

Jozimine A2, a Dimeric Naphthylisoquinoline (NIQ) Alkaloid, Shows In Vitro Cytotoxic Effects against Leukemia Cells through NF-κB Inhibition

 and
and
Abstract
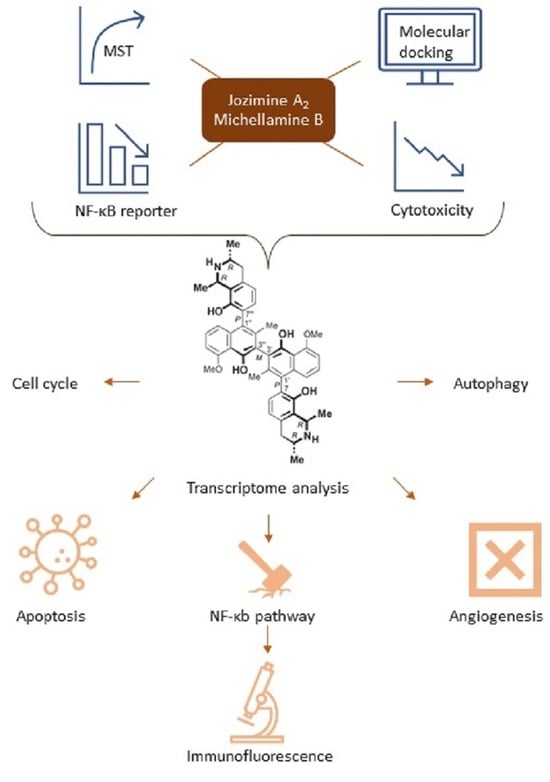
1. Introduction
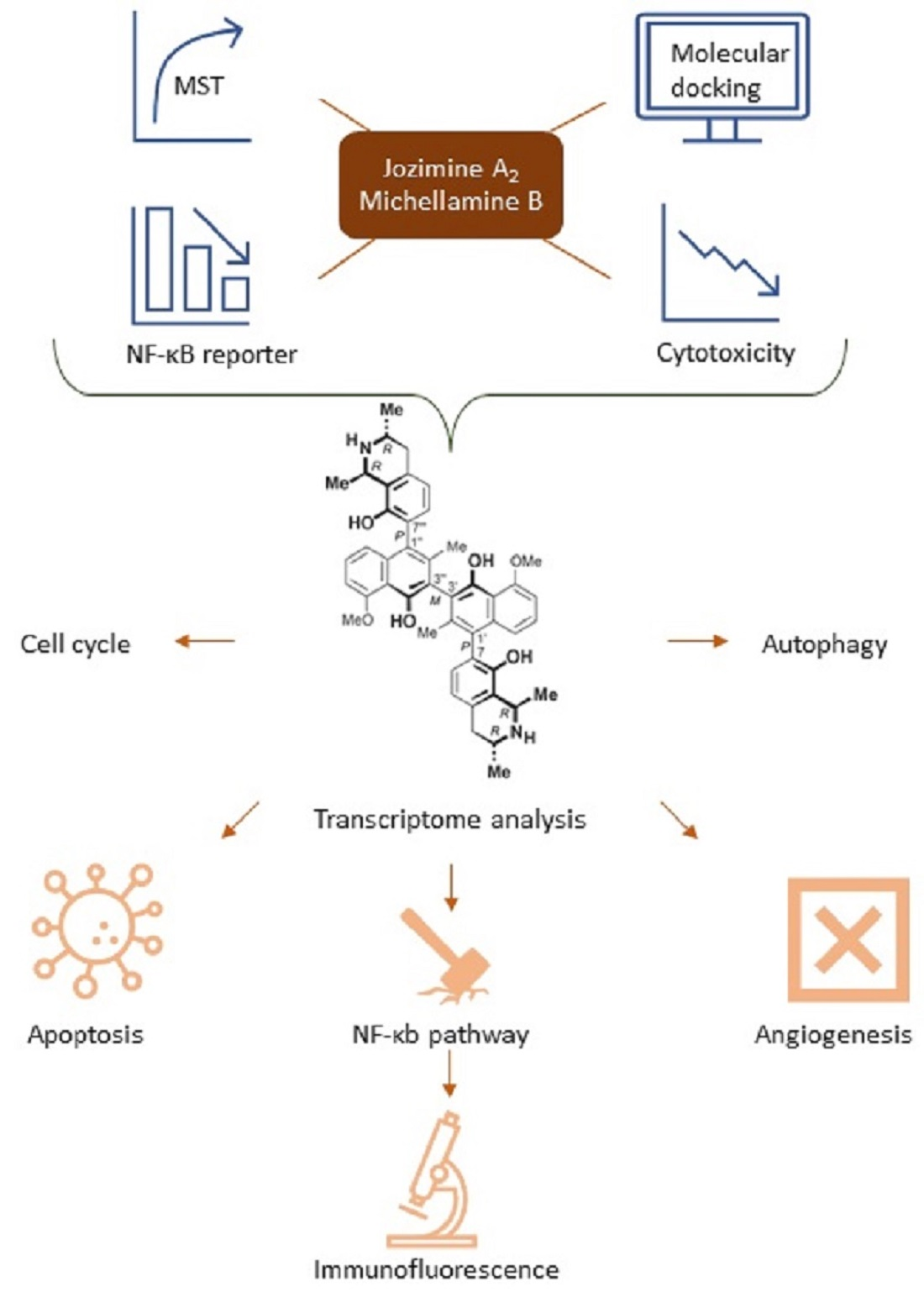
2. Results
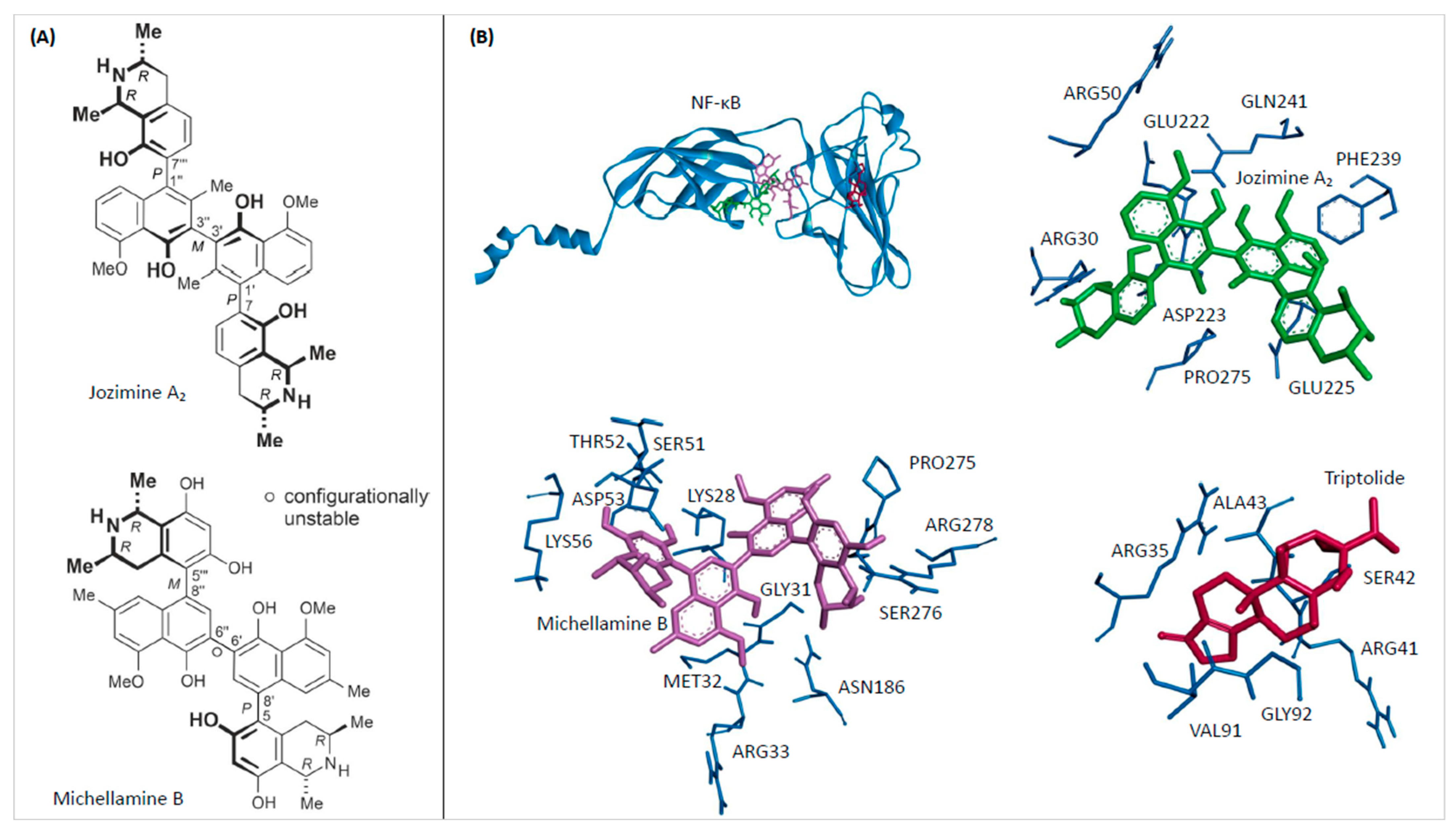
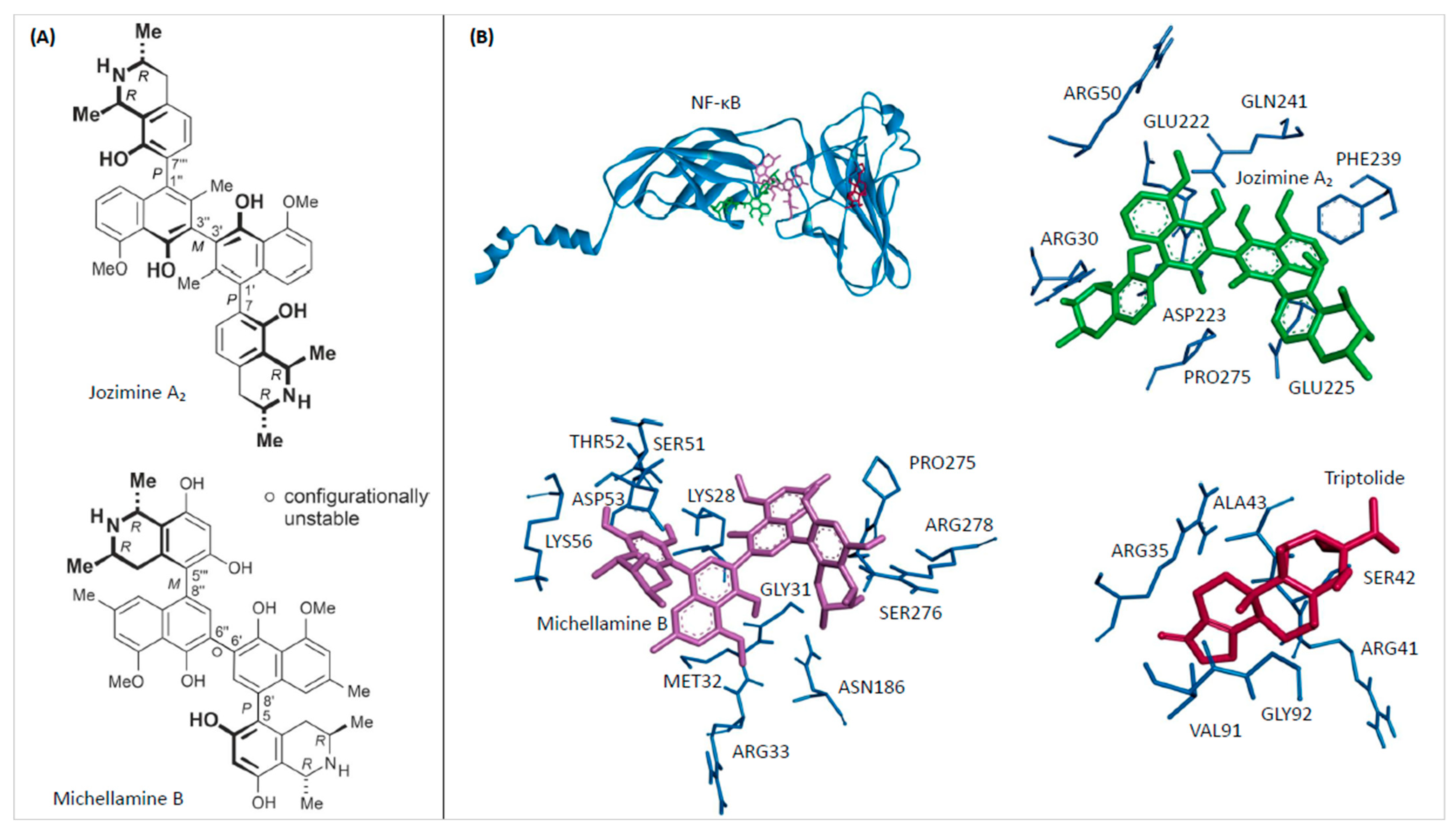
2.1. In Silico Binding of Jozimine A2 and Michellamine B to NF-κB
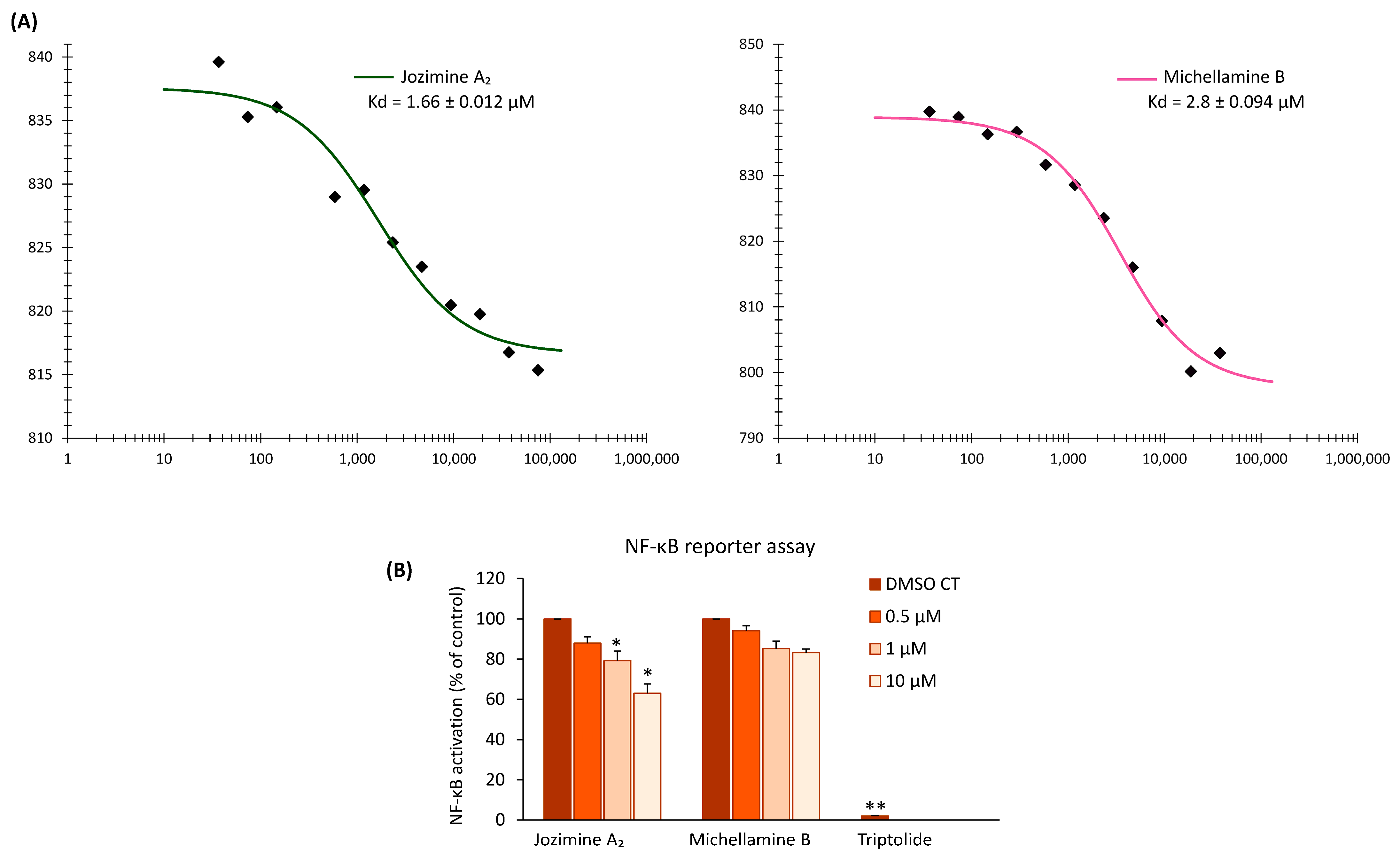
2.2. Jozimine A2 and Michellamine B Both Bind to NF-κB In Vitro, but Only Jozimine A2 Inhibits NF-κB Activity
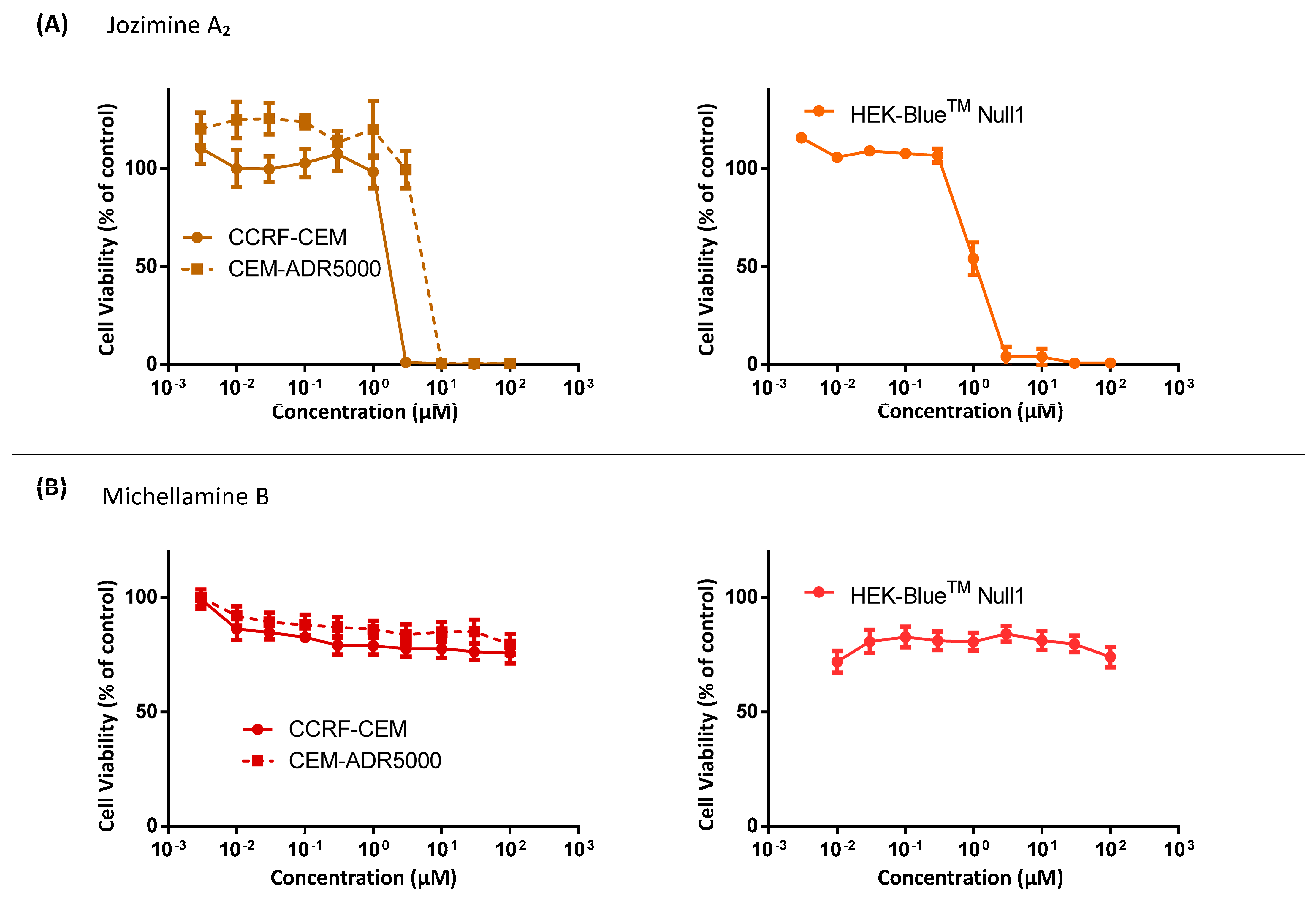
2.3. Cytotoxicity of Jozimine A2 and Michellamine B
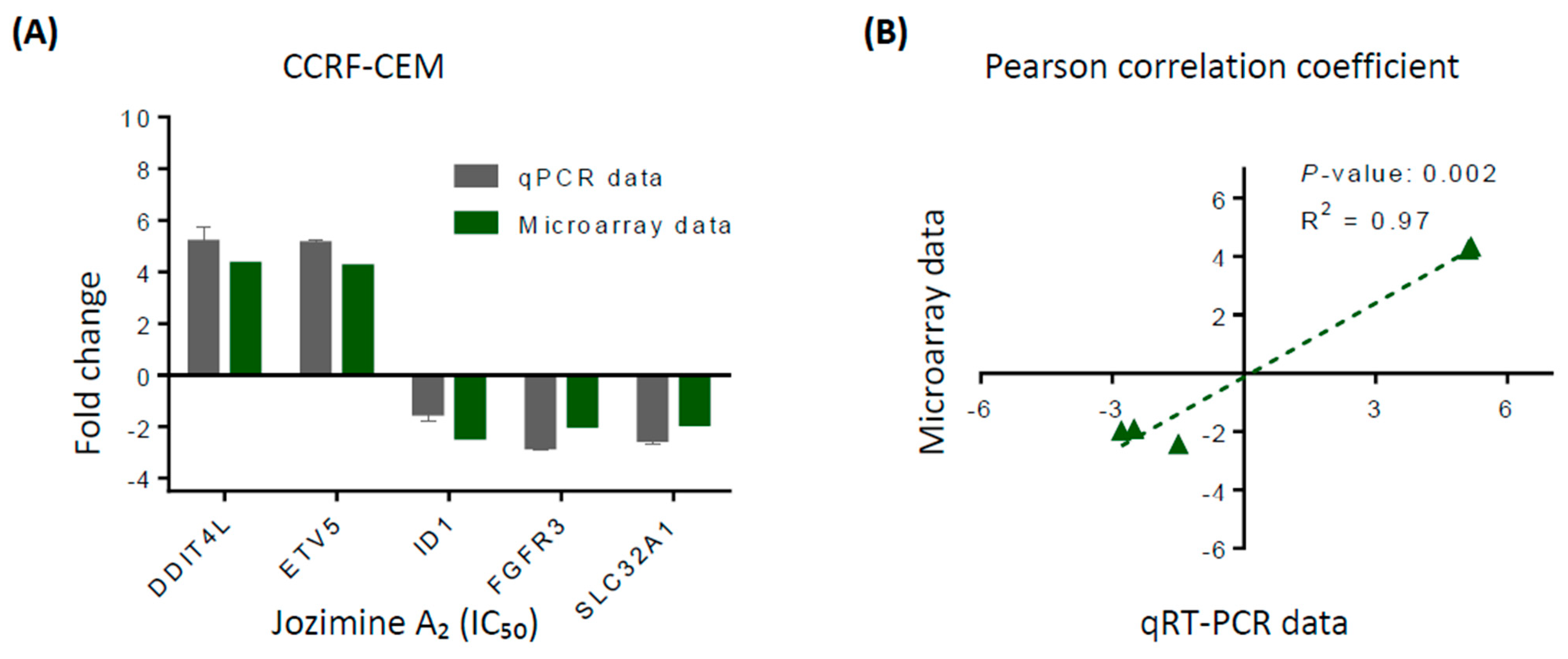
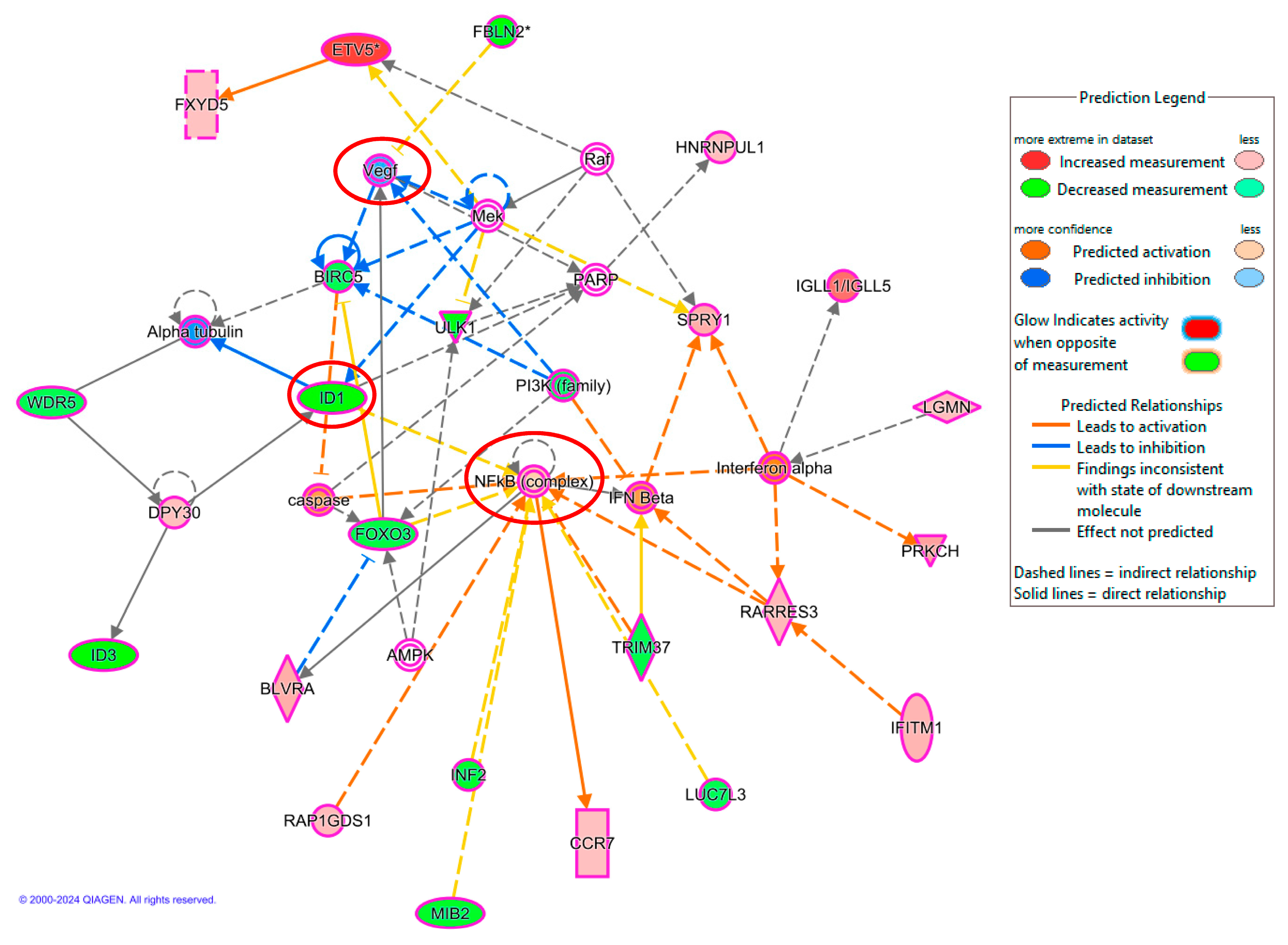
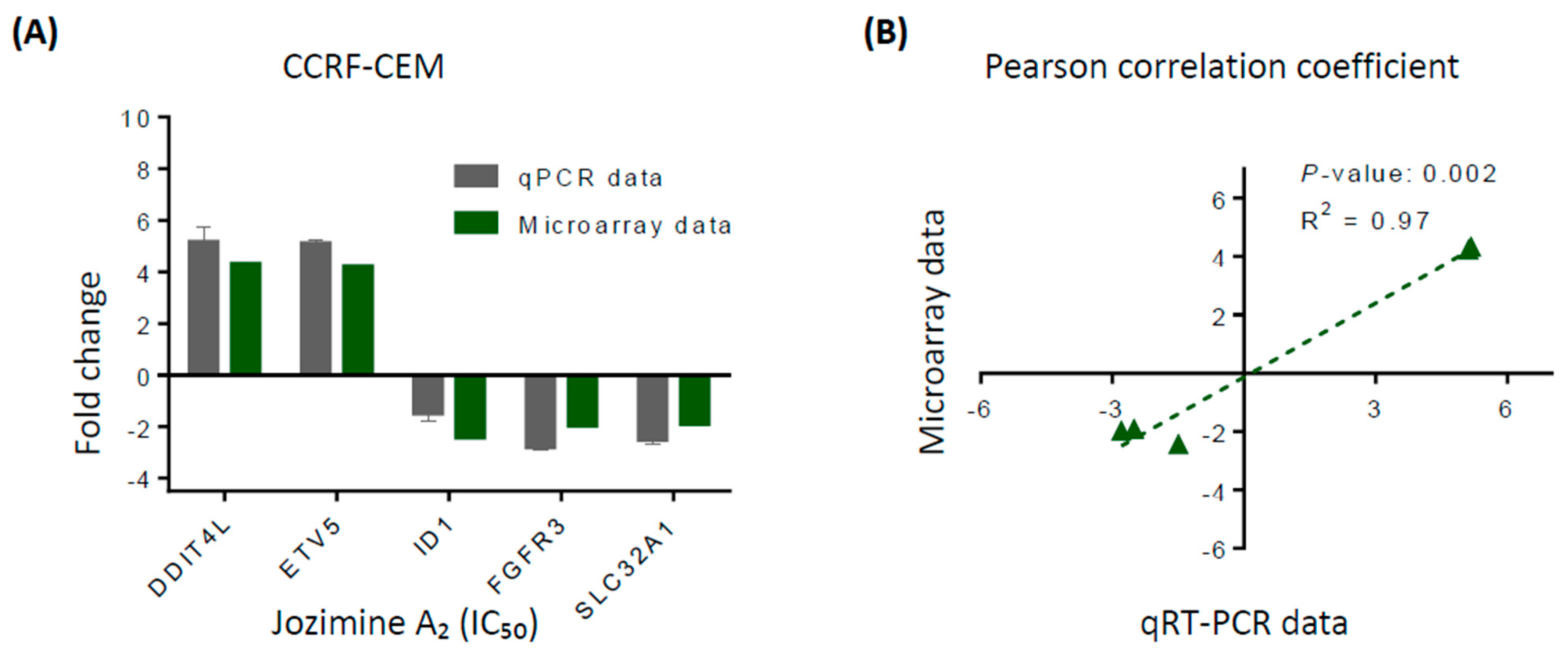
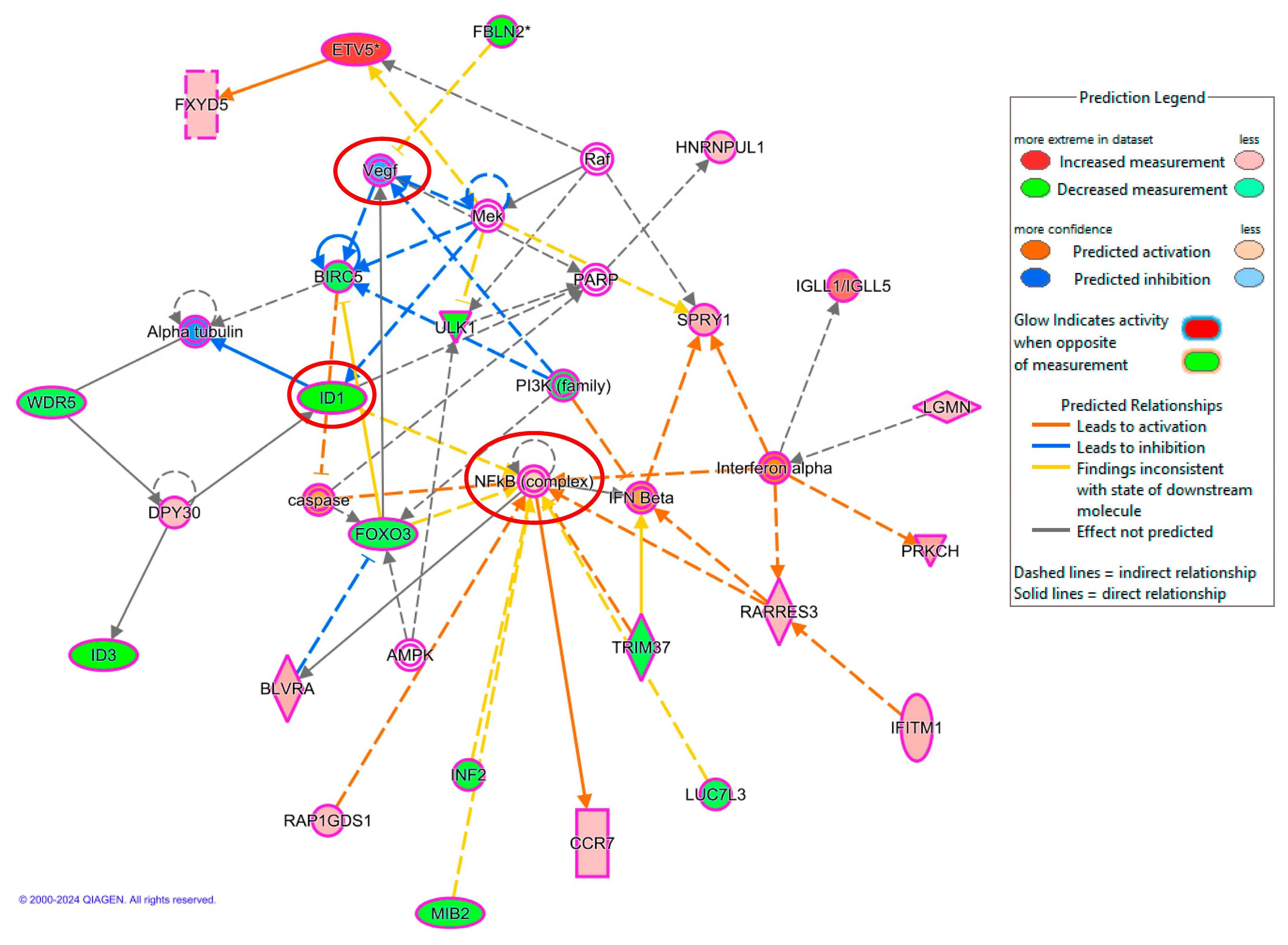
2.4. Validation of Microarray Analysis Using qRT-PCR
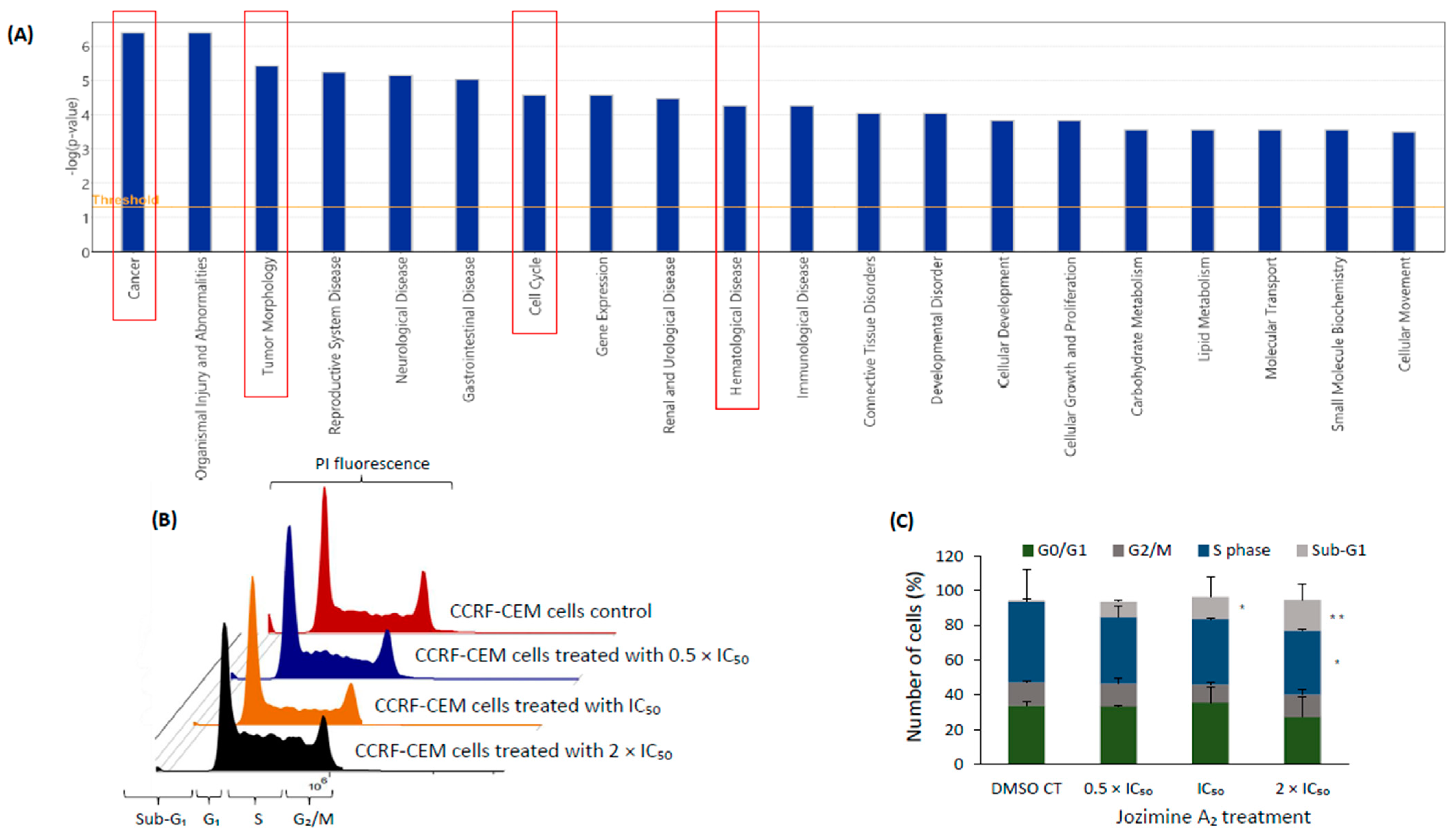
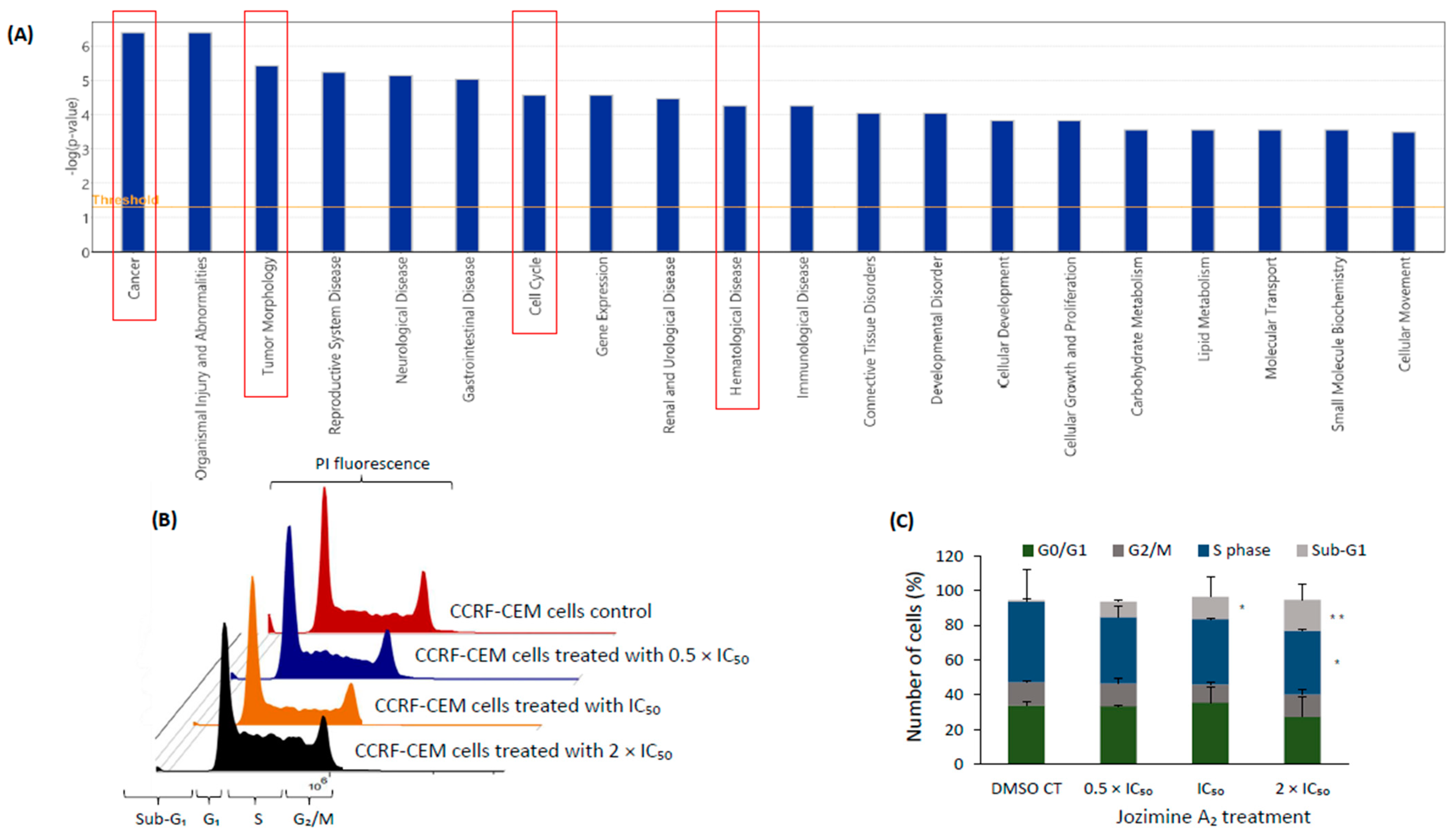
2.5. The Effect of Jozimine A2 on the Cell Cycle
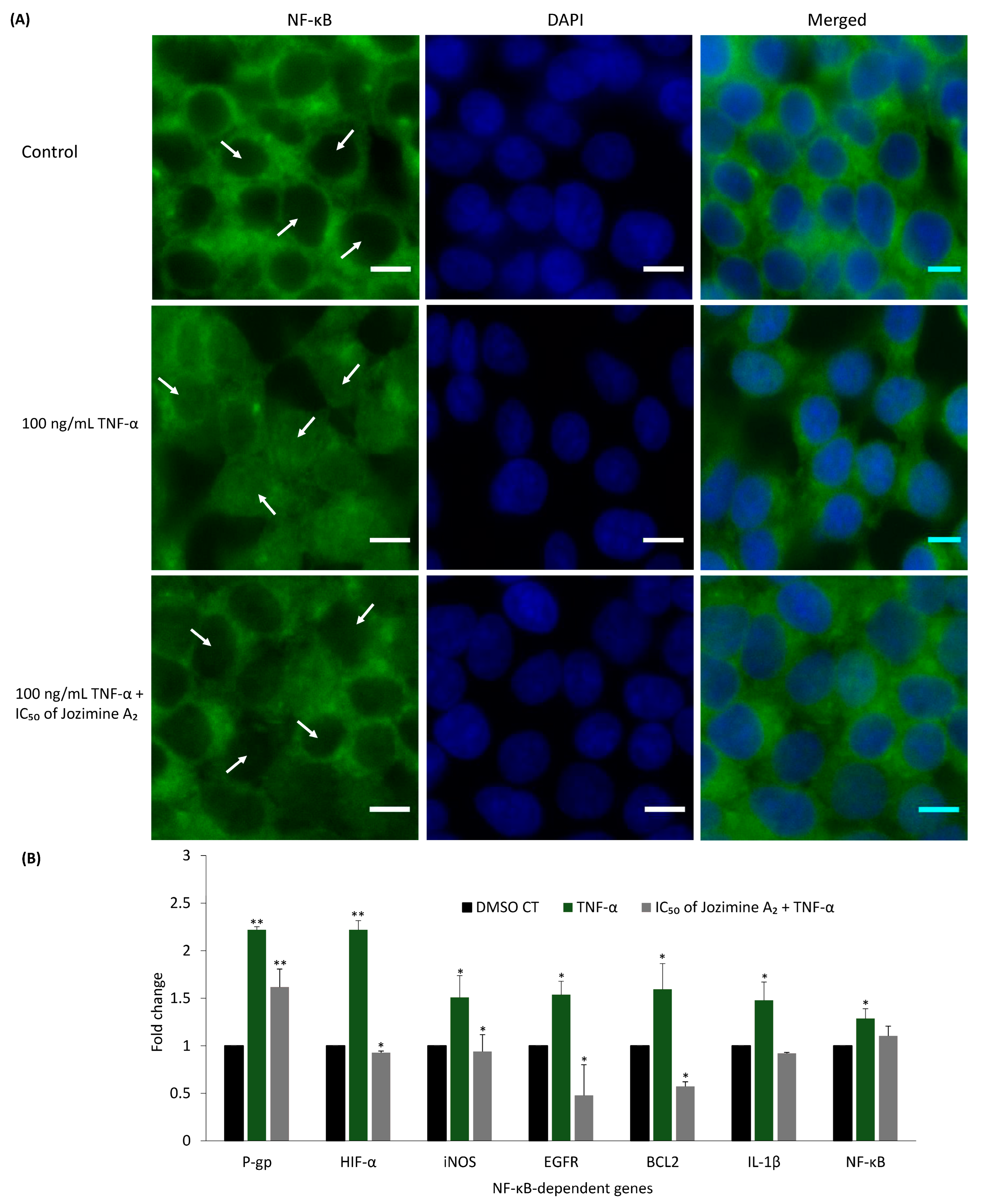
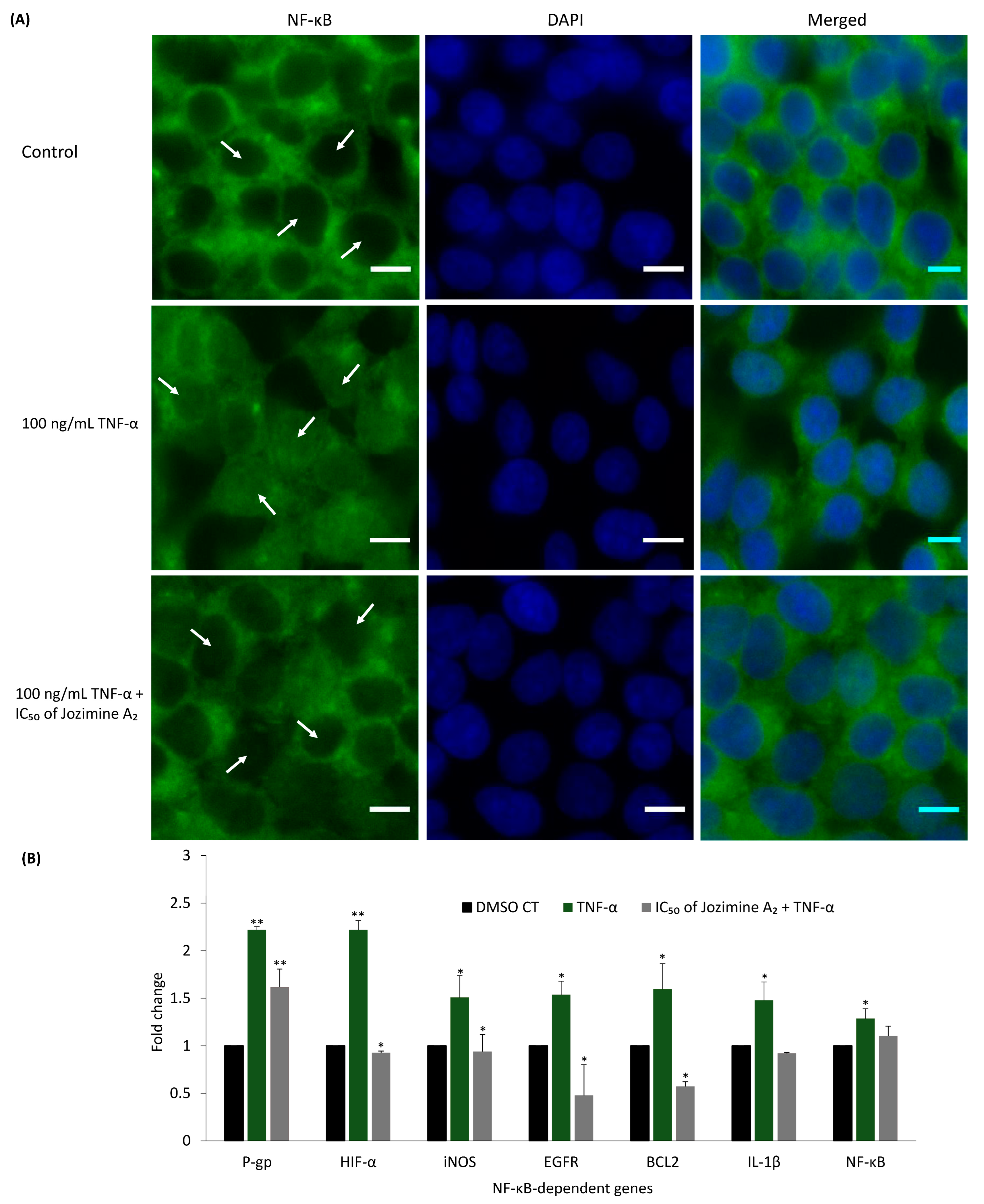
2.6. Jozimine A2 Prevents NF-κB Translocation and Suppresses the Expression of NF-κB-Dependent Genes
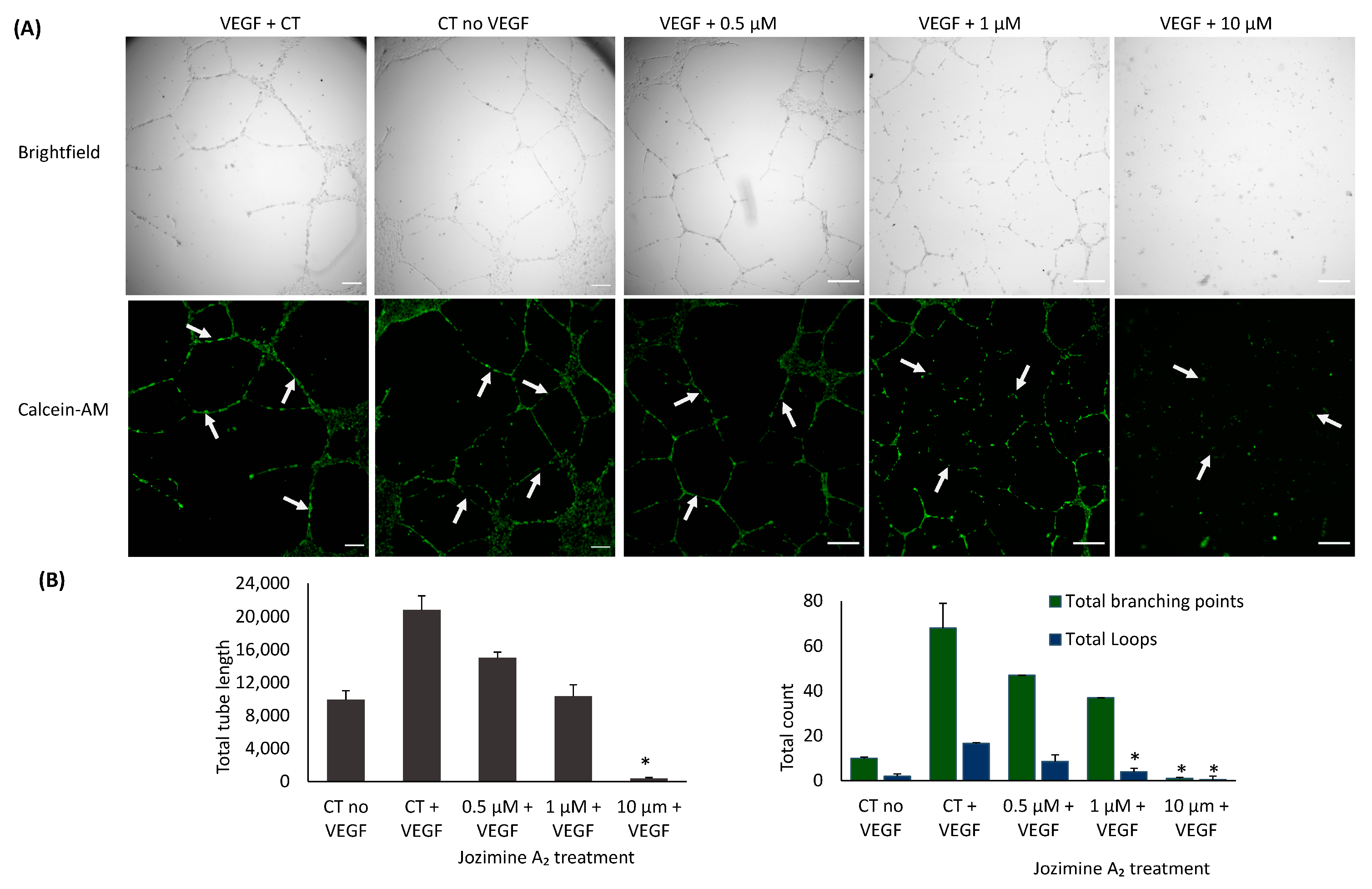
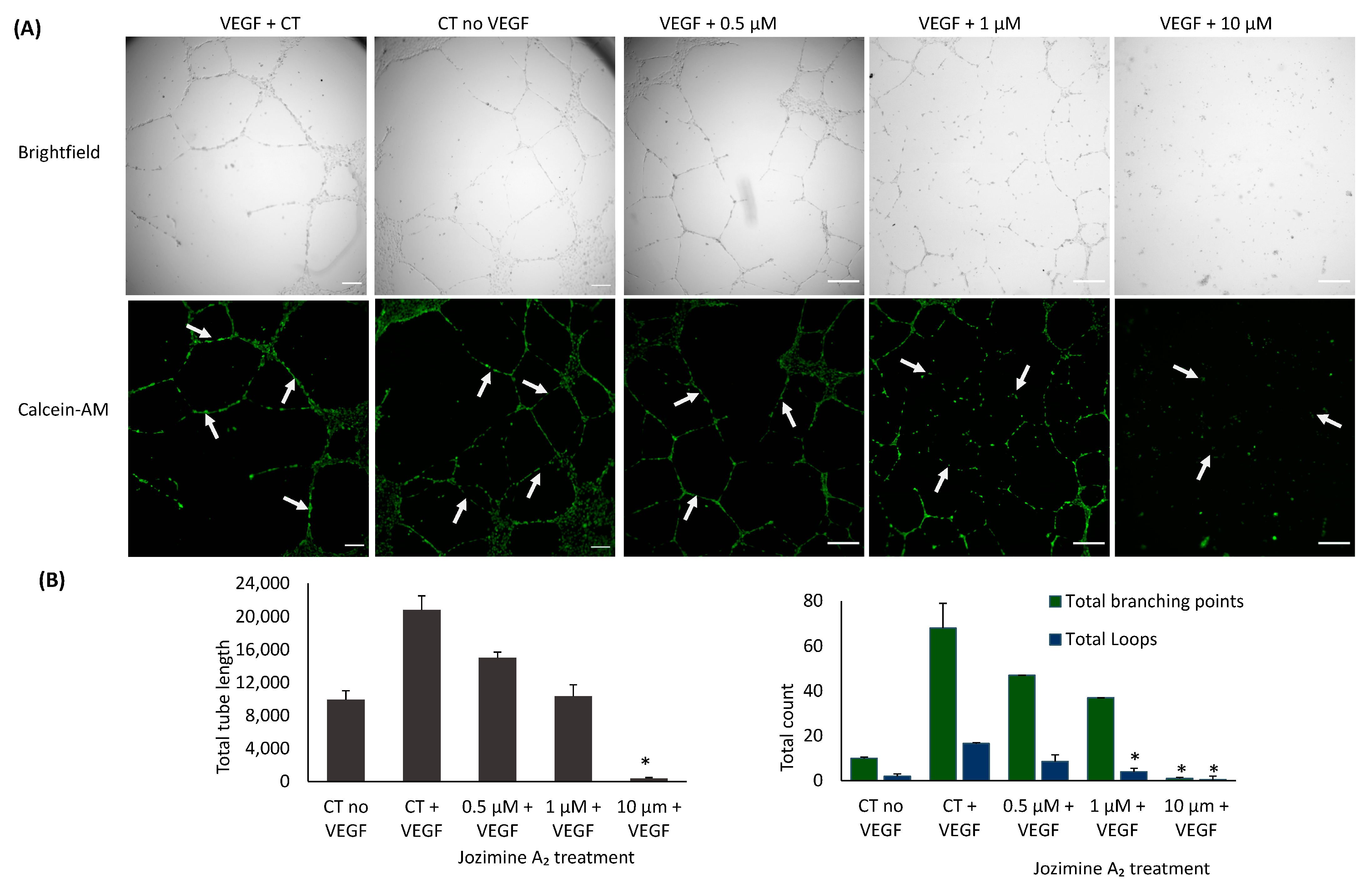
2.7. Suppression of Endothelial Tube Formation by Jozimine A2
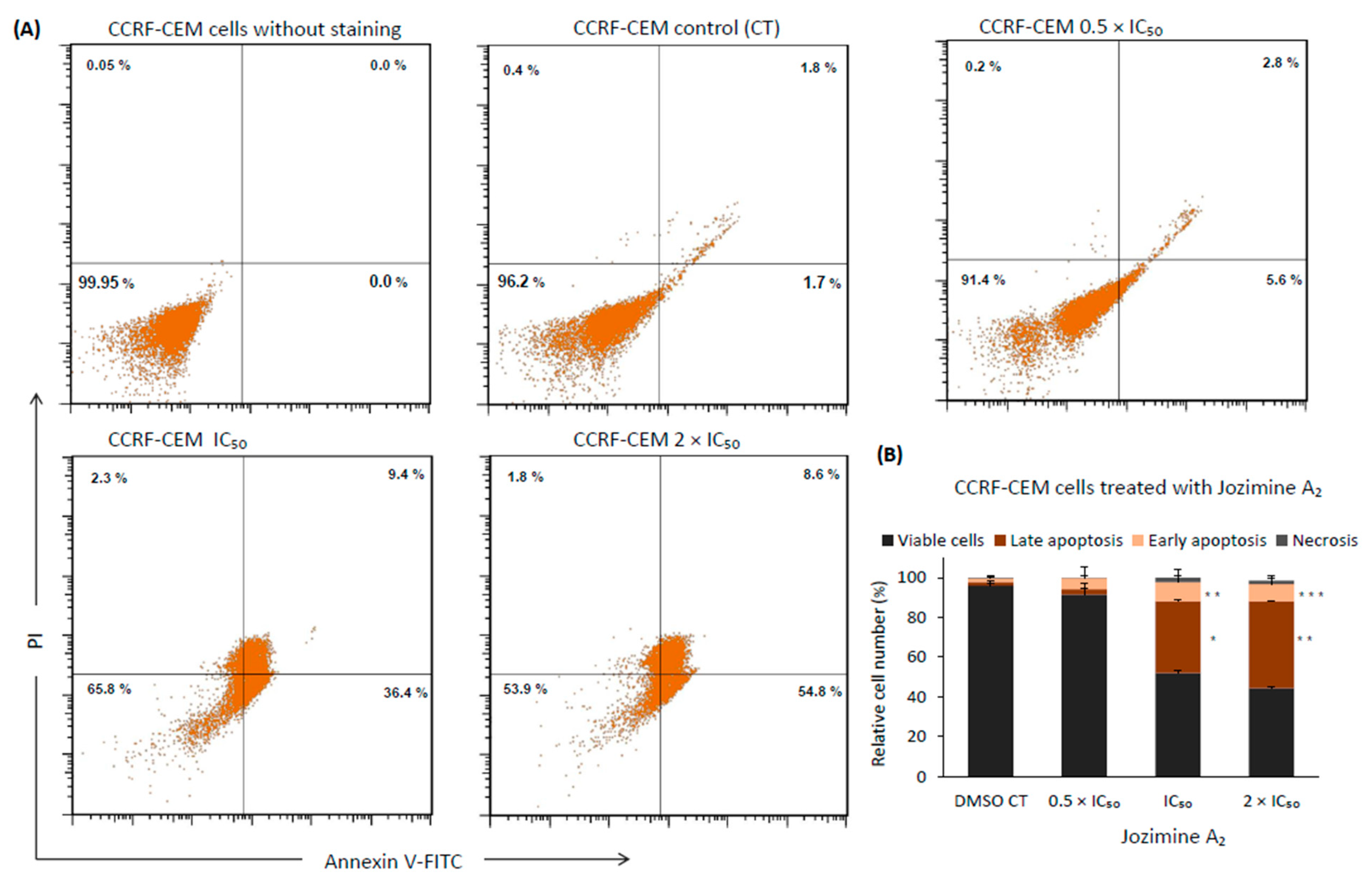
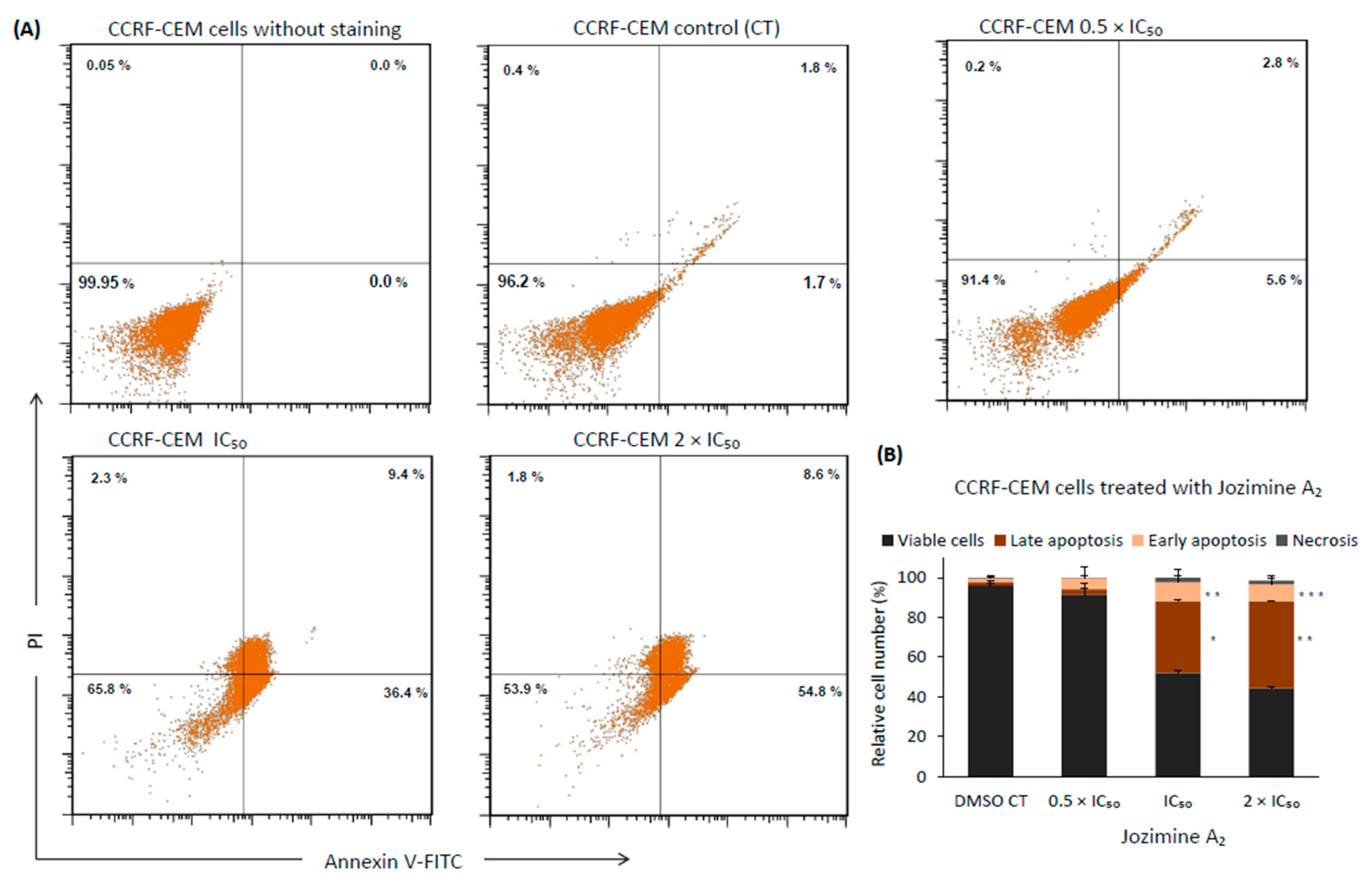
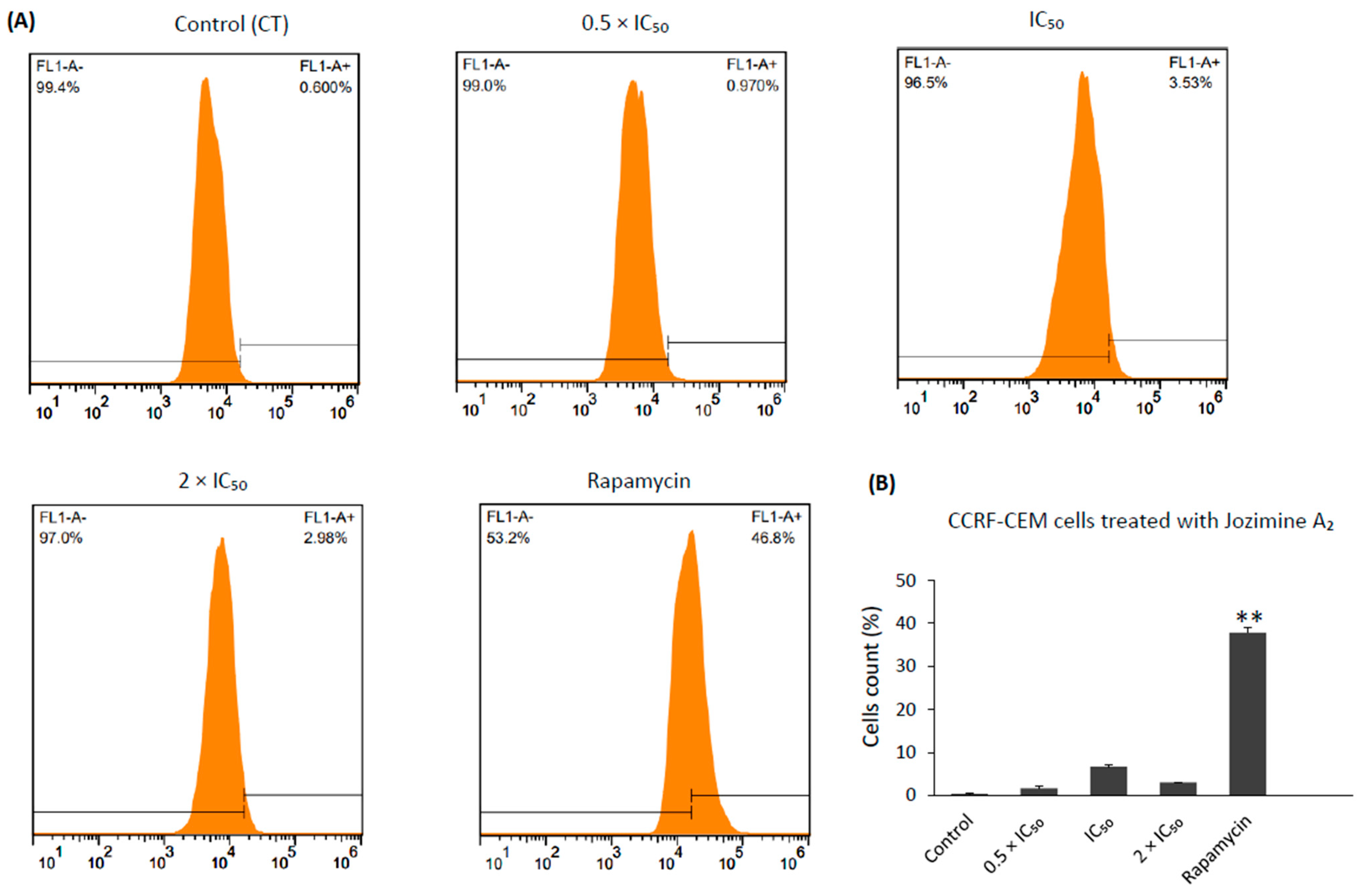
2.8. Jozimine A2 Induces Cell Death through Apoptosis by Autophagy
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.2. Microscale Thermophoresis
4.3. NF-κB Reporter Assay
4.4. Cytotoxicity Assay
4.5. Microarray Hybridization
4.6. Quantitative Real-Time PCR
4.7. Cell Cycle
4.8. Immunofluorescence
4.9. Endothelial Tube Formation
4.10. Apoptosis
4.11. Autophagy
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feineis, D.; Bringmann, G. Asian Ancistrocladus Lianas as Creative Producers of Naphthylisoquinoline Alkaloids. In Progress in the Chemistry of Organic Natural Products; Springer: Berlin/Heidelberg, Germany, 2023; Volume 119. [Google Scholar]
- Lombe, B.K.; Feineis, D.; Bringmann, G. Dimeric Naphthylisoquinoline Alkaloids: Polyketide-Derived Axially Chiral Bioactive Quateraryls. Nat. Prod. Rep. 2019, 36, 1513–1545. [Google Scholar] [CrossRef]
- Yücer, R.; Fayez, S.; Feineis, D.; Klauck, S.M.; Shan, L.; Bringmann, G.; Efferth, T.; Dawood, M. Cytotoxicity of Dioncophylline A and Related Naphthylisoquinolines in Leukemia Cells, Mediated by NF-κB Inhibition, Angiogenesis Suppression, G2/M Cell Cycle Arrest, and Autophagy Induction. Phytomedicine 2023, 2023, 155267. [Google Scholar] [CrossRef]
- Bringmann, G.; Zhang, G.; Büttner, T.; Bauckmann, G.; Kupfer, T.; Braunschweig, H.; Brun, R.; Mudogo, V. Jozimine A2: The First Dimeric Dioncophyllaceae-Type Naphthylisoquinoline Alkaloid, with Three Chiral Axes and High Antiplasmodial Activity. Chem. A Eur. J. 2013, 19, 916–923. [Google Scholar] [CrossRef]
- Fayez, S.; Feineis, D.; Mudogo, V.; Seo, E.J.; Efferth, T.; Bringmann, G. Ancistrolikokine I and Further 5,8′-Coupled Naphthylisoquinoline Alkaloids from the Congolese Liana Ancistrocladus likoko and Their Cytotoxic Activities against Drug-Sensitive and Multidrug Resistant Human Leukemia Cells. Fitoterapia 2018, 129, 114–125. [Google Scholar] [CrossRef]
- Li, J.; Seupel, R.; Bruhn, T.; Feineis, D.; Kaiser, M.; Brun, R.; Mudogo, V.; Awale, S.; Bringmann, G. Jozilebomines A and B, Naphthylisoquinoline Dimers from the Congolese Liana Ancistrocladus ileboensis, with Antiausterity Activities against the PANC-1 Human Pancreatic Cancer Cell Line. J. Nat. Prod. 2017, 80, 2807–2817. [Google Scholar] [CrossRef]
- Boyd, M.R.; Hallock, Y.F.; Cardellina, J.H.; Manfredi, K.P.; Blunt, J.W.; McMahon, J.B.; Buckheit, R.W.; Bringmann, G.; Schäffer, M.; Cragg, G.M.; et al. Anti-HIV Michellamines from Ancistrocladus korupensis. J. Med. Chem. 1994, 37, 1740–1745. [Google Scholar] [CrossRef]
- Orlowski, R.Z.; Baldwin, A.S. NF-κB as a Therapeutic Target in Cancer. Trends. Mol. Med. 2002, 8, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Staudt, L.M. Oncogenic Activation of NF-κB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000109. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. NF-κB as a Critical Link between Inflammation and Cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef]
- Lin, A.; Karin, M. NF-κB in Cancer: A Marked Target. Semin. Cancer Biol. 2003, 13, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, B.; Peng, J.; Tang, H.; Wang, S.; Peng, S.; Ye, F.; Wang, J.; Ouyang, K.; Li, J.; et al. Inhibition of NF-κB Signaling Unveils Novel Strategies to Overcome Drug Resistance in Cancers. Drug Resist. Updates 2024, 73, 101042. [Google Scholar] [CrossRef]
- deGraffenried, L.A.; Chandrasekar, B.; Friedrichs, W.E.; Donzis, E.; Silva, J.; Hidalgo, M.; Freeman, J.W.; Weiss, G.R. NF-κB Inhibition Markedly Enhances Sensitivity of Resistant Breast Cancer Tumor Cells to Tamoxifen. Ann. Oncol. 2004, 15, 885–890. [Google Scholar] [CrossRef]
- Abdin, S.M.; Tolba, M.F.; Zaher, D.M.; Omar, H.A. Nuclear Factor-κB Signaling Inhibitors Revert Multidrug-Resistance in Breast Cancer Cells. Chem. Biol. Interact. 2021, 340, 109450. [Google Scholar] [CrossRef] [PubMed]
- Bentires-Alj, M.; Barbu, V.; Fillet, M.; Chariot, A.; Relic, B.; Jacobs, N.; Gielen, J.; Merville, M.P.; Bours, V. NF-κB Transcription Factor Induces Drug Resistance through MDR1 Expression in Cancer Cells. Oncogene 2003, 22, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Radzka, J.; Łapińska, Z.; Szwedowicz, U.; Gajewska-Naryniecka, A.; Gizak, A.; Kulbacka, J. Alternations of NF-κB Signaling by Natural Compounds in Muscle-Derived Cancers. Int. J. Mol. Sci. 2023, 24, 11900. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The Complexity of NF-κB Signaling in Inflammation and Cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 Signaling Pathways Collaboratively Link Inflammation to Cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-κB Signaling in Inflammation and Cancer. MedComm 2021, 2, 618–653. [Google Scholar] [CrossRef]
- Yu, L.; Li, L.; Medeiros, L.J.; Young, K.H. NF-κB Signaling Pathway and Its Potential as a Target for Therapy in Lymphoid Neoplasms. Blood Rev. 2017, 31, 77–92. [Google Scholar] [CrossRef]
- Bernal-Mizrachi, L.; Lovly, C.M.; Ratner, L. The Role of NF-κB-1 and NF-κB-2-Mediated Resistance to Apoptosis in Lymphomas. Proc. Natl. Acad. Sci. USA 2006, 103, 9220–9225. [Google Scholar] [CrossRef]
- Di Francesco, B.; Verzella, D.; Capece, D.; Vecchiotti, D.; Di Vito Nolfi, M.; Flati, I.; Cornice, J.; Di Padova, M.; Angelucci, A.; Alesse, E.; et al. NF-κB: A Druggable Target in Acute Myeloid Leukemia. Cancers 2022, 14, 3557. [Google Scholar] [CrossRef]
- Braun, T.; Carvalho, G.; Fabre, C.; Grosjean, J.; Fenaux, P.; Kroemer, G. Targeting NF-κB in Hematologic Malignancies. Cell Death Differ. 2006, 13, 748–758. [Google Scholar] [CrossRef]
- Zhou, J.; Ching, Y.Q.; Chng, W.J. Aberrant Nuclear Factor-Kappa B Activity in Acute Myeloid Leukemia: From Molecular Pathogenesis to Therapeutic Target. Oncotarget 2015, 6, 5490. [Google Scholar] [CrossRef]
- Trocoli, A.; Djavaheri-Mergny, M. The Complex Interplay between Autophagy and NF-κB Signaling Pathways in Cancer Cells. Am. J. Cancer Res. 2011, 1, 629. [Google Scholar]
- Ahmadi-Dehlaghi, F.; Mohammadi, P.; Valipour, E.; Pournaghi, P.; Kiani, S.; Mansouri, K. Autophagy: A Challengeable Paradox in Cancer Treatment. Cancer Med. 2023, 12, 11542–11569. [Google Scholar] [CrossRef]
- Luo, M.; Han, L. Autophagy Induction Sensitizes Cancer Cells to Anti-Cancer Drugs. Autophagy 2022, 19, 2393–2394. [Google Scholar] [CrossRef]
- Jung, H.J.; Seo, I.; Casciello, F.; Jacquelin, S.; Lane, S.W.; Suh, S.I.; Suh, M.H.; Lee, J.S.; Baek, W.K. The Anticancer Effect of Chaetocin Is Enhanced by Inhibition of Autophagy. Cell Death Dis. 2016, 7, e2098. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Zhou, X.; Li, H.; Ma, X.; Zhuang, J. NF-κB Inhibitors Gifted by Nature: The Anticancer Promise of Polyphenol Compounds. Biomed. Pharmacother. 2022, 156, 113951. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Mehta, D.K.; Dhanawat, M. Medicinal Plants in Cancer Treatment: Contribution of Nuclear Factor- Kappa B (NF-κB) Inhibitors. Mini-Rev. Med. Chem. 2022, 22, 1938–1962. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, T.S.; Ayob, A.Z.; Myint, H.H.L.; Thiagarajah, S.; Amini, F. Targeting Colorectal Cancer Stem Cells Using Curcumin and Curcumin Analogues: Insights into the Mechanism of the Therapeutic Efficacy. Cancer Cell Int. 2015, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Jobin, C.; Bradham, C.A.; Russo, M.P.; Juma, B.; Narula, A.S.; Brenner, D.A.; Sartor, R.B. Curcumin Blocks Cytokine-Mediated NF-κB Activation and Proinflammatory Gene Expression by Inhibiting Inhibitory Factor I-κB Kinase Activity. J. Immunol. 1999, 163, 3474–3483. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.F.S.; Silva, G.D.B.; Pavan, A.R.; Chiba, D.E.; Chin, C.M.; Dos Santos, J.L. Epigenetic Regulatory Mechanisms Induced by Resveratrol. Nutrients 2017, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.; Ullah, M.; Faisal, M.; Farooqi, A.; Sabitaliyevich, U.; Biersack, B.; Ahmad, A. Differential Methylation and Acetylation as the Epigenetic Basis of Resveratrol’s Anticancer Activity. Medicines 2019, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Wang, L.; Fu, S.; Jiang, B. Resveratrol Is Cytotoxic and Acts Synergistically with NF-κB Inhibition in Osteosarcoma MG-63 Cells. Arch. Med. Sci. 2021, 17, 166. [Google Scholar] [CrossRef] [PubMed]
- Jerabek-Willemsen, M.; Wienken, C.J.; Braun, D.; Baaske, P.; Duhr, S. Molecular Interaction Studies Using Microscale Thermophoresis. Assay Drug Dev. Technol. 2011, 9, 342–353. [Google Scholar] [CrossRef]
- Awale, S.; Dibwe, D.F.; Balachandran, C.; Fayez, S.; Feineis, D.; Lombe, B.K.; Bringmann, G. Ancistrolikokine E3, a 5,8′-Coupled Naphthylisoquinoline Alkaloid, Eliminates the Tolerance of Cancer Cells to Nutrition Starvation by Inhibition of the Akt/MTOR/Autophagy Signaling Pathway. J. Nat. Prod. 2018, 81, 2282–2291. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yi, J.; Li, T.; Wen, J.; Huang, K.; Liu, J.; Wang, G.; Kim, P.; Song, Q.; Zhou, X. DRMref: Comprehensive Reference Map of Drug Resistance Mechanisms in Human Cancer. Nucleic Acids Res. 2024, 52, D1253–D1264. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Welz, L.; Kakavand, N.; Hang, X.; Laue, G.; Ito, G.; Silva, M.G.; Plattner, C.; Mishra, N.; Tengen, F.; Ogris, C.; et al. Epithelial X-Box Binding Protein 1 Coordinates Tumor Protein P53-Driven DNA Damage Responses and Suppression of Intestinal Carcinogenesis. Gastroenterology 2022, 162, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wei, H.; Prabhu, L.; Zhao, W.; Martin, M.; Hartley, A.V.; Lu, T. Role of Novel Serine 316 Phosphorylation of the P65 Subunit of NF-κB in Differential Gene Regulation. J. Biol. Chem. 2015, 290, 20336–20347. [Google Scholar] [CrossRef]
- Shimizu, H.; Horii, A.; Sunamura, M.; Motoi, F.; Egawa, S.; Unno, M.; Fukushige, S. Identification of Epigenetically Silenced Genes in Human Pancreatic Cancer by a Novel Method “Microarray Coupled with Methyl-CpG Targeted Transcriptional Activation” (MeTA-Array). Biochem. Biophys. Res. Commun. 2011, 411, 162–167. [Google Scholar] [CrossRef]
- Li, L.; Zhang, S.; Li, H.; Chou, H. FGFR3 Promotes the Growth and Malignancy of Melanoma by Influencing EMT and the Phosphorylation of ERK, AKT, and EGFR. BMC Cancer 2019, 19, 963. [Google Scholar] [CrossRef]
- Wu, W.B.; Chen, L.; Jia, G.Z.; Tang, Q.L.; Han, B.M.; Xia, S.J.; Jiang, Q.; Liu, H.T. Inhibition of FGFR3 Upregulates MHC-I and PD-L1 via TLR3/NF-κB Pathway in Muscle-Invasive Bladder Cancer. Cancer Med. 2023, 12, 15676–15690. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar]
- Brünnert, D.; Seupel, R.; Goyal, P.; Bach, M.; Schraud, H.; Kirner, S.; Köster, E.; Feineis, D.; Bargou, R.C.; Schlosser, A.; et al. Ancistrocladinium A Induces Apoptosis in Proteasome Inhibitor-Resistant Multiple Myeloma Cells: A Promising Therapeutic Agent Candidate. Pharmaceuticals 2023, 16, 1181. [Google Scholar] [CrossRef]
- Stucki, M. Histone H2A.X Tyr142 Phosphorylation: A Novel SWItCH for Apoptosis? DNA Repair. 2009, 8, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.; Moridnia, A. Apoptosis-Inducing and Antiproliferative Effect by Inhibition of MiR-182-5p through the Regulation of CASP9 Expression in Human Breast Cancer. Cancer Gene Ther. 2017, 24, 75–82. [Google Scholar] [CrossRef]
- Li, L.; Liu, W.; Sun, Q.; Zhu, H.; Hong, M.; Qian, S. Decitabine Downregulates TIGAR to Induce Apoptosis and Autophagy in Myeloid Leukemia Cells. Oxid. Med. Cell Longev. 2021, 2021, 8877460. [Google Scholar] [CrossRef]
- Yinjun, L.; Jie, J.; Yungui, W. Triptolide Inhibits Transcription Factor NF-κB and Induces Apoptosis of Multiple Myeloma Cells. Leuk. Res. 2005, 29, 99–105. [Google Scholar] [CrossRef]
- Saeed, M.E.M.; Yücer, R.; Dawood, M.; Hegazy, M.E.F.; Drif, A.; Ooko, E.; Kadioglu, O.; Seo, E.J.; Kamounah, F.S.; Titinchi, S.J.; et al. In Silico and In Vitro Screening of 50 Curcumin Compounds as EGFR and NF-κB Inhibitors. Int. J. Mol. Sci. 2022, 23, 3966. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (Resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Konkimalla, V.B.; Wang, Y.F.; Sauerbrey, A.; Meinhardt, S.; Zintl, F.; Mattern, J.; Volm, M. Prediction of Broad Spectrum Resistance of Tumors towards Anticancer Drugs. Clin. Cancer Res. 2008, 14, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Seo, E.J.; Khelifi, D.; Fayez, S.; Feineis, D.; Bringmann, G.; Efferth, T.; Dawood, M. Molecular Determinants of the Response of Cancer Cells towards Geldanamycin and Its Derivatives. Chem. Biol. Interact. 2023, 383, 110677. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar]
- Biosciences, B.D. White Paper The Essential Cell Analysis Tool The Essential Cell Analysis Tool BD Biosciences. 2013. Available online: https://www.bdbiosciences.com/content/dam/bdb/marketing-documents/Accuri-WP-Essential-Cell-Analysis-Tool.pdf (accessed on 1 March 2023).
- Wimasis WimTube: Tube Formation Assay Image Analysis Solution. Release 4.0. Available online: https://www.wimasis.com/citations (accessed on 26 February 2024).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mean Binding Energy (kcal/mol) | pKi (µM) | Interacting Residues |
|---|---|---|---|
| Michellamine B | −11.11 ± 0.02 | 0.007 ± 0.000 | LYS28, GLY31, MET32, ARG33, SER51, THR52, ASP53, LYS56, ASN186, PRO275, SER276, ARG278 |
| Jozimine A2 | −8.20 ± 0.00 | 0.979 ± 0.005 | ARG30, ARG50, GLU222, ASP223, GLU225, PHE239, GLN241, PRO275 |
| Triptolide | −6.15 ± 0.00 | 31.130 ± 0.070 | GLY32, ARG35, ARG41, SER42, ALA43, VAL91 |
| Cell Lines | IC50 of Jozimine A2 (µM) | IC50 of Michellamine B (µM) |
|---|---|---|
| CCRF-CEM | 0.44 ± 0.001 | >100 |
| CEM/ADR5000 | 0.69 ± 0.04 | >100 |
| HEK-BlueTM Null1 | 1.09 ± 0.003 | >100 |
| Gene | Forward Primer (5′ ⇒ 3′) | Reverse Primer (5′ ⇒ 3′) |
|---|---|---|
| DDIT4L | TCCTGAACCCAACCTCAACG | AAAGCCGCAGGACATCTTGA |
| ETV5 | AGCTGTCTCTGGATTGGCAC | TTTGGTGGTTTTCTGCCCCT |
| ID1 | GTGCCTAAGGAGCCTGGAAA | CCGCCTGTGAAAACGAGAAG |
| FGFR3 | GGTGGGCTTCTTCCTGTTCA | GGGACACCTGTCGCTTGAG |
| SLC32A1 | CGGCAGCTCCGCAGT | GCGTTCGAGGCTCTCTCAG |
| Pgp | CTGGTGTTTGGAGAA | GCCAGTGAAAAATGTTGCCAT |
| HIF1A | GTCTGAGGGGACAGGAGGAT | CTCCTCAGGTGGCTTGTCAG |
| I NOS | GCCATAGAGATGGCCTGTCC | GTGTCACTGGACTGGAGGTG |
| EGFR | GGTGAGTGGCTTGTCTGGAA | GTTTCCCCCTCTGGAGATGC |
| BCL2 | GGATAACGGAGGCTGGGATG | TGACTTCACTTGTGGCCCAG |
| IL-1B | TCGCCAGTGAAATGATGGCT | GGTCGGAGATTCGTAGCTGG |
| NF-ĸB | CAATCACGATCGTCACCGGA | GGTCCGCTGAAAGGACTCTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damiescu, R.; Yücer, R.; Klauck, S.M.; Bringmann, G.; Efferth, T.; Dawood, M. Jozimine A2, a Dimeric Naphthylisoquinoline (NIQ) Alkaloid, Shows In Vitro Cytotoxic Effects against Leukemia Cells through NF-κB Inhibition. Int. J. Mol. Sci. 2024, 25, 3087. https://doi.org/10.3390/ijms25063087
Damiescu R, Yücer R, Klauck SM, Bringmann G, Efferth T, Dawood M. Jozimine A2, a Dimeric Naphthylisoquinoline (NIQ) Alkaloid, Shows In Vitro Cytotoxic Effects against Leukemia Cells through NF-κB Inhibition. International Journal of Molecular Sciences. 2024; 25(6):3087. https://doi.org/10.3390/ijms25063087
Chicago/Turabian StyleDamiescu, Roxana, Rümeysa Yücer, Sabine M. Klauck, Gerhard Bringmann, Thomas Efferth, and Mona Dawood. 2024. "Jozimine A2, a Dimeric Naphthylisoquinoline (NIQ) Alkaloid, Shows In Vitro Cytotoxic Effects against Leukemia Cells through NF-κB Inhibition" International Journal of Molecular Sciences 25, no. 6: 3087. https://doi.org/10.3390/ijms25063087
APA StyleDamiescu, R., Yücer, R., Klauck, S. M., Bringmann, G., Efferth, T., & Dawood, M. (2024). Jozimine A2, a Dimeric Naphthylisoquinoline (NIQ) Alkaloid, Shows In Vitro Cytotoxic Effects against Leukemia Cells through NF-κB Inhibition. International Journal of Molecular Sciences, 25(6), 3087. https://doi.org/10.3390/ijms25063087