
Integrated Computational Approaches for Drug Design Targeting Cruzipain




Abstract
1. Introduction
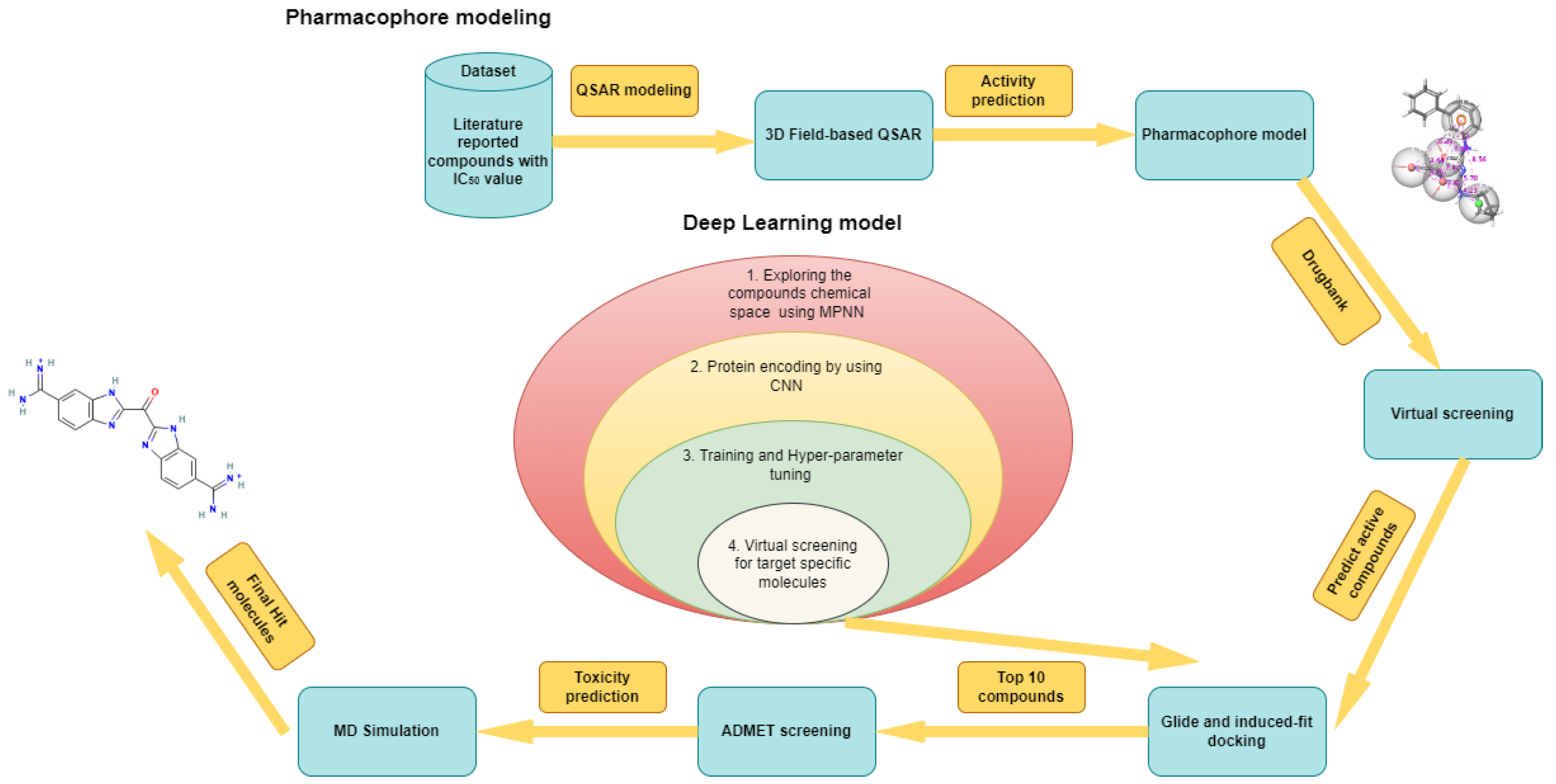
2. Results
2.1. Three-Dimensional Field-Based QSAR Model
2.2. Pharmacophore Model
2.3. Validation
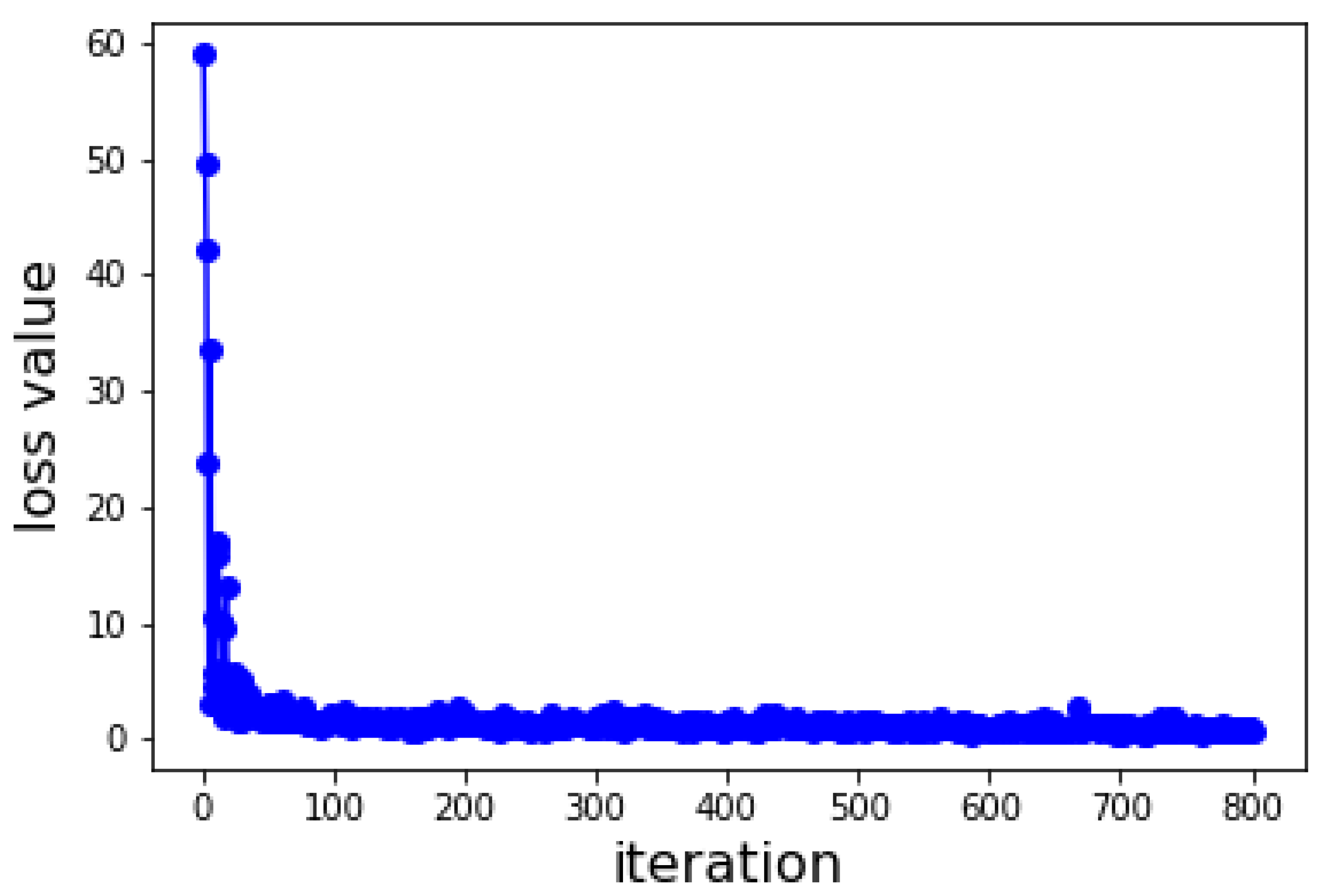
2.4. Deep Learning Model
2.5. Virtual Screening
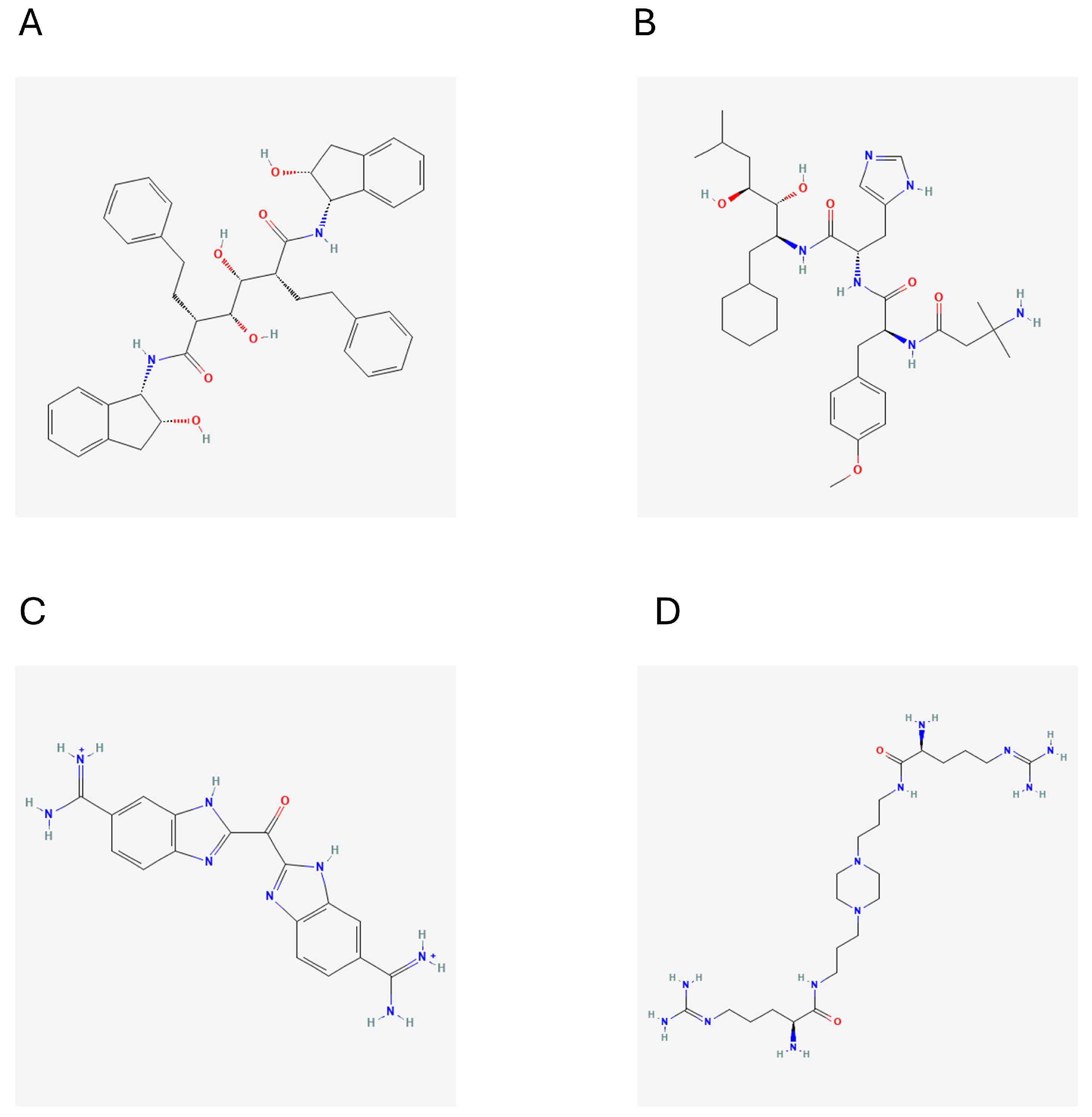
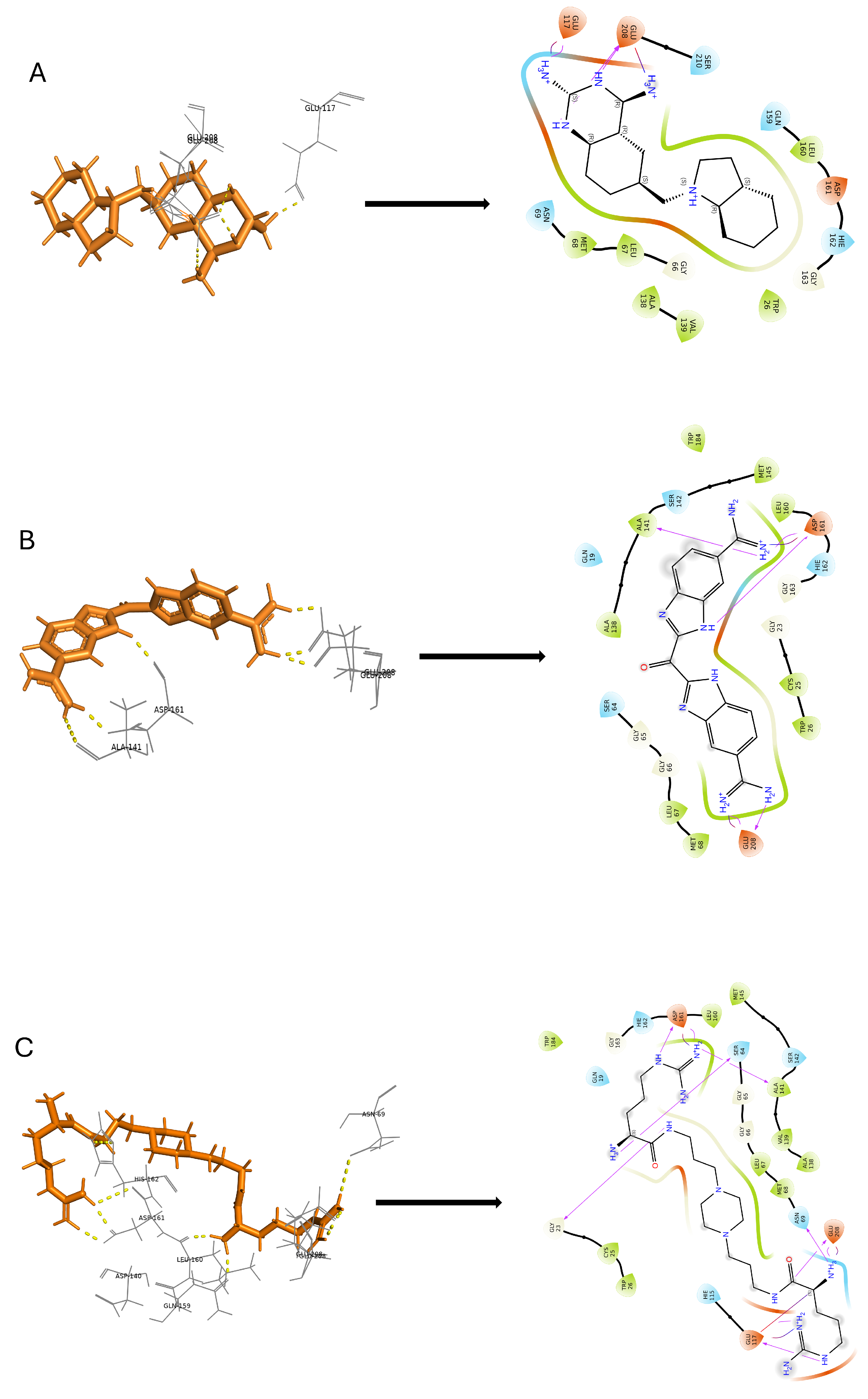
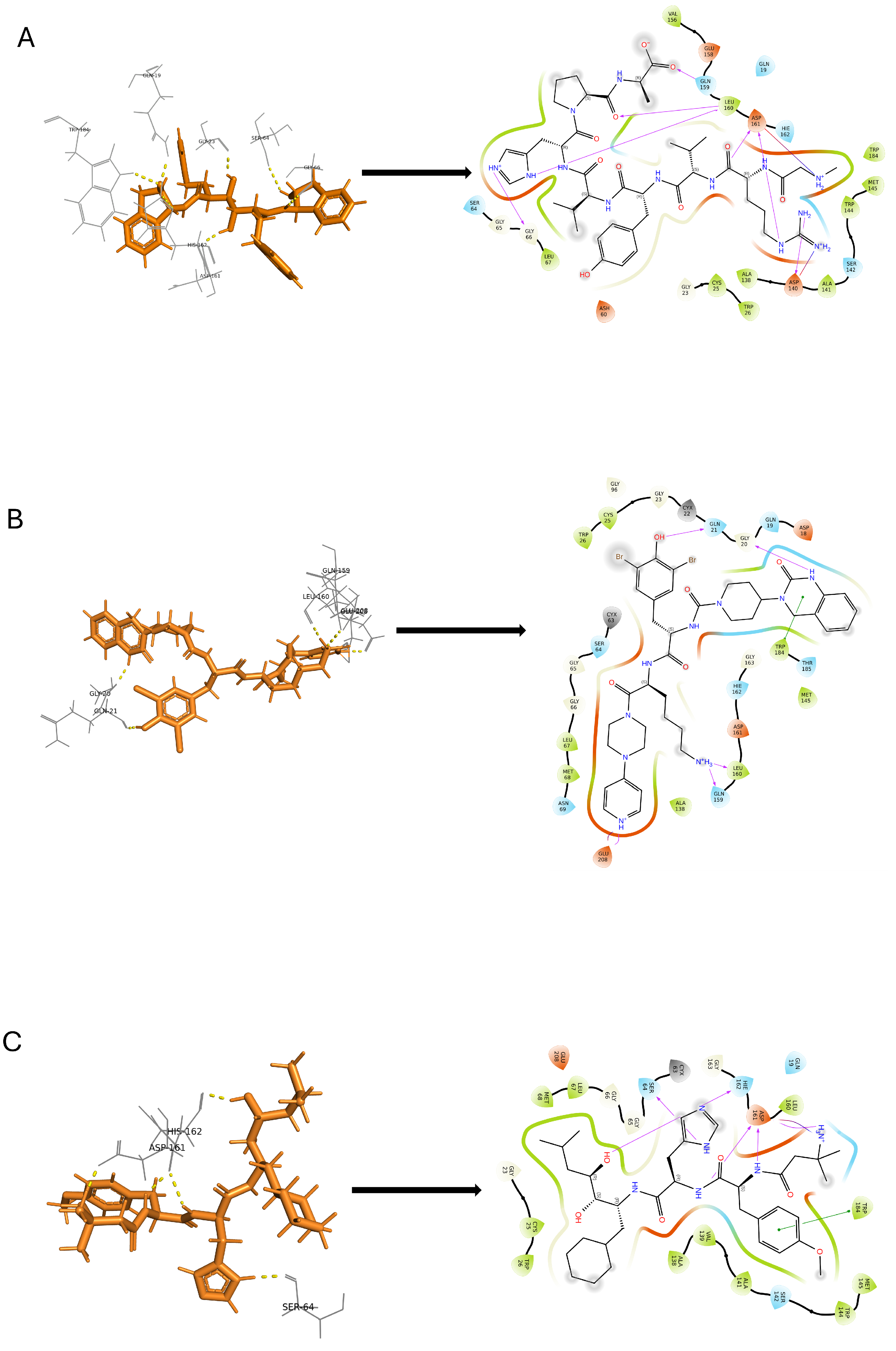
2.6. Molecular Docking Studies
2.6.1. Glide SP (Standard Precision) Docking
2.6.2. Induced Fit Docking
2.7. In Silico Predicted Physicochemical Parameters
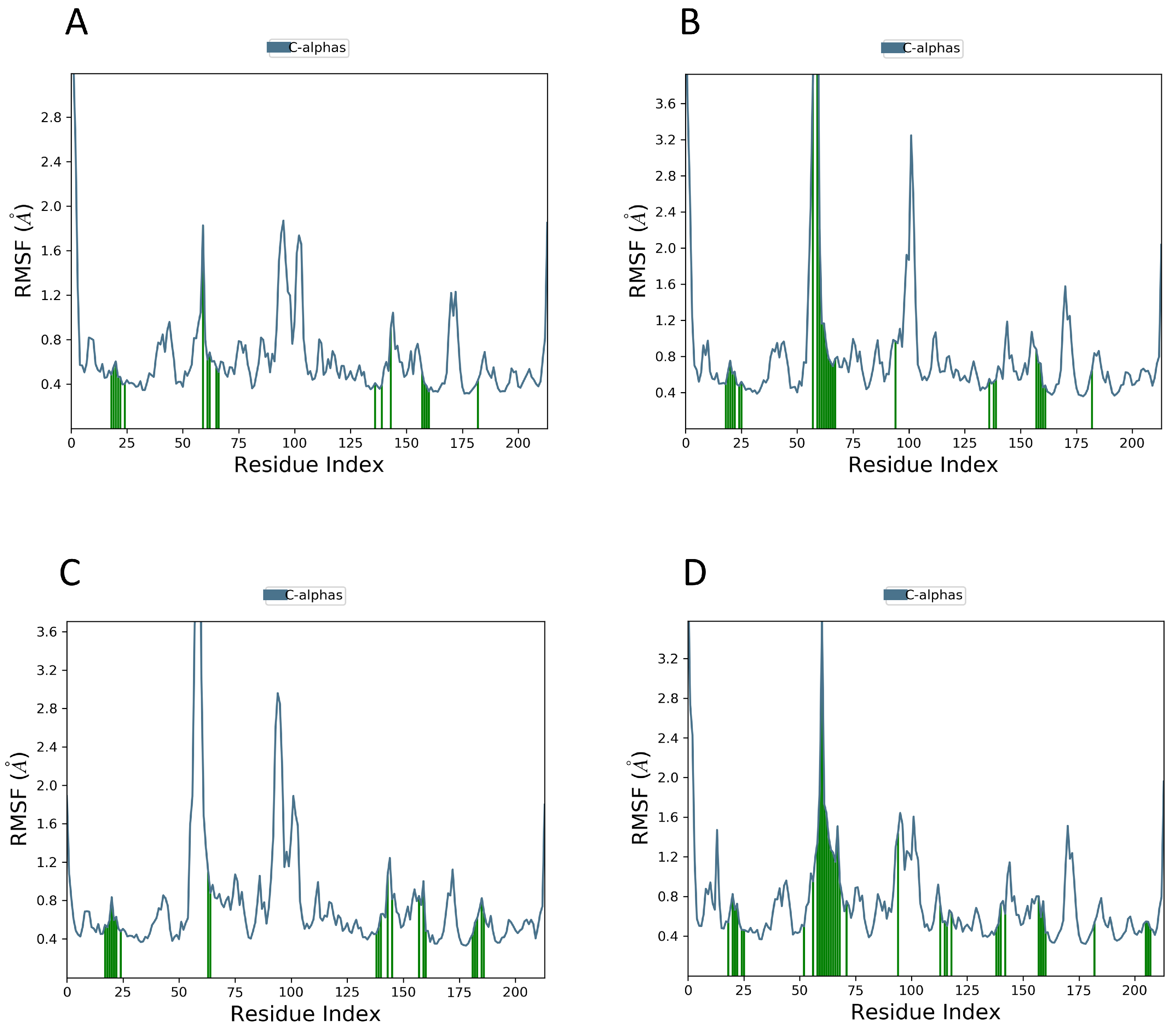
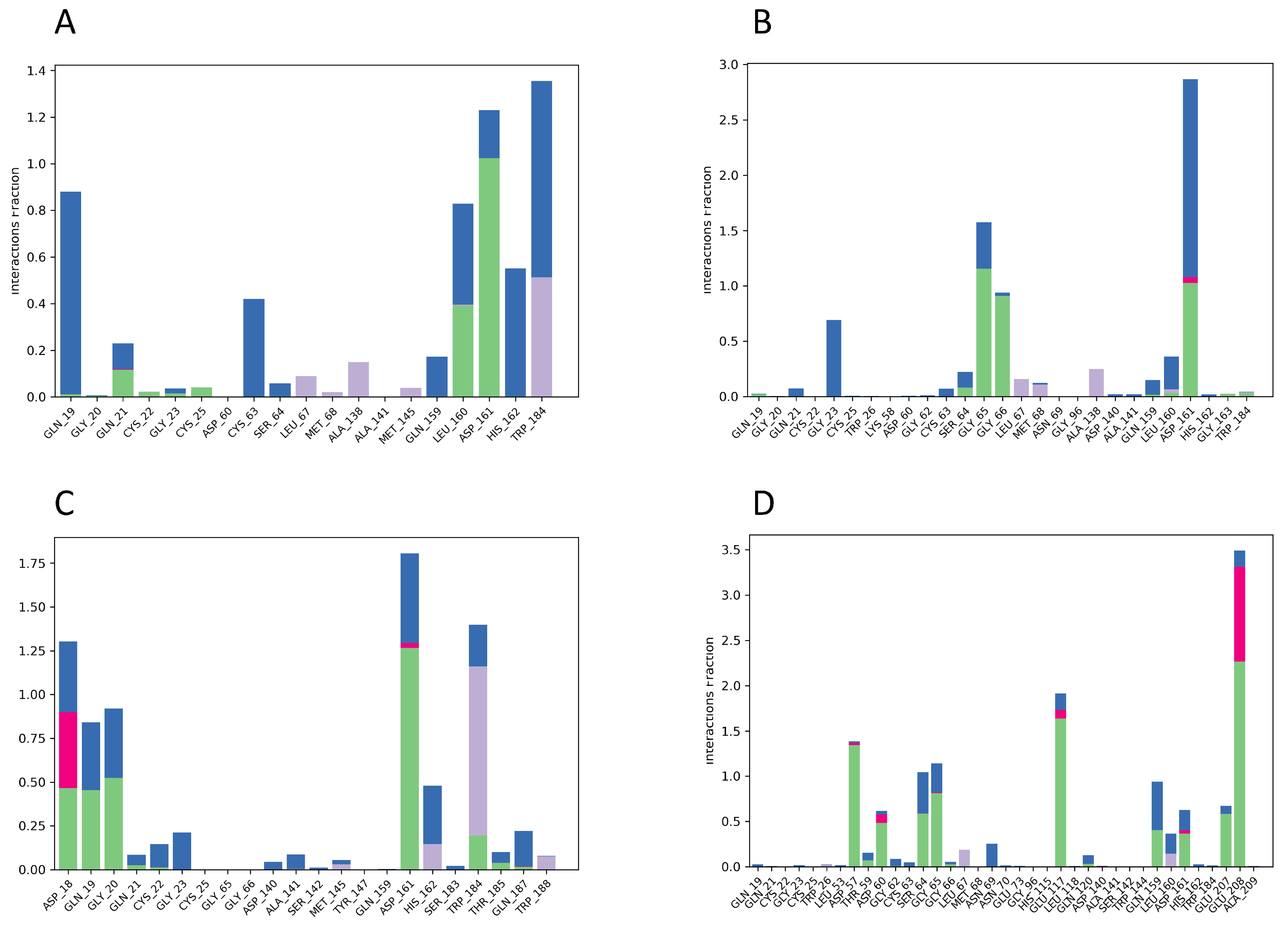
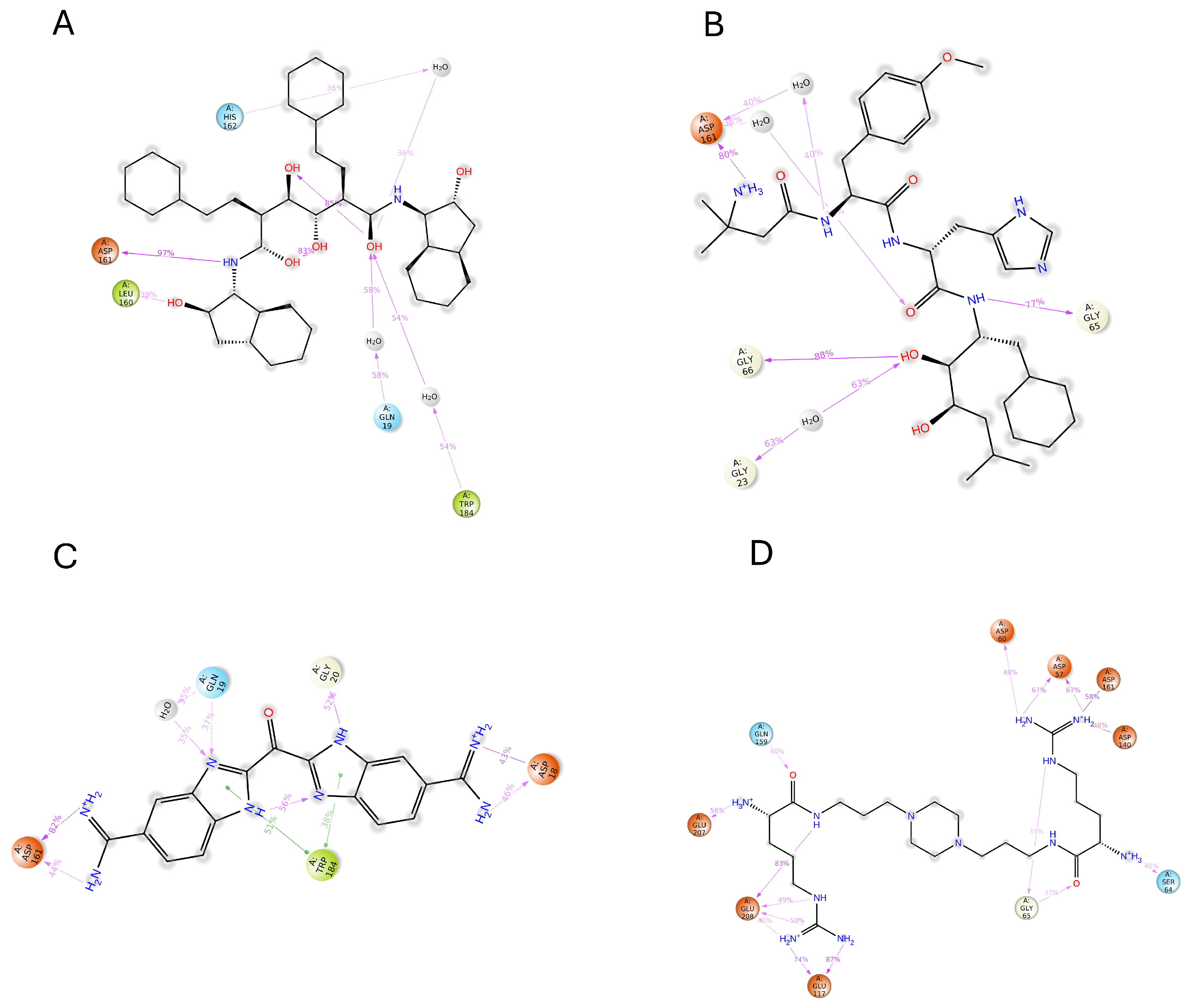
2.8. MD Simulations
3. Discussion
4. Materials and Methods
4.1. Data Retrieval for Deep Learning
4.2. Data Retrieval for QSAR
4.3. Building 3D-QSAR Model
4.4. Ligand-Based Pharmacophore Generation
4.5. Pharmacophore Validation
4.6. Deep Learning Model
4.7. Hyper Parameterization
4.8. Virtual Screening
4.8.1. Drugbank Database Preparation
4.8.2. Deep Learning-Based Virtual Screening
4.8.3. Pharmacophore-Based Virtual Screening
4.9. Docking Investigation
4.9.1. Glide SP (Standard Precision) Docking
4.9.2. Induced Fit Docking
4.10. Absorption, Distribution, Metabolism, Excretion (ADME), and Toxicity
4.11. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| T. cruzi | Trypansoma cruzi |
| CZP | Cruzipain |
| QSAR | Quantitative structure–activity relationship |
| CNN | Convolutional neural networks |
| MPNN | Message-passing neural networks |
| AAC | Amino acid composition |
| ADME | Absorption, distribution, metabolism, and excretion |
| SMILES | Simplified Molecular Input Line Entry System |
| DTI | Drug target interaction |
| AlogP | Octanol–water partition coefficient |
| FASTA | Fast alignment |
References
- Herrera, L.; Morocoima, A.; Lozano-Arias, D.; García-Alzate, R.; Viettri, M.; Lares, M.; Ferrer, E. Infections and coinfections by Trypanosomatid parasites in a rural community of Venezuela. Acta Parasitol. 2022, 67, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- González-Martin, G.; Thambo, S.; Paulos, C.; Vásquez, I.; Paredes, J. The pharmacokinetics of nifurtimox in chronic renal failure. Eur. J. Clin. Pharmacol. 1992, 42, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.H.P.d.; Silva, R.M.d.; Fonseca, K.d.S.; Cardoso, J.M.d.O.; Mathias, F.A.S.; Reis, L.E.S.; Molina, I.; Oliveira, R.C.d.; Vieira, P.M.d.A.; Carneiro, C.M. Pharmacokinetic and tissue distribution of benznidazole after oral administration in mice. Antimicrob. Agents Chemother. 2017, 61, e02410-16. [Google Scholar]
- Echeverría, L.E.; Marcus, R.; Novick, G.; Sosa-Estani, S.; Ralston, K.; Zaidel, E.J.; Forsyth, C.; Ribeiro, A.L.P.; Mendoza, I.; Falconi, M.L.; et al. WHF IASC roadmap on Chagas disease. Glob. Heart 2020, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Lidani, K.; Andrade, F.; Bavia, L.; Damasceno, F.; Beltrame, M.; Messias-Reason, I.; Lucas-Sandri, T. Chagas disease: From discovery to a worldwide health problem. Front. Public Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Jasinski, G.; Salas-Sarduy, E.; Vega, D.; Fabian, L.; Martini, M.F.; Moglioni, A.G. Thiosemicarbazone derivatives: Evaluation as cruzipain inhibitors and molecular modeling study of complexes with cruzain. Bioorg. Med. Chem. 2022, 61, 116708. [Google Scholar] [CrossRef] [PubMed]
- Duschak, V.G. Major kinds of drug targets in Chagas disease or American Trypanosomiasis. Curr. Drug Targets 2019, 20, 1203–1216. [Google Scholar] [CrossRef]
- Duschak, V.G.; Couto, A.S. Cruzipain, the major cysteine protease of Trypanosoma cruzi: A sulfated glycoprotein antigen as relevant candidate for vaccine development and drug target. A review. Curr. Med. Chem. 2009, 16, 3174–3202. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.C.; Doyle, P.S.; Hsieh, I.; McKerrow, J.H. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J. Exp. Med. 1998, 188, 725–734. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Gan, F.; Cao, B.; Wu, D.; Chen, Z.; Hou, T.; Mao, X. Exploring old drugs for the treatment of hematological malignancies. Curr. Med. Chem. 2011, 18, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A.; Castro, E.A.; Bruno-Blanch, L.E. Virtual Screening: An Emergent, Key Methodology for Drug Development in an Emergent Continent. A Bridge Towards Patentability. In Advanced Methods and Applications in Chemoinformatics: Research Progress and New Applications; IGI Global: Pennsylvania, PA, USA, 2012; pp. 229–245. [Google Scholar]
- Asmare, M.M.; Nitin, N.; Yun, S.I.; Mahapatra, R.K. QSAR and deep learning model for virtual screening of potential inhibitors against Inosine 5’Monophosphate dehydrogenase (IMPDH) of Cryptosporidium parvum. J. Mol. Graph. Model. 2022, 111, 108108. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.N.; Andronis, C.; Friedla, E.J.; Persidis, A.; Persidis, A. Drug repurposing and adverse event prediction using high-throughput literature analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Lussier, Y.A.; Chen, J.L. The emergence of genome-based drug repositioning. Sci. Transl. Med. 2011, 3, 96ps35. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Williams, A.J.; Krasowski, M.D.; Freundlich, J.S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today 2011, 16, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Sardana, D.; Zhu, C.; Zhang, M.; Gudivada, R.C.; Yang, L.; Jegga, A.G. Drug repositioning for orphan diseases. Briefings Bioinform. 2011, 12, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Pollastri, M.; Campbell, R. Target repurposing for neglected diseases. Future Med. Chem. 2011, 3, 1307–1315. [Google Scholar] [CrossRef]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. DeepPurpose: A deep learning library for drug–target interaction prediction. Bioinformatics 2020, 36, 5545–5547. [Google Scholar] [CrossRef]
- Gaulton, A.; Kale, N.; van Westen, G.J.; Bellis, L.J.; Bento, A.P.; Davies, M.; Hersey, A.; Papadatos, G.; Forster, M.; Wege, P.; et al. The ChEMBL bioactivity database: An update. Sci. Data 2013, 2, 150032. [Google Scholar] [CrossRef]
- Du, X.; Guo, C.; Hansell, E.; Doyle, P.S.; Caffrey, C.R.; Holler, T.P.; McKerrow, J.H.; Cohen, F.E. Synthesis and structure- activity relationship study of potent trypanocidal thio semicarbazone inhibitors of the trypanosomal cysteine protease cruzain. J. Med. Chem. 2002, 45, 2695–2707. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Filho, G.B.; de Oliveira Cardoso, M.V.; Espíndola, J.W.P.; e Silva, D.A.O.; Ferreira, R.S.; Coelho, P.L.; Dos Anjos, P.S.; de Souza Santos, E.; Meira, C.S.; Moreira, D.R.M.; et al. Structural design, synthesis and pharmacological evaluation of thiazoles against Trypanosoma cruzi. Eur. J. Med. Chem. 2017, 141, 346–361. [Google Scholar] [CrossRef] [PubMed]
- de Moraes Gomes, P.A.T.; de Oliveira Barbosa, M.; Santiago, E.F.; de Oliveira Cardoso, M.V.; Costa, N.T.C.; Hernandes, M.Z.; Moreira, D.R.M.; da Silva, A.C.; Dos Santos, T.A.R.; Pereira, V.R.A.; et al. New 1, 3-thiazole derivatives and their biological and ultrastructural effects on Trypanosoma cruzi. Eur. J. Med. Chem. 2016, 121, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Royo, S.; Schirmeister, T.; Kaiser, M.; Jung, S.; Rodriguez, S.; Bautista, J.M.; Gonzalez, F.V. Antiprotozoal and cysteine proteases inhibitory activity of dipeptidyl enoates. Bioorg. Med. Chem. 2018, 26, 4624–4634. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Cardoso, M.V.; de Siqueira, L.R.P.; da Silva, E.B.; Costa, L.B.; Hernandes, M.Z.; Rabello, M.M.; Ferreira, R.S.; da Cruz, L.F.; Moreira, D.R.M.; Pereira, V.R.A.; et al. 2-Pyridyl thiazoles as novel anti-Trypanosoma cruzi agents: Structural design, synthesis and pharmacological evaluation. Eur. J. Med. Chem. 2014, 86, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Mott, B.T.; Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Ang, K.K.H.; Leister, W.; Shen, M.; Silveira, J.T.; Doyle, P.S.; Arkin, M.R.; et al. Identification and optimization of inhibitors of Trypanosomal cysteine proteases: Cruzain, rhodesain, and TbCatB. J. Med. Chem. 2010, 53, 52–60. [Google Scholar] [CrossRef]
- Beaulieu, C.; Isabel, E.; Fortier, A.; Massé, F.; Mellon, C.; Méthot, N.; Ndao, M.; Nicoll-Griffith, D.; Lee, D.; Park, H.; et al. Identification of potent and reversible cruzipain inhibitors for the treatment of Chagas disease. Bioorg. Med. Chem. Lett. 2010, 20, 7444–7449. [Google Scholar] [CrossRef] [PubMed]
- Chiyanzu, I.; Hansell, E.; Gut, J.; Rosenthal, P.J.; McKerrow, J.H.; Chibale, K. Synthesis and evaluation of isatins and thiosemicarbazone derivatives against cruzain, falcipain-2 and rhodesain. Bioorg. Med. Chem. Lett. 2003, 13, 3527–3530. [Google Scholar] [CrossRef]
- Hernandes, M.Z.; Rabello, M.M.; Leite, A.C.L.; Cardoso, M.V.O.; Moreira, D.R.M.; Brondani, D.J.; Simone, C.A.; Reis, L.C.; Souza, M.A.; Pereira, V.R.A.; et al. Studies toward the structural optimization of novel thiazolylhydrazone-based potent antitrypanosomal agents. Bioorg. Med. Chem. 2010, 18, 7826–7835. [Google Scholar] [CrossRef]
- Siles, R.; Chen, S.E.; Zhou, M.; Pinney, K.G.; Trawick, M.L. Design, synthesis, and biochemical evaluation of novel cruzain inhibitors with potential application in the treatment of Chagas’ disease. Bioorg. Med. Chem. Lett. 2006, 16, 4405–4409. [Google Scholar] [CrossRef]
- Greenbaum, D.C.; Mackey, Z.; Hansell, E.; Doyle, P.; Gut, J.; Caffrey, C.R.; Lehrman, J.; Rosenthal, P.J.; McKerrow, J.H.; Chibale, K. Synthesis and structure- activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J. Med. Chem. 2004, 47, 3212–3219. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.S.; Dessoy, M.A.; Pauli, I.; Souza, M.L.; Krogh, R.; Sales, A.I.; Oliva, G.; Dias, L.C.; Andricopulo, A.D. Synthesis, biological evaluation, and structure–activity relationships of potent noncovalent and nonpeptidic cruzain inhibitors as anti-Trypanosoma cruzi agents. J. Med. Chem. 2014, 57, 2380–2392. [Google Scholar] [CrossRef] [PubMed]
- Neitz, R.J.; Bryant, C.; Chen, S.; Gut, J.; Caselli, E.H.; Ponce, S.; Chowdhury, S.; Xu, H.; Arkin, M.R.; Ellman, J.A.; et al. Tetrafluorophenoxymethyl ketone cruzain inhibitors with improved pharmacokinetic properties as therapeutic leads for Chagas’ disease. Bioorg. Med. Chem. Lett. 2015, 25, 4834–4837. [Google Scholar] [CrossRef] [PubMed]
- Espíndola, J.W.P.; de Oliveira Cardoso, M.V.; de Oliveira Filho, G.B.; e Silva, D.A.O.; Moreira, D.R.M.; Bastos, T.M.; de Simone, C.A.; Soares, M.B.P.; Villela, F.S.; Ferreira, R.S.; et al. Synthesis and structure–activity relationship study of a new series of antiparasitic aryloxyl thiosemicarbazones inhibiting Trypanosoma Cruzi Cruzain. Eur. J. Med. Chem. 2015, 101, 818–835. [Google Scholar] [CrossRef] [PubMed]
- Cianni, L.; Feldmann, C.W.; Gilberg, E.; Gütschow, M.; Juliano, L.; Leitao, A.; Bajorath, J.; Montanari, C.A. Can cysteine protease cross-class inhibitors achieve selectivity? J. Med. Chem. 2019, 62, 10497–10525. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.; Kerr, I.D.; Debnath, M.; Ang, K.K.; Ratnam, J.; Ferreira, R.S.; Jaishankar, P.; Zhao, D.; Arkin, M.R.; McKerrow, J.H.; et al. Novel non-peptidic vinylsulfones targeting the S2 and S3 subsites of parasite cysteine proteases. Bioorg. Med. Chem. Lett. 2009, 19, 6218–6221. [Google Scholar] [CrossRef]
- González, F.V.; Izquierdo, J.; Rodrı, S.; McKerrow, J.H.; Hansell, E. Dipeptidyl-α, β-epoxyesters as potent irreversible inhibitors of the cysteine proteases cruzain and rhodesain. Bioorg. Med. Chem. Lett. 2007, 17, 6697–6700. [Google Scholar] [CrossRef]
- Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Eidam, O.; Mott, B.T.; Keiser, M.J.; McKerrow, J.H.; Maloney, D.J.; Irwin, J.J.; Shoichet, B.K. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J. Med. Chem. 2010, 53, 4891–4905. [Google Scholar] [CrossRef]
- dos Santos Filho, J.M.; Moreira, D.R.M.; de Simone, C.A.; Ferreira, R.S.; McKerrow, J.H.; Meira, C.S.; Guimarães, E.T.; Soares, M.B.P. Optimization of anti-Trypanosoma cruzi oxadiazoles leads to identification of compounds with efficacy in infected mice. Bioorg. Med. Chem. 2012, 20, 6423–6433. [Google Scholar] [CrossRef]
- Silva, L.R.; Guimaraes, A.S.; do Nascimento, J.; do Santos Nascimento, I.J.; da Silva, E.B.; McKerrow, J.H.; Cardoso, S.H.; da Silva-Junior, E.F. Computer-aided design of 1, 4-naphthoquinone-based inhibitors targeting cruzain and rhodesain cysteine proteases. Bioorg. Med. Chem. 2021, 41, 116213. [Google Scholar] [CrossRef]
- Barbosa da Silva, E.; Rocha, D.A.; Fortes, I.S.; Yang, W.; Monti, L.; Siqueira-Neto, J.L.; Caffrey, C.R.; McKerrow, J.; Andrade, S.F.; Ferreira, R.S. Structure-based optimization of quinazolines as cruzain and Tbr CATL inhibitors. J. Med. Chem. 2021, 64, 13054–13071. [Google Scholar] [CrossRef] [PubMed]
- Barbosa Da Silva, E.; Sharma, V.; Hernandez-Alvarez, L.; Tang, A.H.; Stoye, A.; O’Donoghue, A.J.; Gerwick, W.H.; Payne, R.J.; McKerrow, J.H.; Podust, L.M. Intramolecular interactions enhance the potency of gallinamide A analogues against Trypanosoma cruzi. J. Med. Chem. 2022, 65, 4255–4269. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Mallari, J.P.; Hansell, E.J.; Mackey, Z.; Doyle, P.; Zhou, Y.; Gut, J.; Rosenthal, P.J.; McKerrow, J.H.; Guy, R.K. Discovery of potent thiosemicarbazone inhibitors of rhodesain and cruzain. Bioorg. Med. Chem. Lett. 2005, 15, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.A.; Feitosa, L.O.; Soares, M.; Costa, T.E.; Henriques, M.G.; Salomão, K.; de Castro, S.L.; Kaiser, M.; Brun, R.; Wardell, J.L.; et al. Design and synthesis of new (E)-cinnamic N-acylhydrazones as potent antitrypanosomal agents. Eur. J. Med. Chem. 2012, 54, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Kryshchyshyn, A.; Kaminskyy, D.; Grellier, P.; Lesyk, R. Trends in research of antitrypanosomal agents among synthetic heterocycles. Eur. J. Med. Chem. 2014, 85, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. Examination of multiple Trypanosoma cruzi targets in a new drug discovery approach for Chagas disease. Bioorg. Med. Chem. 2022, 58, 116577. [Google Scholar] [CrossRef] [PubMed]
- Chenna, B.C.; Li, L.; Mellott, D.M.; Zhai, X.; Siqueira-Neto, J.L.; Calvet Alvarez, C.; Bernatchez, J.A.; Desormeaux, E.; Alvarez Hernandez, E.; Gomez, J.; et al. Peptidomimetic vinyl heterocyclic inhibitors of cruzain effect antitrypanosomal activity. J. Med. Chem. 2020, 63, 3298–3316. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Imrie, F.; Bradley, A.R.; Deane, C.M. Generating property-matched decoy molecules using deep learning. Bioinformatics 2021, 37, 2134–2141. [Google Scholar] [CrossRef]
- Brak, K.; Kerr, I.D.; Barrett, K.T.; Fuchi, N.; Debnath, M.; Ang, K.; Engel, J.C.; McKerrow, J.H.; Doyle, P.S.; Brinen, L.S.; et al. Nonpeptidic tetrafluorophenoxymethyl ketone cruzain inhibitors as promising new leads for Chagas disease chemotherapy. J. Med. Chem. 2010, 53, 1763–1773. [Google Scholar] [CrossRef]
- ödinger Release, S. 2: Protein Preparation Wizard, Epik, Schrödinger, LLC, New York, NY, 2021; Impact, Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J.-Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Release, S. 1: QikProp; Schrödinger, LLC: New York, NY, USA, 2023; Volume 2023. [Google Scholar]
- Bastos, R.S.; de Lima, L.R.; Neto, M.F.; Maryam; Yousaf, N.; Cruz, J.N.; Campos, J.M.; Kimani, N.M.; Ramos, R.S.; Santos, C.B. Design and Identification of Inhibitors for the Spike-ACE2 Target of SARS-CoV-2. Int. J. Mol. Sci. 2023, 24, 8814. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84-es. [Google Scholar]
- Yousaf, N.; Alharthy, R.D.; Kamal, I.; Saleem, M.; Muddassar, M. Identification of human phosphoglycerate mutase 1 (PGAM1) inhibitors using hybrid virtual screening approaches. PeerJ 2023, 11, e14936. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | SD | Scramble | F | P | RMSE | Pearson-R | |
|---|---|---|---|---|---|---|---|
| 1 | 0.4866 | 0.4710 | 0.2810 | 24.0 | 3.96 | 0.46 | 0.7224 |
| 2 | 0.4514 | 0.5614 | 0.4323 | 16.6 | 2.22 | 0.41 | 0.8994 |
| 3 | 0.4245 | 0.6272 | 0.5292 | 14.0 | 1.46 | 0.42 | 0.8665 |
| 4 | 0.3900 | 0.6978 | 0.6215 | 13.9 | 5.44 | 0.41 | 0.8790 |
| 5 | 0.3753 | 0.7318 | 0.6854 | 12.6 | 5.98 | 0.40 | 0.8564 |
| Hypo ID | Phase Hypo Score | Vector Score | Volume Score | BEDROC Score | Survival Score |
|---|---|---|---|---|---|
| AAAHR_1 | 1.279 | 1.000 | 0.872 | 0.950 | 5.490 |
| AAAHR_3 | 1.259 | 0.976 | 0.872 | 0.938 | 5.347 |
| AAAHR_2 | 1.256 | 0.976 | 0.872 | 0.935 | 5.357 |
| AAADHR_3 | 1.226 | 0.983 | 0.872 | 0.886 | 5.672 |
| AADHR_2 | 1.223 | 0.998 | 0.872 | 0.895 | 5.467 |
| S. No | Drugbank ID | Compound Name | H-Bond Interaction | IFD Score (Kcal/mol) |
|---|---|---|---|---|
| 1 | DB03213 | Bis(5-Amidino-2-Benzimidazolyl) Methane Ketone | ASP-161, GLU-208 | −10.167 |
| 2 | DB02559 | 6-(Octahydro-1h-Indol-1-Ylmethyl) Decahydroquinazoline-2,4-Diamine | GLU-208 | −10.207 |
| 3 | DB15199 | Ciraparantag | ASP-161, GLY-23, SER-64, GLU-117, GLU-208, ASN-69 | −9.253 |
| 4 | DB02704 | (2R,3R,4R,5R)-3,4-Dihydroxy-N,N’-bis [(1S,2R)-2-hydroxy-2,3-dihydro-1H- inden-1-yl]-2,5-bis(2-phenylethyl) hexanediamide | ASP-161, SER-64, GLY-66, GLY-23, GLY-19 | −11.177 |
| 5 | DB03395 | Enalkiren | ASP-161, HIE-162, SER-64 | −10.856 |
| 6 | DB04869 | Olcegepant | GLN-159, GLN-21, GLY-20 | −13.285 |
| Drugbank ID | Stars | Mol. MW | Dipole | SASA | Donor HB | Accpt HB | QPlog o/w | QPlogS | QPlog Khsa | No. of Metabolites | QP Log BB | %Human Oral Absorption |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DB03213 | 4 | 346.351 | 5.974 | 613.698 | 8 | 8 | −0.602 | −2.828 | −0.731 | 0 | −3.358 | 24.921 |
| DB15199 | 13 | 512.7 | 3.277 | 1008.917 | 14 | 15 | −4.661 | 2 | −1.966 | 12 | −6.172 | 0 |
| DB01705 | 1 | 332.367 | 8.009 | 612.184 | 8 | 6 | 0.24 | −2.932 | −0.606 | 1 | −2.903 | 37.431 |
| DB02559 | 2 | 307.481 | 1.904 | 589.065 | 6 | 7 | −1.371 | 2 | −0.169 | 3 | −0.237 | 19.966 |
| DB00183 | 11 | 767.896 | 9.505 | 1157.206 | 4.75 | 13.25 | 3.256 | −5.197 | −0.768 | 9 | −4.437 | 2.862 |
| DB02704 | 10 | 648.797 | 3.572 | 1056.131 | 4 | 9.8 | 5.862 | −7.727 | 0.569 | 12 | −2.4 | 63.601 |
| DB03395 | 7 | 656.864 | 1.331 | 1066.353 | 5.5 | 11.65 | 3.509 | −4.657 | −0.137 | 9 | −2.333 | 34.644 |
| DB04593 | 8 | 587.693 | 6.042 | 984.581 | 8.5 | 13 | 0.868 | −4.599 | −1.142 | 6 | −5.243 | 0 |
| DB04869 | 9 | 869.655 | 6.464 | 1147.692 | 4.5 | 12.25 | 4.49 | −7.99 | 0.52 | 10 | −2.443 | 29.794 |
| DB06763 | 17 | 912.057 | 14.239 | 1258.121 | 9.5 | 21.25 | −3.498 | −2.531 | −2.55 | 14 | −5.912 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parvez, A.; Lee, J.-S.; Alam, W.; Tayara, H.; Chong, K.T. Integrated Computational Approaches for Drug Design Targeting Cruzipain. Int. J. Mol. Sci. 2024, 25, 3747. https://doi.org/10.3390/ijms25073747
Parvez A, Lee J-S, Alam W, Tayara H, Chong KT. Integrated Computational Approaches for Drug Design Targeting Cruzipain. International Journal of Molecular Sciences. 2024; 25(7):3747. https://doi.org/10.3390/ijms25073747
Chicago/Turabian StyleParvez, Aiman, Jeong-Sang Lee, Waleed Alam, Hilal Tayara, and Kil To Chong. 2024. "Integrated Computational Approaches for Drug Design Targeting Cruzipain" International Journal of Molecular Sciences 25, no. 7: 3747. https://doi.org/10.3390/ijms25073747
APA StyleParvez, A., Lee, J.-S., Alam, W., Tayara, H., & Chong, K. T. (2024). Integrated Computational Approaches for Drug Design Targeting Cruzipain. International Journal of Molecular Sciences, 25(7), 3747. https://doi.org/10.3390/ijms25073747