Sizemore, A.; Lawlor, M.A.; Goodale, A.; Cowley, G.S.; Vazquez, F.; et al. Small-Molecule Targeting of Brachyury Transcription Factor Addiction in Chordoma. Nat. Med. 2019, 25, 292–300. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, S.; Nie, D.; Lai, P.; Li, Y.; Li, Y.; Jin, Y.; Pan, J. Super-Enhancer Landscape Reveals Leukemia Stem Cell Reliance on X-Box Binding Protein 1 as a Therapeutic Vulnerability. Sci. Transl. Med. 2021, 13, eabh3462. [Google Scholar] [CrossRef]
- Gryder, B.E.; Pomella, S.; Sayers, C.; Wu, X.S.; Song, Y.; Chiarella, A.M.; Bagchi, S.; Chou, H.-C.; Sinniah, R.S.; Walton, A.; et al. Histone Hyperacetylation Disrupts Core Gene Regulatory Architecture in Rhabdomyosarcoma. Nat. Genet. 2019, 51, 1714–1722. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.M.; Rudek, M.A.; Piekarz, R. Entinostat: A Promising Treatment Option for Patients with Advanced Breast Cancer. Future Oncology 2017, 13, 1137–1148. [Google Scholar] [CrossRef]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical Genomics Reveals Histone Deacetylases Are Required for Core Regulatory Transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the Class and Isoform Selectivity of Small-Molecule Histone Deacetylase Inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef]
- Hong, J.; Luesch, H. Largazole: From Discovery to Broad-Spectrum Therapy. Nat. Prod. Rep. 2012, 29, 449. [Google Scholar] [CrossRef]
- Mayr, C.; Kiesslich, T.; Erber, S.; Bekric, D.; Dobias, H.; Beyreis, M.; Ritter, M.; Jäger, T.; Neumayer, B.; Winkelmann, P.; et al. HDAC Screening Identifies the HDAC Class I Inhibitor Romidepsin as a Promising Epigenetic Drug for Biliary Tract Cancer. Cancers 2021, 13, 3862. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective Gene Dependencies in MYCN-Amplified Neuroblastoma Include the Core Transcriptional Regulatory Circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; You, X.; Dai, C.; Xu, Q.; Li, F.; Wang, L.; Huang, X.; Wang, J.; Li, S.; Gao, Z.; et al. Targeting Super-Enhancers via Nanoparticle-Facilitated BRD4 and CDK7 Inhibitors Synergistically Suppresses Pancreatic Ductal Adenocarcinoma. Adv. Sci. 2020, 7, 1902926. [Google Scholar] [CrossRef]
- Zhang, W.; Ge, H.; Jiang, Y.; Huang, R.; Wu, Y.; Wang, D.; Guo, S.; Li, S.; Wang, Y.; Jiang, H.; et al. Combinational Therapeutic Targeting of BRD4 and CDK7 Synergistically Induces Anticancer Effects in Head and Neck Squamous Cell Carcinoma. Cancer Lett. 2020, 469, 510–523. [Google Scholar] [CrossRef]
- Bolin, S.; Borgenvik, A.; Persson, C.U.; Sundström, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.-J.; Weishaupt, H.; Swartling, F.J. Combined BET Bromodomain and CDK2 Inhibition in MYC-Driven Medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; Hamdan, F.H.; Kubiak, K.; Diederichs, C.; Gielen, G.H.; Nussbaumer, G.; Carcaboso, A.M.; Hulleman, E.; Johnsen, S.A.; Kramm, C.M. Combined Treatment with CBP and BET Inhibitors Reverses Inadvertent Activation of Detrimental Super Enhancer Programs in DIPG Cells. Cell Death Dis. 2020, 11, 673. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cao, Z.; Wang, Y.; Zhang, X.; Xu, L.; Wang, Y.; Chen, Y.; Yang, C.; Ding, J.; Meng, L. Repressing MYC by Targeting BET Synergizes with Selective Inhibition of PI3Kα against B Cell Lymphoma. Cancer Lett. 2022, 524, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.E.; Ciampa, O.C.; Wong, M.; Fletcher, J.I.; Kamili, A.; Chen, J.; Ho, N.; Sun, Y.; Carter, D.R.; Cheung, B.B.; et al. Combination Therapy with the CDK7 Inhibitor and the Tyrosine Kinase Inhibitor Exerts Synergistic Anticancer Effects against MYCN-amplified Neuroblastoma. Int. J. Cancer 2020, 147, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- Shang, E.; Nguyen, T.T.T.; Shu, C.; Westhoff, M.-A.; Karpel-Massler, G.; Siegelin, M.D. Epigenetic Targeting of Mcl-1 Is Synthetically Lethal with Bcl-xL/Bcl-2 Inhibition in Model Systems of Glioblastoma. Cancers 2020, 12, 2137. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652.e6. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Sun, Y.; Xi, Z.; Milazzo, G.; Poulos, R.C.; Bartenhagen, C.; Bell, J.L.; Mayoh, C.; Ho, N.; Tee, A.E.; et al. JMJD6 Is a Tumorigenic Factor and Therapeutic Target in Neuroblastoma. Nat. Commun. 2019, 10, 3319. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC Inhibitors Elicit Metabolic Reprogramming by Targeting Super-Enhancers in Glioblastoma Models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef]
- Zanellato, I.; Colangelo, D.; Osella, D. JQ1, a BET Inhibitor, Synergizes with Cisplatin and Induces Apoptosis in Highly Chemoresistant Malignant Pleural Mesothelioma Cells. Curr. Cancer Drug Targets 2018, 18, 816–828. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the Roles of Histone-Modifying Enzymes and Their Associated Chromatin Modifications in Transcriptional Regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef]
- Shi, Y.-J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 Histone Demethylase Activity by Its Associated Factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, M.; Han, W.; Wang, Z.; Han, D.; Patalano, S.; Macoska, J.A.; Balk, S.P.; He, H.H.; Corey, E.; et al. LSD1 Inhibition Disrupts Super-Enhancer–Driven Oncogenic Transcriptional Programs in Castration-Resistant Prostate Cancer. Cancer Res. 2023, 83, 1684–1698. [Google Scholar] [CrossRef]
- Thandapani, P. Super-Enhancers in Cancer. Pharmacol. Ther. 2019, 199, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Belloucif, Y.; Lobry, C. Super-Enhancers Dysregulations in Hematological Malignancies. Cells 2022, 11, 196. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Long, W.; Liu, Q. Targeting Super-Enhancers as a Therapeutic Strategy for Cancer Treatment. Front. Pharmacol. 2019, 10, 361. [Google Scholar] [CrossRef] [PubMed]
- Dębek, S.; Juszczyński, P. Super Enhancers as Master Gene Regulators in the Pathogenesis of Hematologic Malignancies. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2022, 1877, 188697. [Google Scholar] [CrossRef]
- Liu, Q.; Guo, L.; Lou, Z.; Xiang, X.; Shao, J. Super-Enhancers and Novel Therapeutic Targets in Colorectal Cancer. Cell Death Dis. 2022, 13, 228. [Google Scholar] [CrossRef]
- Spirin, P.V.; Lebedev, T.D.; Orlova, N.N.; Gornostaeva, A.S.; Prokofjeva, M.M.; Nikitenko, N.A.; Dmitriev, S.E.; Buzdin, A.A.; Borisov, N.M.; Aliper, A.M.; et al. Silencing AML1-ETO Gene Expression Leads to Simultaneous Activation of Both pro-Apoptotic and Proliferation Signaling. Leukemia 2014, 28, 2222–2228. [Google Scholar] [CrossRef]
- Lebedev, T.D.; Spirin, P.V.; Orlova, N.N.; Prokofjeva, M.M.; Prassolov, V.S. Comparative Analysis of Gene Expression: Targeted Antitumor Therapy in Neuroblastoma Cell Lines. Mol. Biol. 2015, 49, 939–942. [Google Scholar] [CrossRef]
- Mitkevich, V.A.; Orlova, N.N.; Petrushanko, I.Y.; Simonenko, O.V.; Spirin, P.V.; Prokof’eva, M.M.; Gornostaeva, A.S.; Stocking, C.; Makarov, A.A.; Prassolov, V.S. Expression of the FLT3-ITD Oncogene Sensitizes Murine Progenitor B-Cell Line BAF3 to Cytotoxic Action of Binase. Mol. Biol. 2013, 47, 248–251. [Google Scholar] [CrossRef]
- Orlova, N.N.; Lebedev, T.D.; Spirin, P.V.; Prassolov, V.S. Key Molecular Mechanisms Associated with Cell Malignant Transformation in Acute Myeloid Leukemia. Mol Biol 2016, 50, 344–352. [Google Scholar] [CrossRef]
- Zhang, C.; Wei, S.; Sun, W.-P.; Teng, K.; Dai, M.-M.; Wang, F.-W.; Chen, J.-W.; Ling, H.; Ma, X.-D.; Feng, Z.-H.; et al. Super-Enhancer-Driven AJUBA Is Activated by TCF4 and Involved in Epithelial-Mesenchymal Transition in the Progression of Hepatocellular Carcinoma. Theranostics 2020, 10, 9066–9082. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.E.; Pushkarev, A.V.; Orlov, A.V.; Nikitin, M.P.; Nikitin, P.I. Interferometric Detection of Chloramphenicol via Its Immunochemical Recognition at Polymer-Coated Nano-Corrugated Surfaces. Sens. Actuators B Chem. 2019, 282, 984–991. [Google Scholar] [CrossRef]
- Novichikhin, D.O.; Orlov, A.V.; Antopolsky, M.L.; Znoyko, S.L.; Nikitin, P.I. Specific and Sensitive Determination of Folic Acid by Label-Free Chemosensors with Microscope Glass Slips as Single-Use Consumables. Chemosensors 2022, 11, 17. [Google Scholar] [CrossRef]
- Orlov, A.V.; Novichikhin, D.O.; Pushkarev, A.V.; Malkerov, Y.A.; Znoiko, S.L.; Guteneva, N.V.; Orlova, N.N.; Gorshkov, B.G.; Nikitin, P.I. Registering the Kinetics of Intermolecular Interactions by Low-Coherence Interferometry for the Development of Biomarker Immunoassays for Cardiovascular Diseases. Dokl. Phys. 2022, 67, 193–196. [Google Scholar] [CrossRef]
- Orlov, A.V.; Burenin, A.G.; Skirda, A.M.; Nikitin, P.I. Kinetic Analysis of Prostate-Specific Antigen Interaction with Monoclonal Antibodies for Development of a Magnetic Immunoassay Based on Nontransparent Fiber Structures. Molecules 2022, 27, 8077. [Google Scholar] [CrossRef]
- Bragina, V.A.; Orlov, A.V.; Znoyko, S.L.; Pushkarev, A.V.; Novichikhin, D.O.; Guteneva, N.V.; Nikitin, M.P.; Gorshkov, B.G.; Nikitin, P.I. Nanobiosensing Based on Optically Selected Antibodies and Superparamagnetic Labels for Rapid and Highly Sensitive Quantification of Polyvalent Hepatitis B Surface Antigen. Anal. Methods 2021, 13, 2424–2433. [Google Scholar] [CrossRef]
- Pushkarev, A.V.; Orlov, A.V.; Znoyko, S.L.; Bragina, V.A.; Nikitin, P.I. Rapid and Easy-to-Use Method for Accurate Characterization of Target Binding and Kinetics of Magnetic Particle Bioconjugates for Biosensing. Sensors 2021, 21, 2802. [Google Scholar] [CrossRef]
- Orlov, A.V.; Malkerov, J.A.; Novichikhin, D.O.; Znoyko, S.L.; Nikitin, P.I. Express High-Sensitive Detection of Ochratoxin A in Food by a Lateral Flow Immunoassay Based on Magnetic Biolabels. Food Chem. 2022, 383, 132427. [Google Scholar] [CrossRef]
- Orlov, A.V.; Malkerov, J.A.; Novichikhin, D.O.; Znoyko, S.L.; Nikitin, P.I. Multiplex Label-Free Kinetic Characterization of Antibodies for Rapid Sensitive Cardiac Troponin I Detection Based on Functionalized Magnetic Nanotags. Int. J. Mol. Sci. 2022, 23, 4474. [Google Scholar] [CrossRef]
- Zolotova, M.O.; Znoyko, S.L.; Orlov, A.V.; Nikitin, P.I.; Sinolits, A.V. Efficient Chlorostannate Modification of Magnetite Nanoparticles for Their Biofunctionalization. Materials 2024, 17, 349. [Google Scholar] [CrossRef]
- Orlov, A.V.; Znoyko, S.L.; Malkerov, J.A.; Skirda, A.M.; Novichikhin, D.O.; Rakitina, A.S.; Zaitseva, Z.G.; Nikitin, P.I. Quantitative Rapid Magnetic Immunoassay for Sensitive Toxin Detection in Food: Non-Covalent Functionalization of Nanolabels vs. Covalent Immobilization. Toxins 2024, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Orlov, A.V.; Malkerov, Y.A.; Skirda, A.M.; Novichikhin, D.O.; Znoyko, S.L.; Bragina, V.A.; Nikitin, P.I. Supersensitive Registration of Polyfunctional Magnetic Nanomaterials for the Rapid Detection of Molecular Markers of Diseases. Dokl. Phys. 2023, 68, 214–218. [Google Scholar] [CrossRef]
- Bragina, V.A.; Khomyakova, E.; Orlov, A.V.; Znoyko, S.L.; Mochalova, E.N.; Paniushkina, L.; Shender, V.O.; Erbes, T.; Evtushenko, E.G.; Bagrov, D.V.; et al. Highly Sensitive Nanomagnetic Quantification of Extracellular Vesicles by Immunochromatographic Strips: A Tool for Liquid Biopsy. Nanomaterials 2022, 12, 1579. [Google Scholar] [CrossRef] [PubMed]
- Burenin, A.G.; Nikitin, M.P.; Orlov, A.V.; Ksenevich, T.I.; Nikitin, P.I. Detection of Pyrethroids by Spectral Correlation Interferometry. Appl. Biochem. Microbiol. 2013, 49, 306. [Google Scholar] [CrossRef]
- Nekrasov, N.; Kudriavtseva, A.; Orlov, A.V.; Gadjanski, I.; Nikitin, P.I.; Bobrinetskiy, I.; Knežević, N.Ž. One-Step Photochemical Immobilization of Aptamer on Graphene for Label-Free Detection of NT-proBNP. Biosensors 2022, 12, 1071. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, N.; Yakunina, N.; Pushkarev, A.V.; Orlov, A.V.; Gadjanski, I.; Pesquera, A.; Centeno, A.; Zurutuza, A.; Nikitin, P.I.; Bobrinetskiy, I. Spectral-Phase Interferometry Detection of Ochratoxin a via Aptamer-Functionalized Graphene Coated Glass. Nanomaterials 2021, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Jarić, S.; Kudriavtseva, A.; Nekrasov, N.; Orlov, A.V.; Komarov, I.A.; Barsukov, L.A.; Gadjanski, I.; Nikitin, P.I.; Bobrinetskiy, I. Femtomolar Detection of the Heart Failure Biomarker NT-proBNP in Artificial Saliva Using an Immersible Liquid-Gated Aptasensor with Reduced Graphene Oxide. Microchem. J. 2024, 196, 109611. [Google Scholar] [CrossRef]
- Orlov, A.V.; Pushkarev, A.V.; Mochalova, E.N.; Nikitin, P.I.; Nikitin, M.P. Development and Label-Free Investigation of Logic-Gating Biolayers for Smart Biosensing. Sens. Actuators B Chem. 2018, 257, 971–979. [Google Scholar] [CrossRef]
- Orlova, N.N.; Bogatova, O.V.; Orlov, A.V. High-Performance Method for Identification of Super Enhancers from ChIP-Seq Data with Configurable Cloud Virtual Machines. MethodsX 2020, 7, 101165. [Google Scholar] [CrossRef]
- Orlova, N.N.; Nikitina, A.S.; Orlov, A.V. Identification and Analysis of Super-Enhancers as Novel Biomarkers and Potential Therapeutic Targets for Age-Associated Diseases. FEBS Open Bio 2019, 9, 394–395. [Google Scholar] [CrossRef]
- Piipponen, M.; Riihilä, P.; Knuutila, J.S.; Kallajoki, M.; Kähäri, V.-M.; Nissinen, L. Super Enhancer-Regulated LINC00094 (SERLOC) Upregulates the Expression of MMP-1 and MMP-13 and Promotes Invasion of Cutaneous Squamous Cell Carcinoma. Cancers 2022, 14, 3980. [Google Scholar] [CrossRef]
- Ma, Q.; Zhao, M.; Long, B.; Li, H. Super-Enhancer-Associated Gene CAPG Promotes AML Progression. Commun. Biol. 2023, 6, 622. [Google Scholar] [CrossRef]
- Kiehlmeier, S.; Rafiee, M.-R.; Bakr, A.; Mika, J.; Kruse, S.; Müller, J.; Schweiggert, S.; Herrmann, C.; Sigismondo, G.; Schmezer, P.; et al. Identification of Therapeutic Targets of the Hijacked Super-Enhancer Complex in EVI1-Rearranged Leukemia. Leukemia 2021, 35, 3127–3138. [Google Scholar] [CrossRef] [PubMed]
- Sang, X.; Zhang, Y.; Fang, F.; Gao, L.; Tao, Y.; Li, X.; Zhang, Z.; Wang, J.; Tian, Y.; Li, Z.; et al. BRD4 Inhibitor GNE-987 Exerts Anticancer Effects by Targeting Super-Enhancer-Related Gene LYL1 in Acute Myeloid Leukemia. J. Immunol. Res. 2022, 2022, 1–18. [Google Scholar] [CrossRef]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator Kinase Inhibition Further Activates Super-Enhancer-Associated Genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, X.; Hui, S.; Ni, J.; Yin, Y.; Cao, W.; Zhang, Y.; Wang, X.; Ma, X.; Cao, P.; et al. Ectopia Associated MN1 Fusions and Aberrant Activation in Myeloid Neoplasms with t(12;22)(P13;Q12). Cancer Gene Ther. 2020, 27, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Lu, J.; Sang, X.; Tao, Y.-F.; Wang, J.-W.; Zhang, Z.-M.; Zhang, Y.-P.; Li, X.-L.; Xie, Y.; Wu, S.-Y.; et al. Super-Enhancer Profiling Identifies Novel Critical and Targetable Cancer Survival Gene LYL1 in Pediatric Acute Myeloid Leukemia. J. Exp. Clin. Cancer Res. 2022, 41, 225. [Google Scholar] [CrossRef]
- Tatsumi, G.; Kawahara, M.; Yamamoto, R.; Hishizawa, M.; Kito, K.; Suzuki, T.; Takaori-Kondo, A.; Andoh, A. LSD1-Mediated Repression of GFI1 Super-Enhancer Plays an Essential Role in Erythroleukemia. Leukemia 2020, 34, 746–758. [Google Scholar] [CrossRef]
- Abraham, B.J.; Hnisz, D.; Weintraub, A.S.; Kwiatkowski, N.; Li, C.H.; Li, Z.; Weichert-Leahey, N.; Rahman, S.; Liu, Y.; Etchin, J.; et al. Small Genomic Insertions Form Enhancers That Misregulate Oncogenes. Nat. Commun. 2017, 8, 14385. [Google Scholar] [CrossRef]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of Proto-Oncogenes by Disruption of Chromosome Neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef]
- Bal, E.; Kumar, R.; Hadigol, M.; Holmes, A.B.; Hilton, L.K.; Loh, J.W.; Dreval, K.; Wong, J.C.H.; Vlasevska, S.; Corinaldesi, C.; et al. Super-Enhancer Hypermutation Alters Oncogene Expression in B Cell Lymphoma. Nature 2022, 607, 808–815. [Google Scholar] [CrossRef]
- Ma, H.; Qu, J.; Luo, J.; Qi, T.; Tan, H.; Jiang, Z.; Zhang, H.; Qu, Q. Super-Enhancer-Associated Hub Genes In Chronic Myeloid Leukemia Identified Using Weighted Gene Co-Expression Network Analysis. Cancer Manag. Res. 2019, 11, 10705–10718. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, Y.-Y.; Xie, J.-J.; Mayakonda, A.; Hazawa, M.; Chen, L.; Xiao, J.-F.; Li, C.-Q.; Huang, M.-L.; Ding, L.-W.; et al. Co-Activation of Super-Enhancer-Driven CCAT1 by TP63 and SOX2 Promotes Squamous Cancer Progression. Nat. Commun. 2018, 9, 3619. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Hoyle, R.G.; Xie, N.; Wang, W.; Cai, H.; Zhang, M.; Ma, Z.; Xiong, G.; Xu, X.; Huang, Z.; et al. A Super-Enhancer Driven by FOSL1 Controls miR-21-5p Expression in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 656628. [Google Scholar] [CrossRef]
- Chen, Q.; Peng, Q.; Cai, J.; Liu, Y.; Lu, X.; Xiong, W.; Zeng, Z.; Li, G.; Li, X.; Li, X.; et al. Super Enhancer Driven Hyaluronan Synthase 3 Promotes Malignant Progression of Nasopharyngeal Carcinoma. J. Cancer 2023, 14, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma Is Composed of Two Super-Enhancer-Associated Differentiation States. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Yuan, C.; Hu, H.; Kuang, M.; Chen, Z.; Tao, X.; Fang, S.; Sun, Y.; Zhang, Y.; Chen, H. Super Enhancer Associated RAI14 Is a New Potential Biomarker in Lung Adenocarcinoma. Oncotarget 2017, 8, 105251–105261. [Google Scholar] [CrossRef]
- Shang, S.; Yang, J.; Jazaeri, A.A.; Duval, A.J.; Tufan, T.; Lopes Fischer, N.; Benamar, M.; Guessous, F.; Lee, I.; Campbell, R.M.; et al. Chemotherapy-Induced Distal Enhancers Drive Transcriptional Programs to Maintain the Chemoresistant State in Ovarian Cancer. Cancer Res. 2019, 79, 4599–4611. [Google Scholar] [CrossRef]
- Guo, W.; Wang, X.; Lu, B.; Yu, J.; Xu, M.; Huang, R.; Cheng, M.; Yang, M.; Zhao, W.; Zou, C. Super-Enhancer-Driven MLX Mediates Redox Balance Maintenance via SLC7A11 in Osteosarcoma. Cell Death Dis. 2023, 14, 439. [Google Scholar] [CrossRef]
- Li, L.; Wang, N.; Zhu, M.; Xiong, Y.; Wang, F.; Guo, G.; Wang, X.; Gu, Y. Aberrant Super-Enhancer-Driven Oncogene ENC1 Promotes the Radio-Resistance of Breast Carcinoma. Cell Death Dis. 2021, 12, 777. [Google Scholar] [CrossRef]
- Deng, R.; Huang, J.-H.; Wang, Y.; Zhou, L.-H.; Wang, Z.-F.; Hu, B.-X.; Chen, Y.-H.; Yang, D.; Mai, J.; Li, Z.-L.; et al. Disruption of Super-Enhancer-Driven Tumor Suppressor Gene RCAN1.4 Expression Promotes the Malignancy of Breast Carcinoma. Mol. Cancer 2020, 19, 122. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Chiang, H.-C.; Hsieh, Y.-P.; Lu, C.; Park, B.H.; Jatoi, I.; Jin, V.X.; Hu, Y.; Li, R. BRCA1 Mutations Attenuate Super-Enhancer Function and Chromatin Looping in Haploinsufficient Human Breast Epithelial Cells. Breast Cancer Res. 2019, 21, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Q.; Liu, G. Genome-Wide Analysis of the FOXA1 Transcriptional Regulatory Network Identifies Super Enhancer Associated LncRNAs in Tamoxifen Resistance. Front. Genet. 2022, 13, 992444. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, Y.; Zhang, S.; Ma, P.; Jin, X.; Wang, Z.; Yao, M.; Zhang, E.; Tao, B.; Qin, Y.; et al. A Tumor-Specific Super-Enhancer Drives Immune Evasion by Guiding Synchronous Expression of PD-L1 and PD-L2. Cell Rep. 2019, 29, 3435–3447.e4. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Yang, N.; Zhang, Y.; Ding, J.; Zhang, W.; Liu, R.; Liu, W.; Chen, C. Inhibition of Super Enhancer Downregulates the Expression of KLF5 in Basal-like Breast Cancers. Int. J. Biol. Sci. 2019, 15, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Cong, Z.; Li, Q.; Yang, Y.; Guo, X.; Cui, L.; You, T. The SNP of Rs6854845 Suppresses Transcription via the DNA Looping Structure Alteration of Super-Enhancer in Colon Cells. Biochem. Biophys. Res. Commun. 2019, 514, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Vidal, E.; Ferreira, H.J.; Vizoso, M.; Sayols, S.; Gomez, A.; Moran, S.; Boque-Sastre, R.; Guil, S.; Martinez-Cardus, A.; et al. Epigenomic Analysis Detects Aberrant Super-Enhancer DNA Methylation in Human Cancer. Genome Biol. 2016, 17, 11. [Google Scholar] [CrossRef]
- Scholz, B.A.; Sumida, N.; De Lima, C.D.M.; Chachoua, I.; Martino, M.; Tzelepis, I.; Nikoshkov, A.; Zhao, H.; Mehmood, R.; Sifakis, E.G.; et al. WNT Signaling and AHCTF1 Promote Oncogenic MYC Expression through Super-Enhancer-Mediated Gene Gating. Nat. Genet. 2019, 51, 1723–1731. [Google Scholar] [CrossRef]
- Cai, C.; Bi, D.; Bick, G.; Wei, Q.; Liu, H.; Lu, L.; Zhang, X.; Qin, H. Hepatocyte Nuclear Factor HNF1A Is a Potential Regulator in Shaping the Super-enhancer Landscape in Colorectal Cancer Liver Metastasis. FEBS Lett. 2021, 595, 3056–3071. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, T.; Zhang, Y.; Chen, H.; Shen, J.; Jin, X.; Wei, J.; Zhang, E.; Xiao, M.; Fan, Y.; et al. A Super-Enhancer Controls TGF-β Signaling in Pancreatic Cancer through Downregulation of TGFBR2. Cell. Signal. 2020, 66, 109470. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered Chromosomal Topology Drives Oncogenic Programs in SDH-Deficient GISTs. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kutschat, A.P.; Yamada, M.; Prokakis, E.; Böttcher, P.; Tanaka, K.; Doki, Y.; Hamdan, F.H.; Johnsen, S.A. Bromodomain Protein BRDT Directs ΔNp63 Function and Super-Enhancer Activity in a Subset of Esophageal Squamous Cell Carcinomas. Cell Death Differ. 2021, 28, 2207–2220. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Jiang, B.; Yuan, X.; Qiu, Y.; Peng, J.; Huang, Y.; Zhang, C.; Zhang, Y.; Lin, Z.; Li, J.; et al. Super-Enhancer–Associated Long Noncoding RNA HCCL5 Is Activated by ZEB1 and Promotes the Malignancy of Hepatocellular Carcinoma. Cancer Res. 2019, 79, 572–584. [Google Scholar] [CrossRef] [PubMed]







| Characteristics | Super-Enhancers | Stretched Enhancers |
|---|---|---|
| Number | x 1 | 10x |
| Genome coverage | x | 2x |
| Average size | 22,812 bp | 5060 bp |
| Distance from TSS | <2 kb (69% of SE) | >10 kb (70% of stretched enhancers) |
| Evolutionary conservation (phastCons scores for 99 vertebrate genomes) | more conserved | less conserved |
| H3K27ac | enriched | depleted |
| H3K4me3 | enriched | depleted |
| H3K4me1 | enriched | enriched |
| H3K27me3 | depleted | enriched |
| DHSs | higher | lower |
| Cohesin (SMC3 and RAD21 components) | more | less |
| CTCF | more | less |
| RNA II | more | less |
| Expression of associated genes | higher | lower |
| eRNA | more | less |
| Comparison between cell types | less shared | significantly shared |
| Database | dbSUPER | Sedb v.2.0 | SEA v.3.0 | Seanalysis 2.0 |
|---|---|---|---|---|
| Link | https://asntech.org/dbsuper/ (accessed on 14 July 2023) | https://bio.liclab.net/sedb/ (accessed on 14 July 2023) | http://sea.edbc.org/ (accessed on 14 July 2023) | https://bio.liclab.net/SEanalysis/ (accessed on 14 July 2023) |
| Last update | 2017 | 2022 | 2019 | 2023 |
| Organisms | Human, mouse | Human, mouse | Human, mouse, Drosophila melanogaster, Caenorhabditis elegans, chicken, chimp, rhesus, sheep, Xenopus tropicalis, stickleback, zebrafish | Human, mouse |
| Data source (ChIP-Seq) | Collected existing data on SEs from published articles + NCBI GEO | NCBI GEO/SRA, ENCODE, Roadmap, Genomics of Gene Regulation Project (GGR), and National Genomics Data Center Genome Sequence Archive (NGDC GSA) | NCBI, GEO/SRA, ENCODE | Data from SEdb |
| Search pipeline (ChIP-Seq data alignment, peak calling, SE search, accordingly) | Varied between articles included in the database, more details in [95] | Bowtie2, MACS2, ROSE | Bowtie2, MACS2, ROSE | |
| Human | ||||
| Input data for SE prediction | H3K27ac | H3K27ac | H3K27ac, p300, BRD4, Med1 | H3K27ac |
| Genome version | hg19 | hg38, hg19 | hg38 | hg19 |
| Mouse | ||||
| Input data for SE prediction | Med1 (for ESCs and pro-B cells), MyoD (myotubes), T-bet (Th cells), C/EBP (macrophages) | H3K24ac | H3K24ac, p300 | H3K27ac |
| Genome version | mm9 | mm39, mm10 | mm10 | — |
| Database completeness | ||||
| Human | ||||
| SEs | 68,729 | 1,167,518 | 109,304 | 1,167,518 |
| Tissue types | Not given | 118 | Not given | Not given |
| Cell lines | Not given | Not given | 133 | Not given |
| Tissue types/cell lines | 99 | Not given | 141 | ∼180 |
| Mouse | ||||
| SEs | 2558 | 550,226 | 23,969 | 550,226 |
| Tissue types | Not given | 636 | Not given | 19 |
| Cell lines | Not given | 1107 | Not given | — |
| Tissue types/cell lines | 5 | Not given | 32 | ∼110 |
| Annotation completeness | ||||
| Tissue type | Yes | Yes | Yes | Yes |
| SE genomic location | Yes | Yes | Yes | Yes |
| SE constituents | Yes | Yes | Yes | Yes |
| SE conservation | No | Yes | Yes | Yes |
| Associated genes | Yes | Yes | Yes | Yes |
| RNA-seq | No | Yes | No | |
| DNase I hypersensitive sites (DHSs) | No | Yes | No | Yes |
| Methylation sites | No | Yes | Yes | No |
| H3K27ac | No | Yes | Yes | Yes |
| Chromatin interactions region | No | Yes | Yes | No |
| TADs | No | Yes | Yes | No |
| TF binding sites (predicted) | No | Yes | Yes | Yes |
| TF binding sites (ChIP-Seq) | No | Yes | Yes | Yes |
| TF binding sites conservation | No | Yes | Yes | Yes |
| SE associated pathways | No | No | Yes | Yes |
| SNPs | No | Yes | Yes | Yes |
| eQTL | No | Yes | No | No |
| CRISPR–Cas9 target site | No | Yes | Yes | No |
| Visualisation | ||||
| Genome browser | UCSC | On the site | On the site | USCS, on the site |
| SE location | Yes | Yes | Yes | No |
| SE constituents’ location | No | Yes | Yes | No |
| Associated genes | No | Yes | Yes | Yes |
| Nearby genes | Yes | Yes | ? | Yes |
| Genes expression | Yes | Yes | Yes | Yes |
| Enhancers | No | Yes | No | No |
| Methylation sites | Yes | Yes | Yes | Yes |
| H3K27ac | Yes | No | Yes | Yes |
| p300 | No | No | Yes | No |
| BRD4 | No | No | Yes | No |
| Med1 | No | No | Yes | No |
| TF binding sites | No | Yes | Yes | No |
| Chromatin interactions region | Yes | Yes | Yes | Yes |
| Regulatory networks formed by SE (SE, TF, gene) | No | Yes | Yes | Yes |
| SE conservation | Yes | Yes | Yes | Yes |
| SNPs | Yes | Yes | Yes | Yes |
| CRISPR–Cas9 target site | Yes | No | Yes | Yes |
| Analytic tools | Overlap analysis, downstream analysis (Galaxy server), Gene Ontology analysis (GREAT server), correlation analysis, gene-expression analysis and motif discovery (Cistrome server) | Differential Overlapping SE analysis, SE-based TF–gene analysis, Gene-SE analysis, SNP-SE analysis, Overlap analysis, Region analysis | GREAT (predicts functions of cis-regulatory regions), Enrichr (gene set enrichment analysis), Specific analysis of H3K27ac status, SE cell-type specificity, TF enrichment analysis, Regulatory network | Pathway downstream analysis, Upstream regulator analysis, Genomic region annotation TF regulatory analysis, Sample Comparative analysis |
| Links with | Galaxy GREAT Cistrome | GREAT | Galaxy Enrichr | GREAT UCSC NCBI Gene GeneCards UniProt |
| Data submission option | Yes | No | No | No |
| Inhibitor Group | Inhibitor | Primary Targets | Secondary Targets |
|---|---|---|---|
| Benzamides | Entinostat (MS-275) | HDAC1/2 | HDAC3 |
| Tacedinaline (C1994) | HDAC1/2/3 | – | |
| Merck60 | HDAC1/2 | – | |
| Mocetinostat (MGCD0103) | HDAC1/2 | HDAC3/11 | |
| 4SC202 | HDAC1/2/3 | – | |
| Hydroxamic acids | Panobinostat | HDAC1/2/3/4/7/9 | HDAC6/8 |
| Vorinostat (SAHA) | HDAC1/2/3/6/10/11 | HDAC8 | |
| Dacinostat (LAQ824) | HDAC1/2/3/6/10/11 | HDAC4/5/8 | |
| Rocilinostat (ACY1215) | HDAC6 | Other HDACs | |
| WT161 | HDAC6 | Other HDACs | |
| Pracinostat (SB939) | All HDACs except 6 | – | |
| MC 1568 | Class IIa, HDAC6 | Other HDACs | |
| OJI-1 | HDAC8 | – | |
| Other | TMP195 | Class IIa | – |
| Selisistat (EX 527) | SIRT1 | – | |
| Largazole | Class I, class IIb, class IV | – | |
| Romidepsin | Class I | – |
| Inhibitor 1 | Inhibitor 2 | Tumour | Effect | Reference(s) |
|---|---|---|---|---|
| JQ1 | THZ1 | Neuroblastoma | Synergetic inhibition of viability of neuroblastoma cell lines by CDK7 inhibition and BET inhibition | [263] |
| JQ1 | THZ1 | Pancreatic ductal adenocarcinoma | G2/M phase cell-cycle arrest, inhibition of cell migration and invasion by CDK7 inhibition and BET inhibition | [264] |
| JQ1 | THZ1 | Head neck squamous cell carcinoma | Antiproliferative and proapoptotic effects by CDK7 inhibition and BET inhibition | [265] |
| JQ1 | YKL-5-124 | Neuroblastoma | Synergistic cytotoxicity, CDK7 inhibition, and BET inhibition | [241] |
| JQ1 | Milciclib | Medulloblastoma | Suppressing MYC-driven tumour, CDK2 inhibition, and BET inhibition | [266] |
| JQ1 | ICG-001 | Glioma | Strong cytotoxic effects, CBP and BET inhibition | [267] |
| JQ1 | Trametinib | Colorectal cancer | Inhibition of cell proliferation by dual targeting of BET proteins and MAPK signaling | [244] |
| OTX015 | CYH33 | B-cell lymphoma | Cell-cycle arrest and apoptosis, BET inhibition combined with PI3Kα-selective inhibition | [268] |
| THZ1 | Ponatinib/ lapatinib | Neuroblastoma | Cell apoptosis: CDK7 inhibition and tyrosine kinase inhibition | [269] |
| THZ1 | BH3-mimetics | Glioblastoma | Growth reduction of tumours by CDK7 inhibition, and antiapoptotic effect | [270] |
| THZ1 | Panobinostat | Diffuse intrinsic pontine glioma | Synergetic SE disruption, CDK7 inhibition, and HDAC inhibition | [271] |
| THZ1 | Panobinostat | Neuroblastoma | Apoptosis induction in cancer cells, CDK7 inhibition and HDAC inhibition | [272] |
| HDAC inhibitors | FAO inhibitors | Glioblastoma | HDAC inhibition and influence lipid metabolism | [273] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasileva, A.V.; Gladkova, M.G.; Ashniev, G.A.; Osintseva, E.D.; Orlov, A.V.; Kravchuk, E.V.; Boldyreva, A.V.; Burenin, A.G.; Nikitin, P.I.; Orlova, N.N. Super-Enhancers and Their Parts: From Prediction Efforts to Pathognomonic Status. Int. J. Mol. Sci. 2024, 25, 3103. https://doi.org/10.3390/ijms25063103
Vasileva AV, Gladkova MG, Ashniev GA, Osintseva ED, Orlov AV, Kravchuk EV, Boldyreva AV, Burenin AG, Nikitin PI, Orlova NN. Super-Enhancers and Their Parts: From Prediction Efforts to Pathognomonic Status. International Journal of Molecular Sciences. 2024; 25(6):3103. https://doi.org/10.3390/ijms25063103
Chicago/Turabian StyleVasileva, Anastasia V., Marina G. Gladkova, German A. Ashniev, Ekaterina D. Osintseva, Alexey V. Orlov, Ekaterina V. Kravchuk, Anna V. Boldyreva, Alexander G. Burenin, Petr I. Nikitin, and Natalia N. Orlova. 2024. "Super-Enhancers and Their Parts: From Prediction Efforts to Pathognomonic Status" International Journal of Molecular Sciences 25, no. 6: 3103. https://doi.org/10.3390/ijms25063103
APA StyleVasileva, A. V., Gladkova, M. G., Ashniev, G. A., Osintseva, E. D., Orlov, A. V., Kravchuk, E. V., Boldyreva, A. V., Burenin, A. G., Nikitin, P. I., & Orlova, N. N. (2024). Super-Enhancers and Their Parts: From Prediction Efforts to Pathognomonic Status. International Journal of Molecular Sciences, 25(6), 3103. https://doi.org/10.3390/ijms25063103