
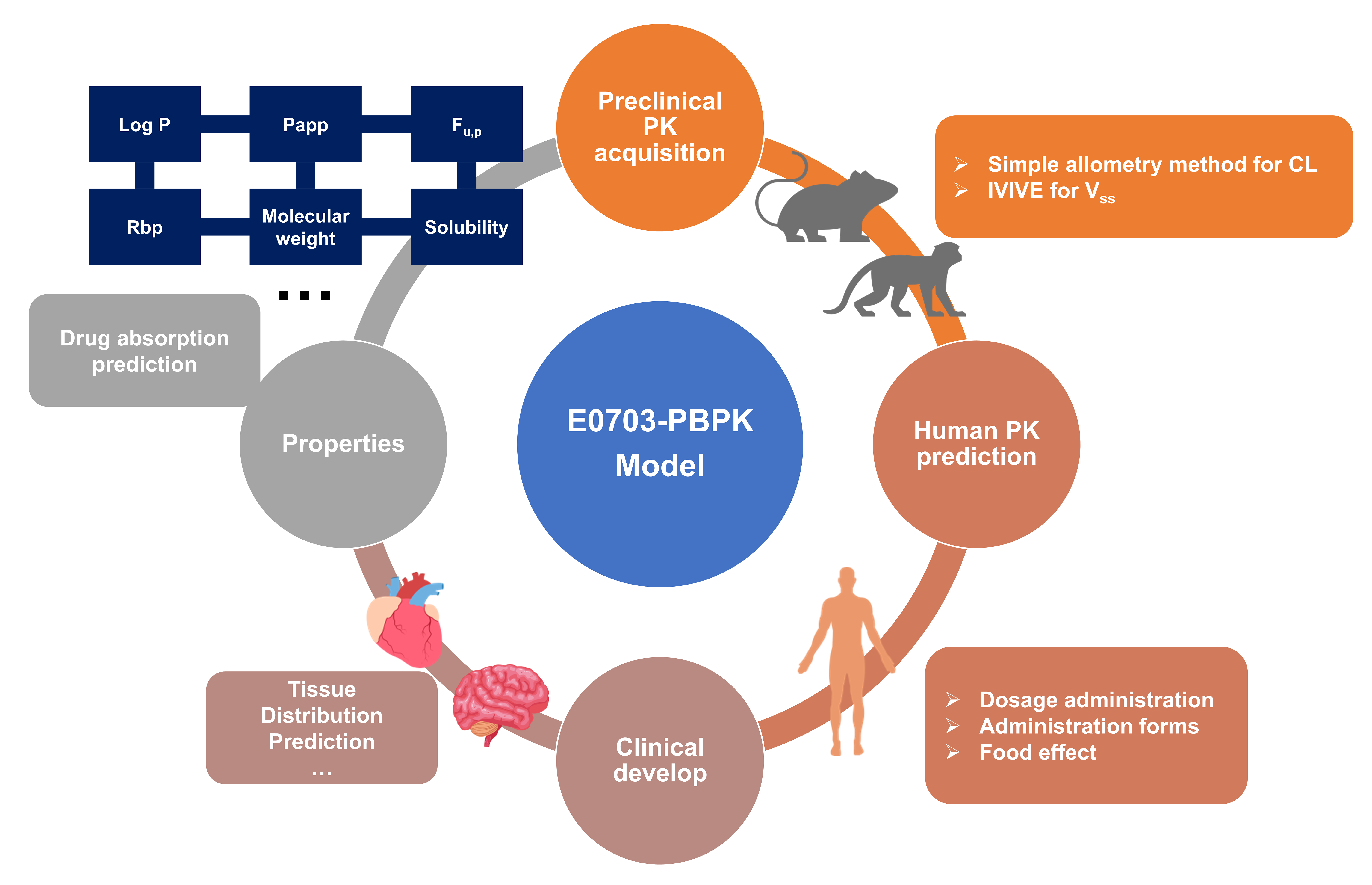
Prediction of Human Pharmacokinetics of E0703, a Novel Radioprotective Agent, Using Physiologically Based Pharmacokinetic Modeling and an Interspecies Extrapolation Approach

 ,
,
Abstract
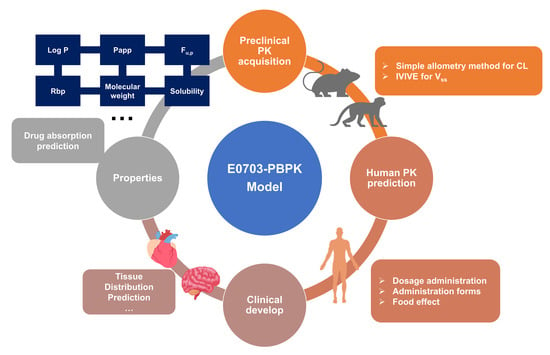
1. Introduction
2. Results
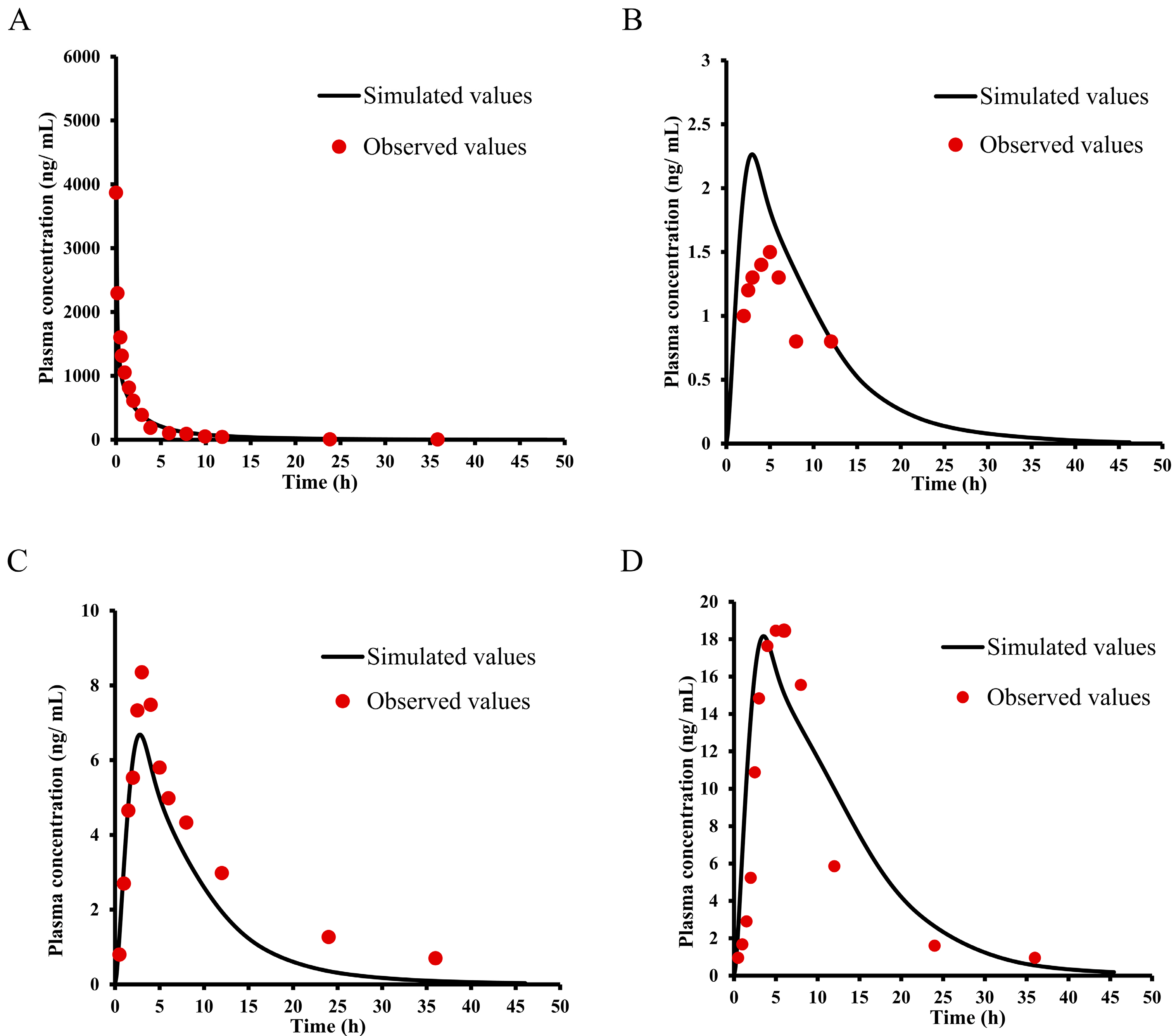
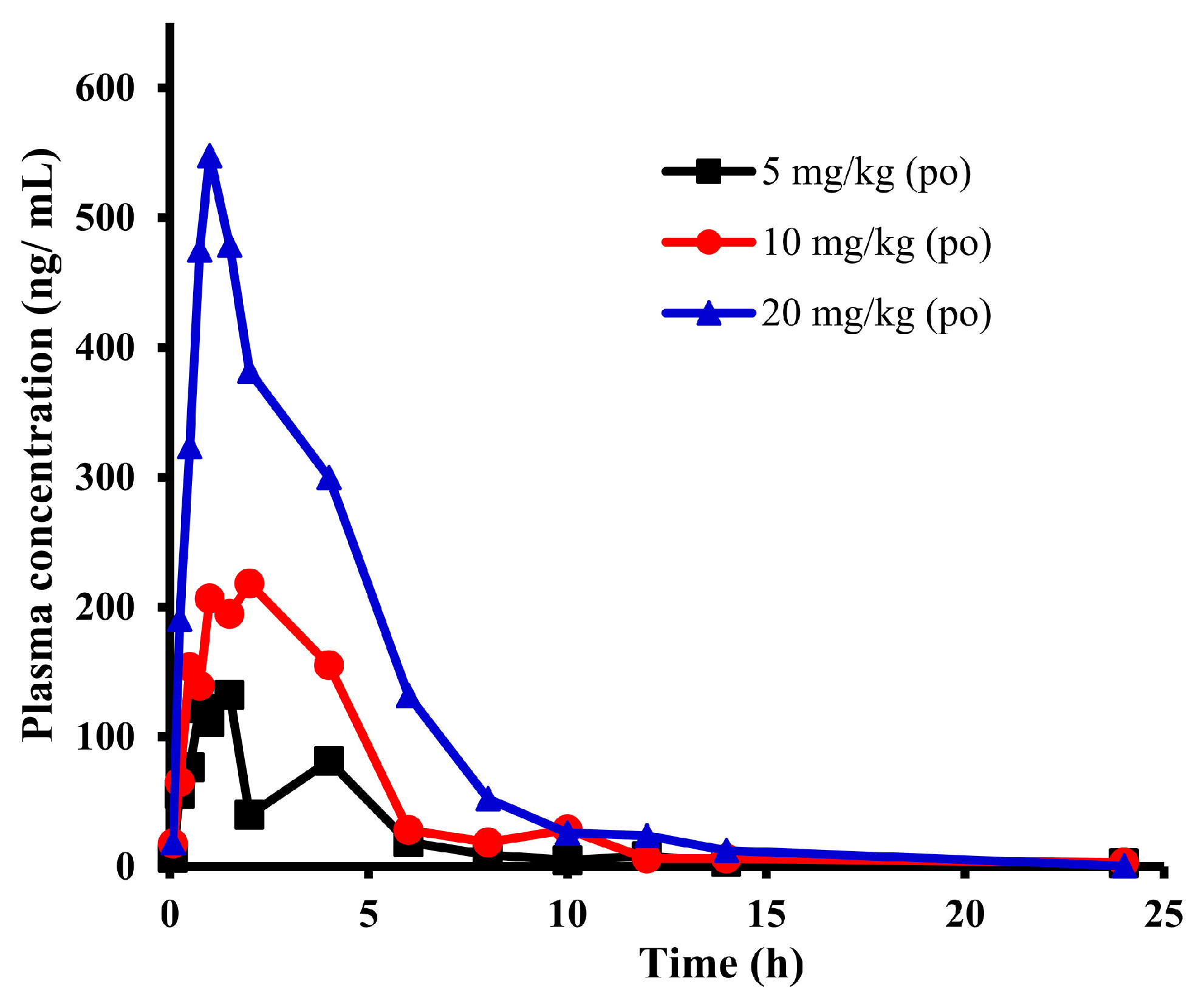
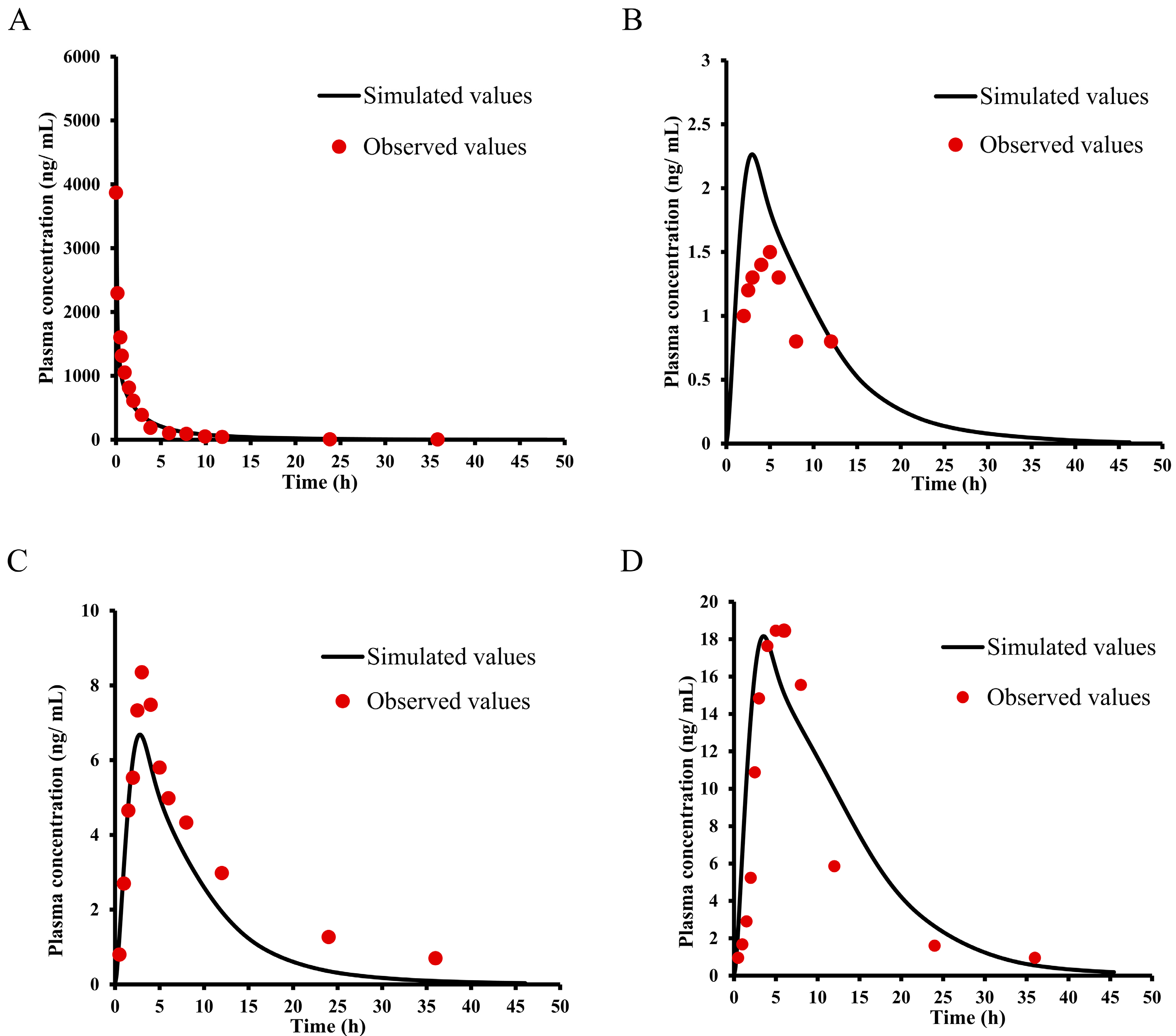
2.1. Pharmacokinetic Study of E0703 in KM Mice
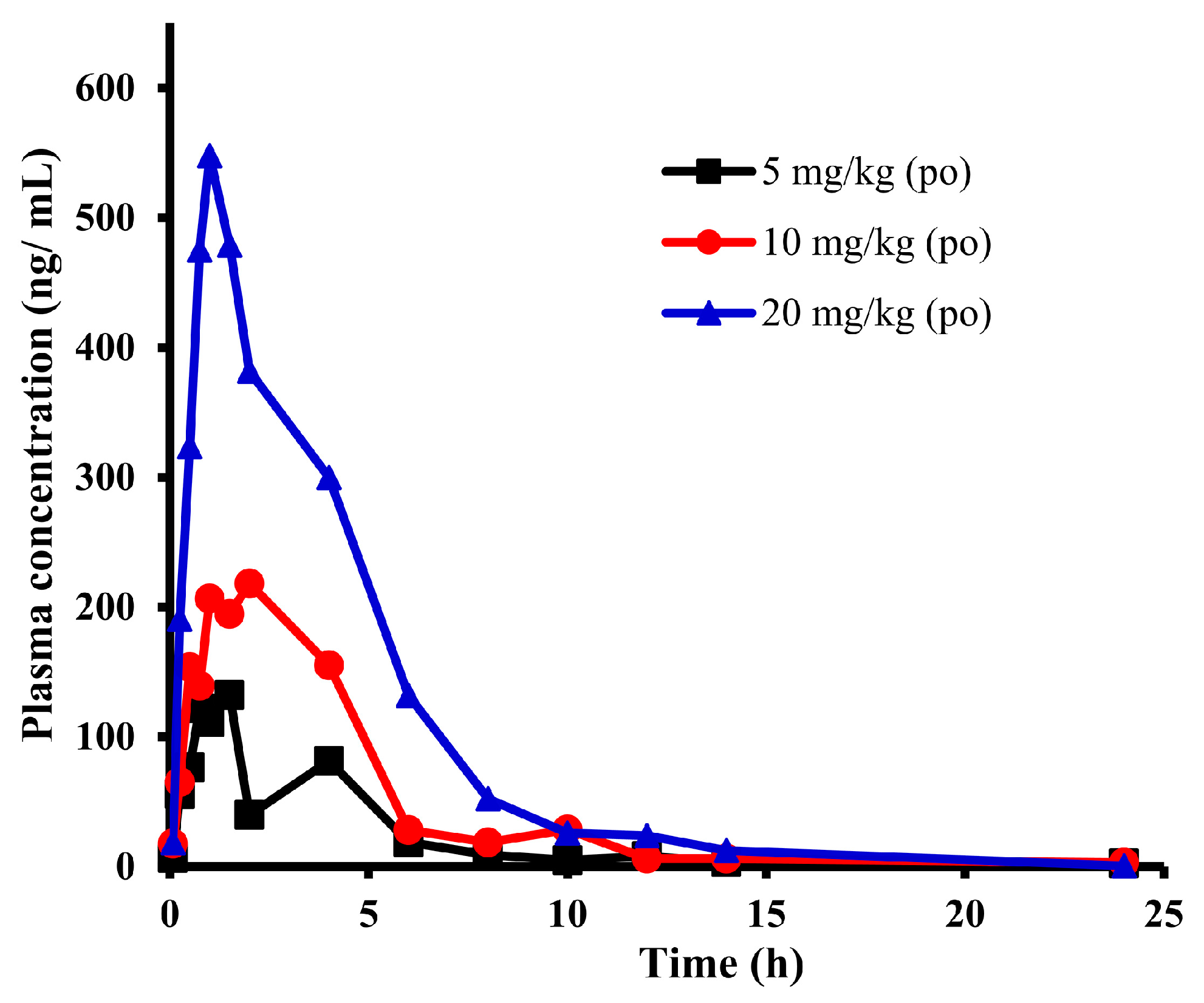
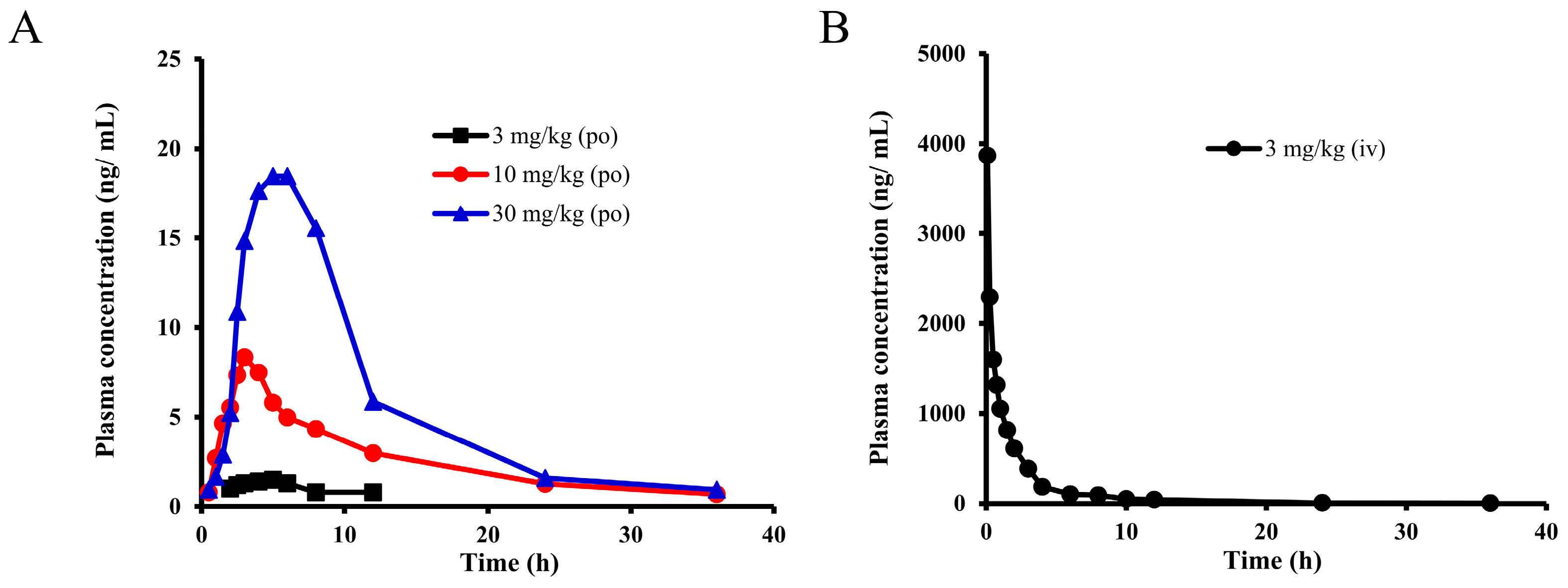
2.2. Pharmacokinetic Study of E0703 in Rhesus Monkeys
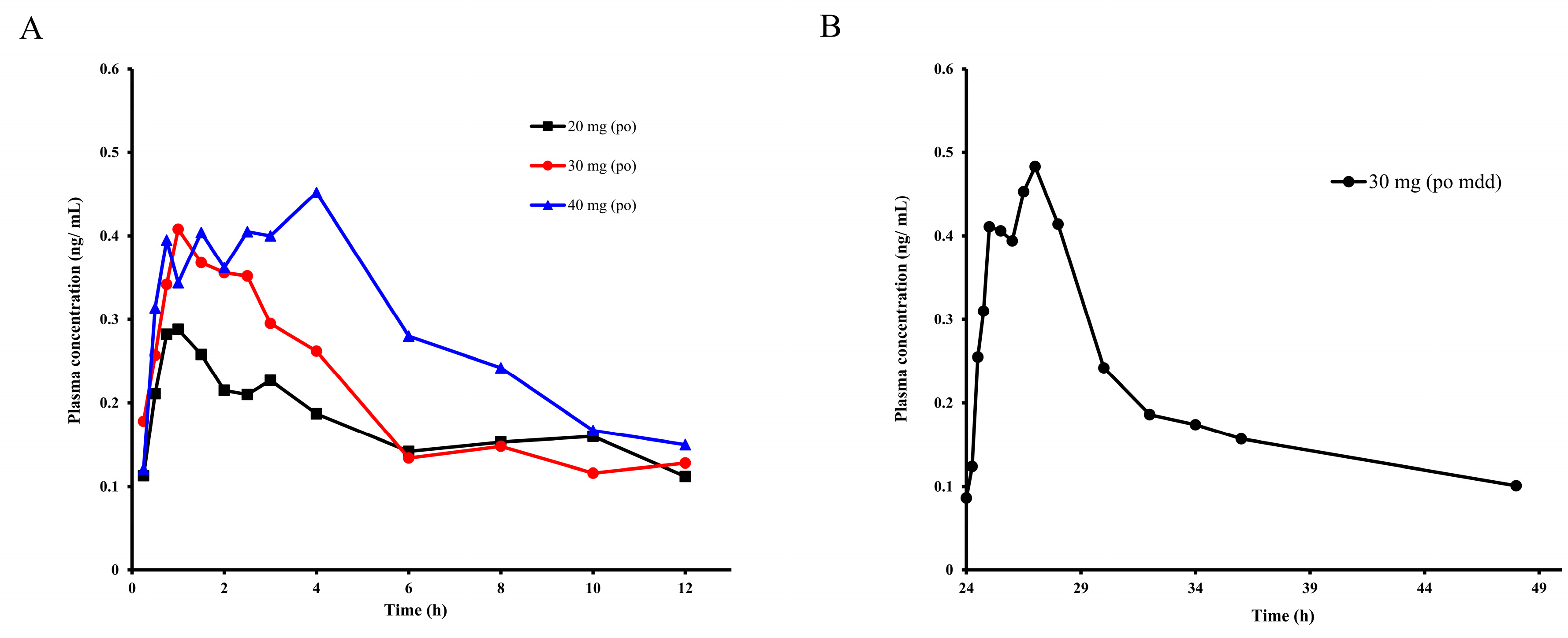
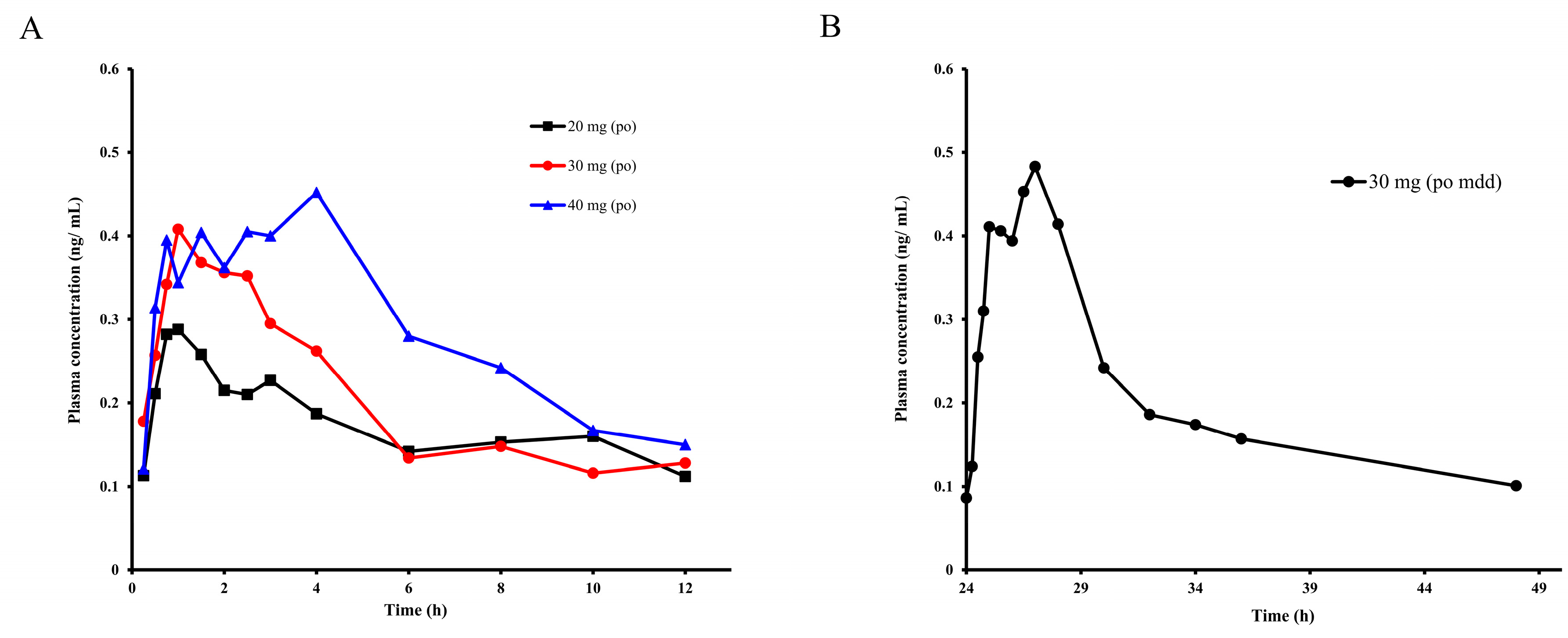
2.3. Pharmacokinetic Study of E0703 in Asian Human
2.4. Unbound Fractions of E0703 in Plasma
2.5. Permeability and Efflux Ratio Determination of E0703 in Caco-2 Cells
2.6. Liver Microsome Metabolism and CYP Enzyme Phenotype
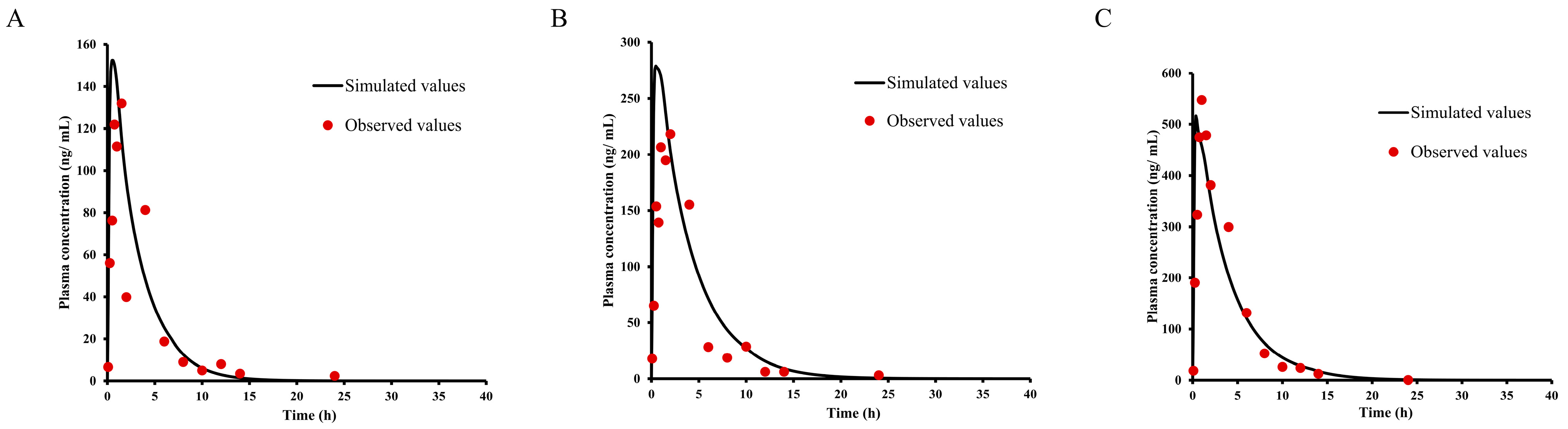
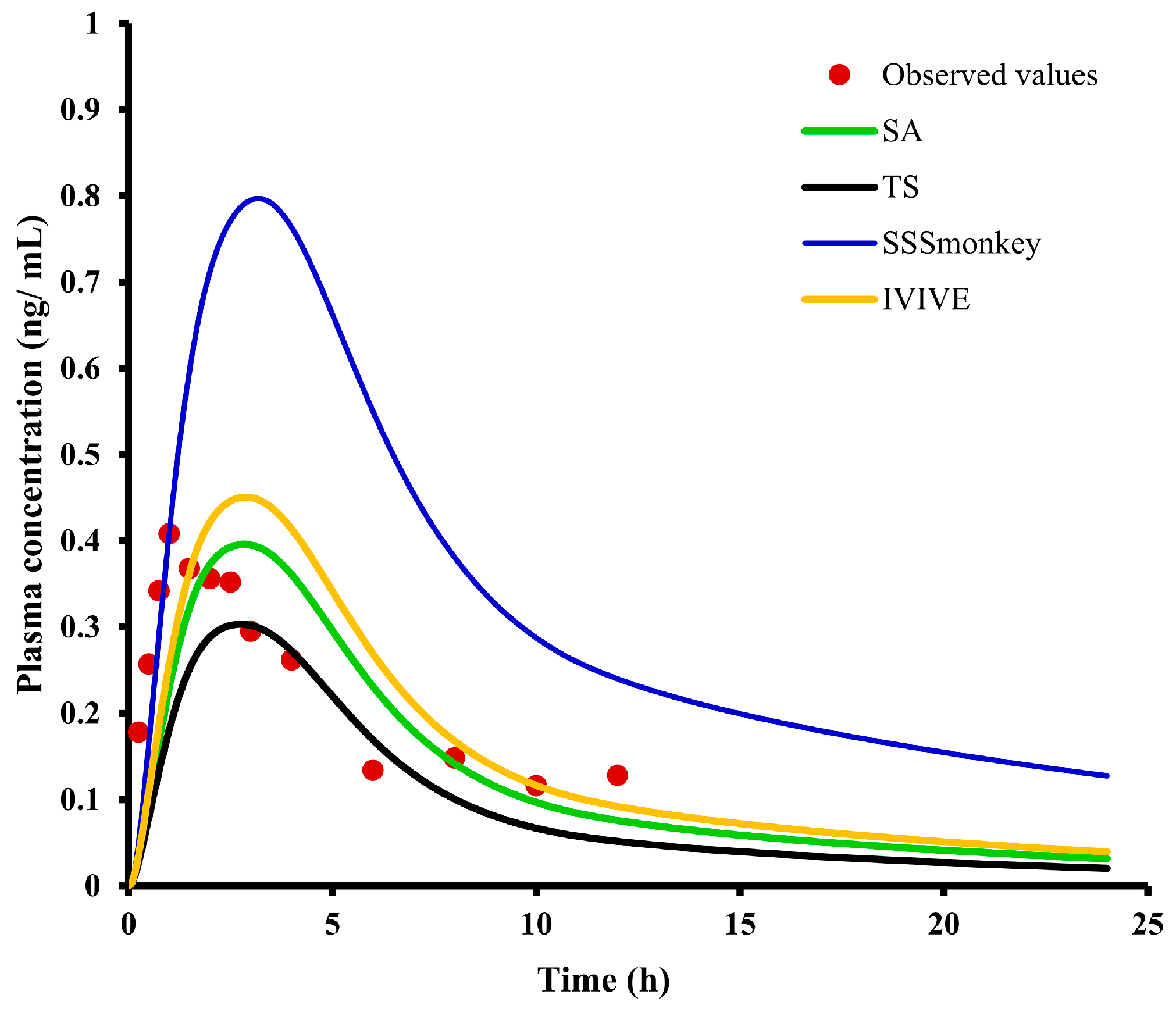
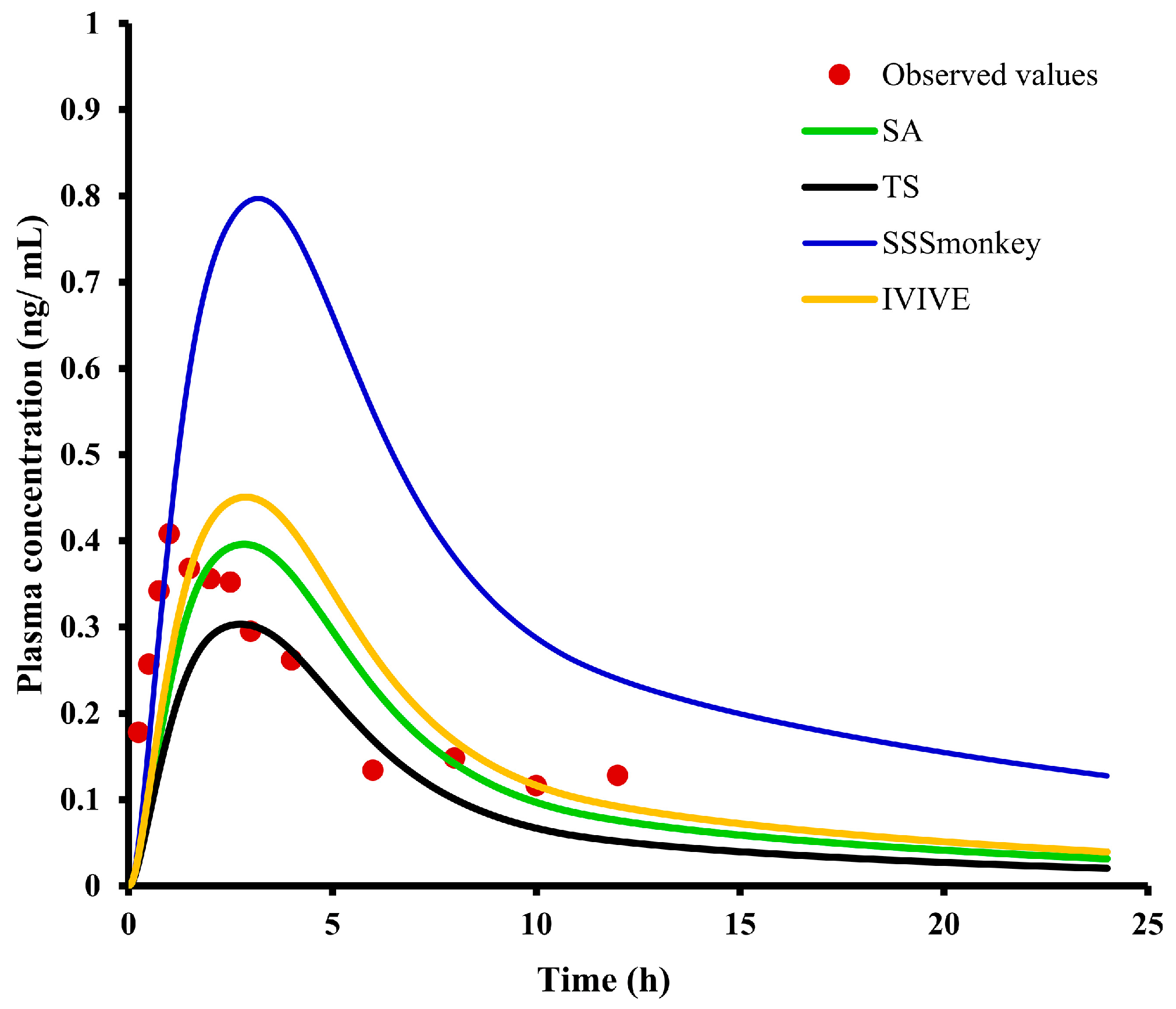
2.7. PBPK Modeling and Species Extrapolation
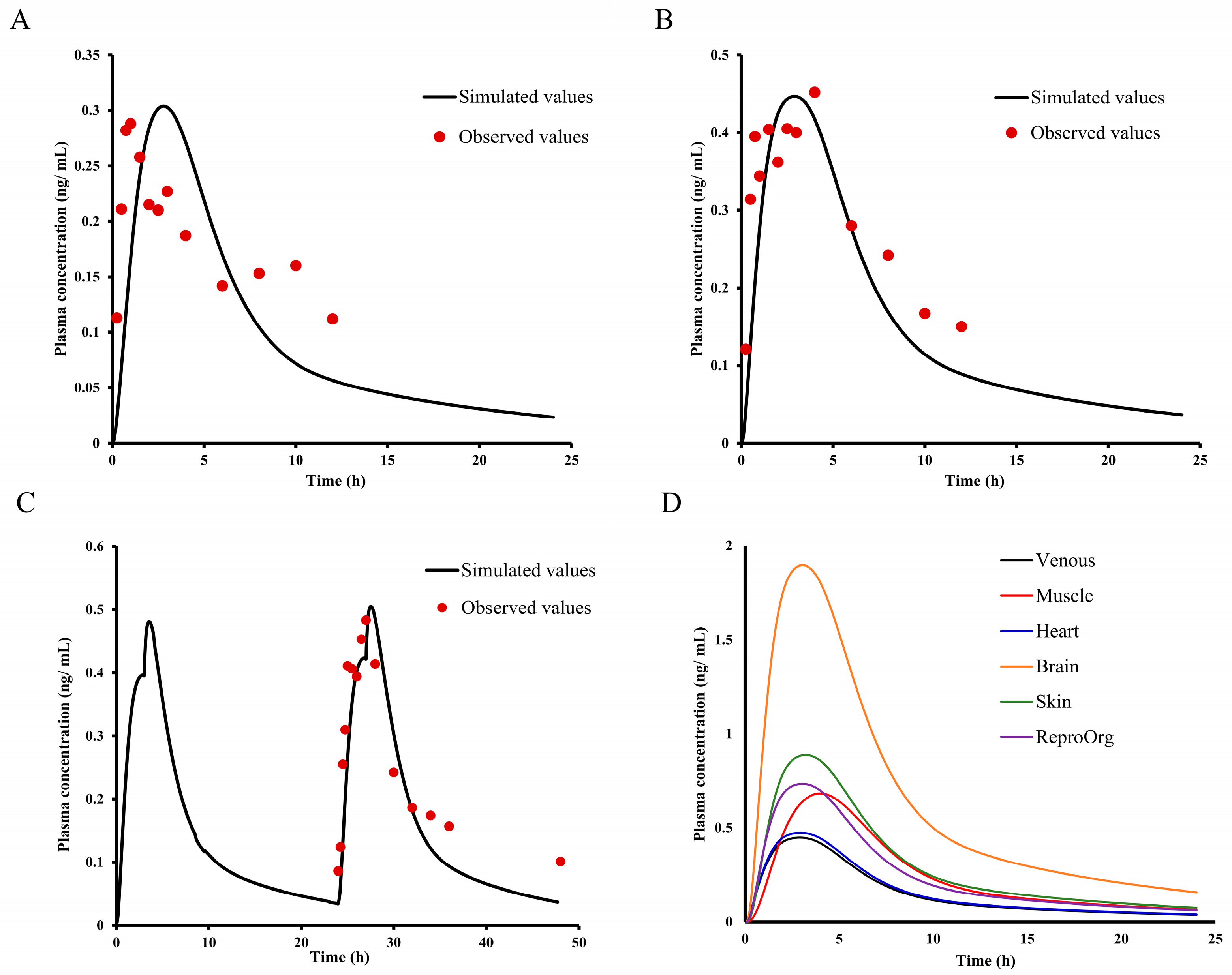
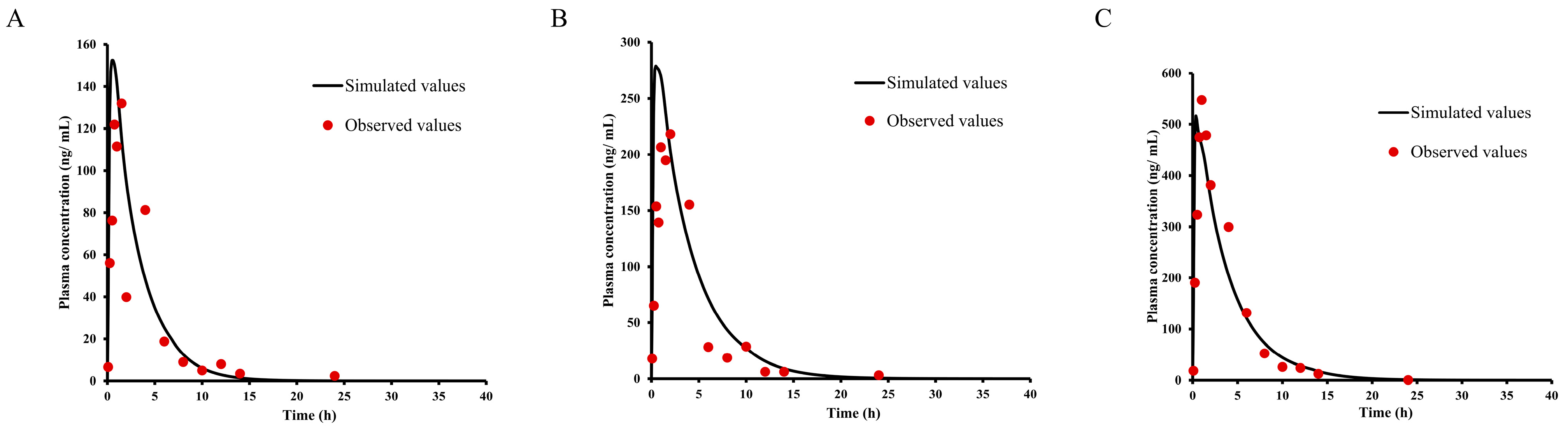
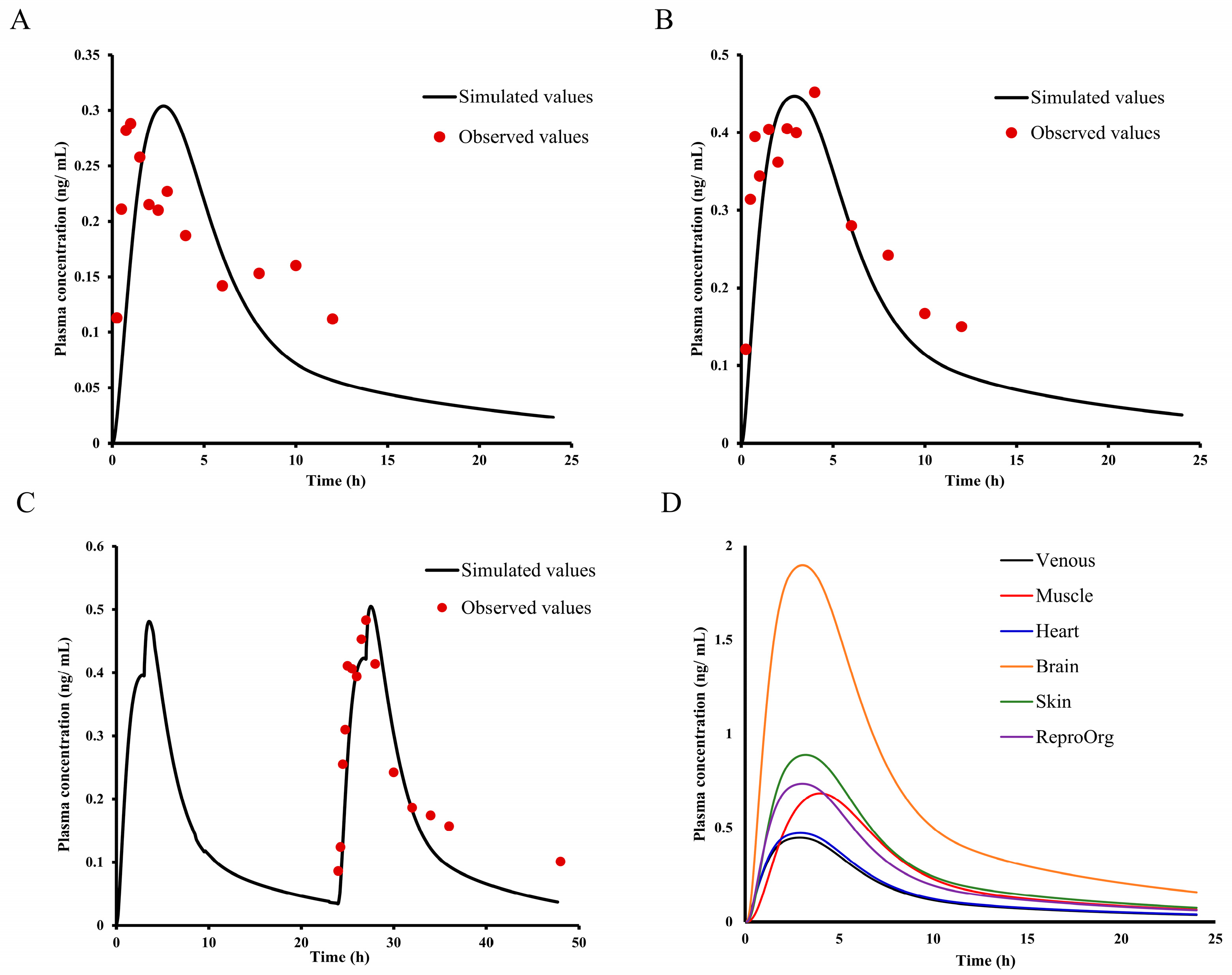
2.8. Asian Human PK and Tissue Distribution Prediction
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. PK Studies in KM Mice, Rhesus Monkeys, and Asian Human
4.3. E0703 Analysis in KM Mice, Rhesus Monkeys, and Asian Human Plasma Samples
4.4. Non-Compartmental PK Analysis
4.5. Permeability of E0703 in Caco-2 Cells
4.6. Plasma Protein Binding
4.7. In Vitro Metabolism of E0703
4.8. Prediction of CL Using Simple Allometry (SA) Method with Body Weight
4.9. Prediction of CLhuman from Single-Species Allometric Scaling (SSSmonkey)
4.10. Prediction of CLhuman Using the Two-Species Scaling Method (TS)
4.11. Prediction of Plasma Concentration–Time Profiles Using the GastroPlus PBPK Model
4.12. Statistical Analysis and Model Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, L.; Liang, Z.; Ma, S.; Li, L.; Liu, X. Radioprotective countermeasures for radiation injury (Review). Mol. Med. Rep. 2023, 27, 66. [Google Scholar] [CrossRef] [PubMed]
- Kamran, M.Z.; Ranjan, A.; Kaur, N.; Sur, S.; Tandon, V. Radioprotective Agents: Strategies and Translational Advances. Med. Res. Rev. 2016, 36, 461–493. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Seed, T.M. The efficacy and safety of amifostine for the acute radiation syndrome. Expert. Opin. Drug Saf. 2019, 18, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- King, M.; Joseph, S.; Albert, A.; Thomas, T.V.; Nittala, M.R.; Woods, W.C.; Vijayakumar, S.; Packianathan, S. Use of Amifostine for Cytoprotection during Radiation Therapy: A Review. Oncology 2020, 98, 61–80. [Google Scholar] [CrossRef]
- Thompson, J.S.; Reilly, R.W.; Crawford, M.; Russe, H.P. The effect of estradiol and estriol on the survival of sublethally and lethally irradiated mice. Radiat. Res. 1965, 26, 567–583. [Google Scholar] [CrossRef]
- Ge, C.; Su, F.; Fu, H.; Wang, Y.; Tian, B.; Liu, B.; Zhu, J.; Ding, Y.; Zheng, X. RNA Profiling Reveals a Common Mechanism of Histone Gene Downregulation and Complementary Effects for Radioprotectants in Response to Ionizing Radiation. Dose Response 2020, 18. [Google Scholar] [CrossRef]
- Li, J.; Meng, Z.; Gan, H.; Gu, R.; Wu, Z.; Dou, G.; Gao, Y. Simultaneous quantitation of E0703, a novel radioprotective agent and its oxidative metabolite M1 in human plasma by UPLC-MS/MS, and application to clinical pharmacokinetics. J. Pharm. Biomed. Anal. 2019, 174, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Madny, M.A.; Deshpande, P.; Tumuluri, V.; Borde, P.; Sangana, R. Physiologically Based Biopharmaceutics Model of Vildagliptin Modified Release (MR) Tablets to Predict In Vivo Performance and Establish Clinically Relevant Dissolution Specifications. AAPS PharmSciTech 2022, 23, 108. [Google Scholar] [CrossRef]
- Wang, Z.; Chan, E.C.Y. Physiologically Based Pharmacokinetic Modelling to Investigate Baricitinib and Tofacitinib Dosing Recommendations for COVID-19 in Geriatrics. Clin. Pharmacol. Ther. 2022, 112, 291–296. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm Res 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Pereva, S.; Nikolova, V.; Angelova, S.; Spassov, T.; Dudev, T. Water inside beta-cyclodextrin cavity: Amount, stability and mechanism of binding. Beilstein J. Org. Chem. 2019, 15, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Alavi, A.; Wong, R.; Akita, S. Radiodermatitis: A Review of Our Current Understanding. Am. J. Clin. Dermatol. 2016, 17, 277–292. [Google Scholar] [CrossRef]
- Yoh, K.; Ikeda, K.; Horie, K.; Inoue, S. Roles of Estrogen, Estrogen Receptors, and Estrogen-Related Receptors in Skeletal Muscle: Regulation of Mitochondrial Function. Int. J. Mol. Sci. 2023, 24, 1853. [Google Scholar] [CrossRef]
- Li, R.; Shen, Y. Estrogen and brain: Synthesis, function and diseases. Front. Biosci. 2005, 10, 257–267. [Google Scholar] [CrossRef]
- Murphy, E.; Korach, K.S. Actions of estrogen and estrogen receptors in nonclassical target tissues. Ernst Schering Found. Symp. Proc. 2006, 13–24. [Google Scholar]
- Tsang, T.Y.; Tsang, S.W.; Lai, K.P.; Tsang, W.P.; Co, N.N.; Kwok, T.T. Facilitation of drug resistance development by gamma-irradiation in human cancer cells. Oncol. Rep. 2009, 22, 921–926. [Google Scholar]
- Sanchez, A.M.; Barrett, J.T.; Schoenlein, P.V. Fractionated ionizing radiation accelerates loss of amplified MDR1 genes harbored by extrachromosomal DNA in tumor cells. Cancer Res. 1998, 58, 3845–3854. [Google Scholar]
- Bottke, D.; Koychev, D.; Busse, A.; Heufelder, K.; Wiegel, T.; Thiel, E.; Hinkelbein, W.; Keilholz, U. Fractionated irradiation can induce functionally relevant multidrug resistance gene and protein expression in human tumor cell lines. Radiat. Res. 2008, 170, 41–48. [Google Scholar] [CrossRef]
- Naritomi, Y.; Sanoh, S.; Ohta, S. Utility of Chimeric Mice with Humanized Liver for Predicting Human Pharmacokinetics in Drug Discovery: Comparison with in Vitro-in Vivo Extrapolation and Allometric Scaling. Biol. Pharm. Bull. 2019, 42, 327–336. [Google Scholar] [CrossRef]
- Tang, H.; Hussain, A.; Leal, M.; Mayersohn, M.; Fluhler, E. Interspecies prediction of human drug clearance based on scaling data from one or two animal species. Drug Metab. Dispos. 2007, 35, 1886–1893. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Amidon, G.L. A compartmental absorption and transit model for estimating oral drug absorption. Int. J. Pharm. 1999, 186, 119–125. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 5 mg/kg | 10 mg/kg | 20 mg/kg | Units |
|---|---|---|---|---|
| Cmax | 132.0 | 218.2 | 547.7 | ng/mL |
| Tmax | 1.5 | 2 | 1 | h |
| T1/2 | 18.37 | 10.00 | 2.28 | h |
| AUC(0-t) | 497.1 | 1063.0 | 2308.8 | ng × h/mL |
| AUC(0-∞) | 560.7 | 1107.7 | 2310.8 | ng × h/mL |
| (CL/F)/kg | 8.92 | 9.03 | 8.66 | L/h/kg |
| MRT | 9.60 | 5.44 | 3.78 | h |
| (Vss/F)/kg | 85.61 | 49.12 | 32.73 | L/kg |
| Label | 3 mg/kg | 10 mg/kg | 30 mg/kg | 3 mg/kg, iv | Units |
|---|---|---|---|---|---|
| Cmax | 1.5 | 8.4 | 18.5 | — | ng/mL |
| Tmax | 5 | 3 | 6 | — | h |
| T1/2 | 2.86 | 13.96 | 15.96 | 13.17 | h |
| AUC(0-t) | 11.7 | 93.8 | 348.8 | 4722.9 | ng × h/mL |
| AUC(0-∞) | 15.0 | 107.9 | 370.6 | 4802.7 | ng × h/mL |
| Bioavailability | 0.35 | 0.76 | 0.87 | — | % |
| CL | — | — | — | 2.811 | L/h |
| (CL/F)/kg | 200.4 | 92.69 | 80.95 | — | L/h/kg |
| MRT | 8.23 | 17.06 | 17.50 | 4.11 | h |
| Vss | — | — | — | 11.55 | L |
| (Vss/F)/kg | 1649.8 | 1581.6 | 1417.0 | — | L/kg |
| Label | 20 mg | 30 mg | 40 mg | 30 mg, mdd | Units |
|---|---|---|---|---|---|
| Cmax | 0.29 | 0.40 | 0.45 | 0.48 | ng/mL |
| Tmax | 1 | 1 | 4 | 27 | h |
| T1/2 | 3.89 | 5.69 | 12.91 | 18.86 | h |
| AUC(0-t) | 2.07 | 2.42 | 3.66 | 5.89 | ng × h/mL |
| AUC(0-∞) | 2.70 | 3.48 | 4.11 | 8.64 | ng × h/mL |
| (CL/F)/kg | 105.8 | 123.3 | 91.83 | 99.26 | L/h/kg |
| MRT | 8.24 | 9.39 | 16.55 | 45.18 | h |
| (Vss/F)/kg | 871.0 | 1157.9 | 1520.1 | 4484.1 | L/kg |
| Parameter | Value | Source |
|---|---|---|
| Molecular weight (g/mol) | 370.54 | Measured in this study |
| Log P | 5.13 | Gastro Plus 9.8 |
| Solubility at pH 7 (mg/mL) | 5 × 10−5 | Assessment value |
| Papp (cm/s) | 1.68 × 10−6 | Measured in this study |
| Fu,p in mouse, rat, Beagle, Rhesus monkey, and human plasma (%) | 1.3, 0.7, 13, 1.2, 0.9 | Measured in this study |
| Rb/p in mice, Rhesus monkeys and Asian humans | 0.82 | Gastro Plus 9.8 |
| Compound | Mean Papp (10−6 cm/s) | Efflux Ratio | Mean Recovery % | Mean Total Recovery % | |||
|---|---|---|---|---|---|---|---|
| A to B | B to A | A to B | B to A | A to B | B to A | ||
| Atenolol | 0.73 | — | — | 90.9 | — | — | — |
| Propranolol | 29.38 | — | — | 81.0 | — | — | — |
| Digoxin | 0.65 | 14.64 | 22.48 | 101.3 | 95.3 | — | — |
| E0703 | 1.68 | 3.74 | 2.22 | 27.8 | 61.1 | 43.7 | 62.7 |
| Species | T1/2 (min) | Clint (mL/min/kg) | CLh (mL/min/kg) | Qh 1 (mL/min/kg) |
|---|---|---|---|---|
| Beagle | 157.5 | 12.7 | 9 | 30.9 |
| mouse | 69.3 | 78.8 | 42 | 90 |
| Rhesus monkey | 83.5 | 22.4 | 14.8 | 43.6 |
| Asian human | 105 | 15.3 | 8.8 | 20.7 |
| rCYP | Metabolic Rate pmol/min (pmolrCYP) | CYPs Content pmol/mg (Protein) | Normalized Metabolic Rate pmol/min mg (Protein) | Ratio (%) |
|---|---|---|---|---|
| 1A2 | 0.58 | 45 | 25.95 | 12.49 |
| 2C9 | 0.06 | 96 | 5.33 | 2.57 |
| 2C19 | 0.09 | 19 | 1.71 | 0.82 |
| 2A6 | 0.06 | 68 | 4.08 | 1.96 |
| 2D6 | 0.83 | 10 | 8.3 | 4 |
| 3A4 | 1.5 | 108 | 162.33 | 78.15 |
| Methods | Poulin and Theil | Berezhkovskiy | Rodgers, Leahy, Rowland | Lokacova (Rodgers-Single) |
|---|---|---|---|---|
| Values (L) | 28.64 | 21.78 | 26.77 | 26.77 |
| Label | Observed | IVIVE, SA, SSSmonkey, TS | Units | Fold Error (O/S) |
|---|---|---|---|---|
| Cmax | 0.40 | 0.45/0.4/0.8/0.3 | ng/mL | 0.89/1.00/0.50/1.33 |
| AUC(0-t) | 2.42 | 3.65/3.14/7.8/2.29 | ng × h/mL | 0.66/0.77/0.31/1.06 |
| AUC(0-∞) | 3.48 | 4.25/3.6/10.5/2.58 | ng × h/mL | 0.82/0.97/0.33/1.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, Y.-X.; Zhang, Z.; Yan, J.-Y.; Ma, Z.-C.; Wang, Y.-G.; Xiao, C.-R.; Zhuang, X.-M.; Gao, Y. Prediction of Human Pharmacokinetics of E0703, a Novel Radioprotective Agent, Using Physiologically Based Pharmacokinetic Modeling and an Interspecies Extrapolation Approach. Int. J. Mol. Sci. 2024, 25, 3047. https://doi.org/10.3390/ijms25053047
Ge Y-X, Zhang Z, Yan J-Y, Ma Z-C, Wang Y-G, Xiao C-R, Zhuang X-M, Gao Y. Prediction of Human Pharmacokinetics of E0703, a Novel Radioprotective Agent, Using Physiologically Based Pharmacokinetic Modeling and an Interspecies Extrapolation Approach. International Journal of Molecular Sciences. 2024; 25(5):3047. https://doi.org/10.3390/ijms25053047
Chicago/Turabian StyleGe, Yun-Xuan, Zhuo Zhang, Jia-Yi Yan, Zeng-Chun Ma, Yu-Guang Wang, Cheng-Rong Xiao, Xiao-Mei Zhuang, and Yue Gao. 2024. "Prediction of Human Pharmacokinetics of E0703, a Novel Radioprotective Agent, Using Physiologically Based Pharmacokinetic Modeling and an Interspecies Extrapolation Approach" International Journal of Molecular Sciences 25, no. 5: 3047. https://doi.org/10.3390/ijms25053047
APA StyleGe, Y.-X., Zhang, Z., Yan, J.-Y., Ma, Z.-C., Wang, Y.-G., Xiao, C.-R., Zhuang, X.-M., & Gao, Y. (2024). Prediction of Human Pharmacokinetics of E0703, a Novel Radioprotective Agent, Using Physiologically Based Pharmacokinetic Modeling and an Interspecies Extrapolation Approach. International Journal of Molecular Sciences, 25(5), 3047. https://doi.org/10.3390/ijms25053047