
Predicting Spin-Dependent Phonon Band Structures of HKUST-1 Using Density Functional Theory and Machine-Learned Interatomic Potentials

Abstract
1. Introduction
2. Results and Discussion
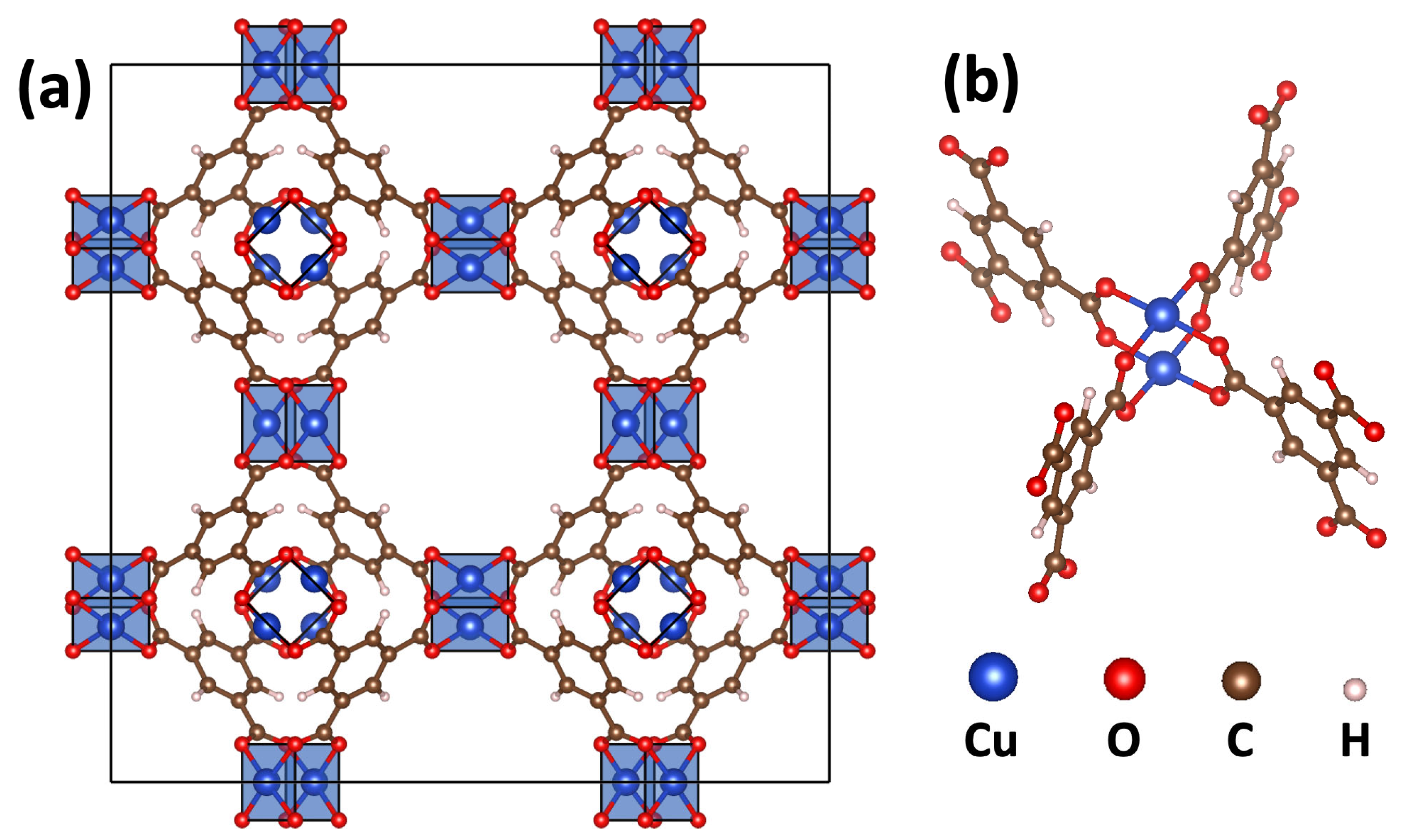
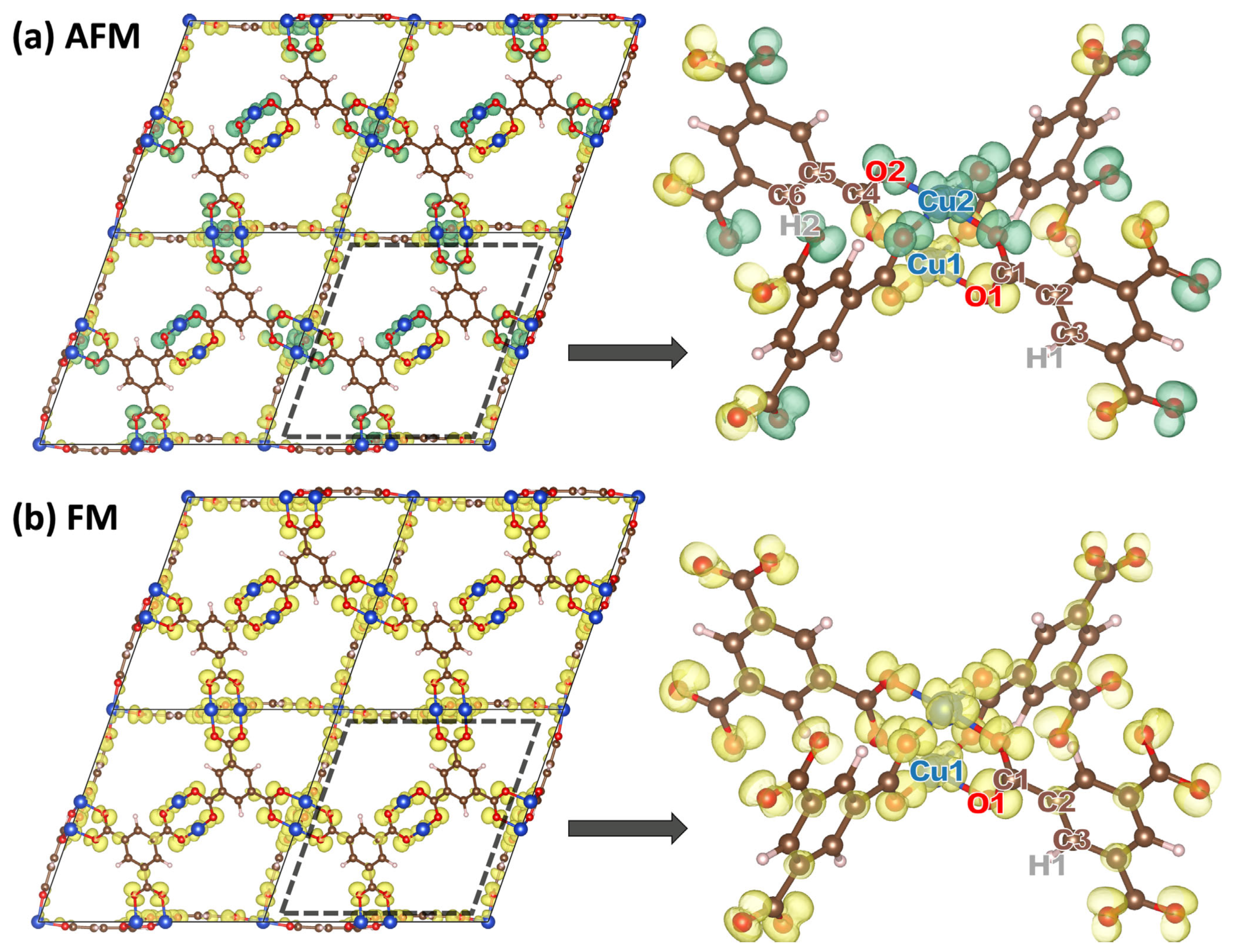
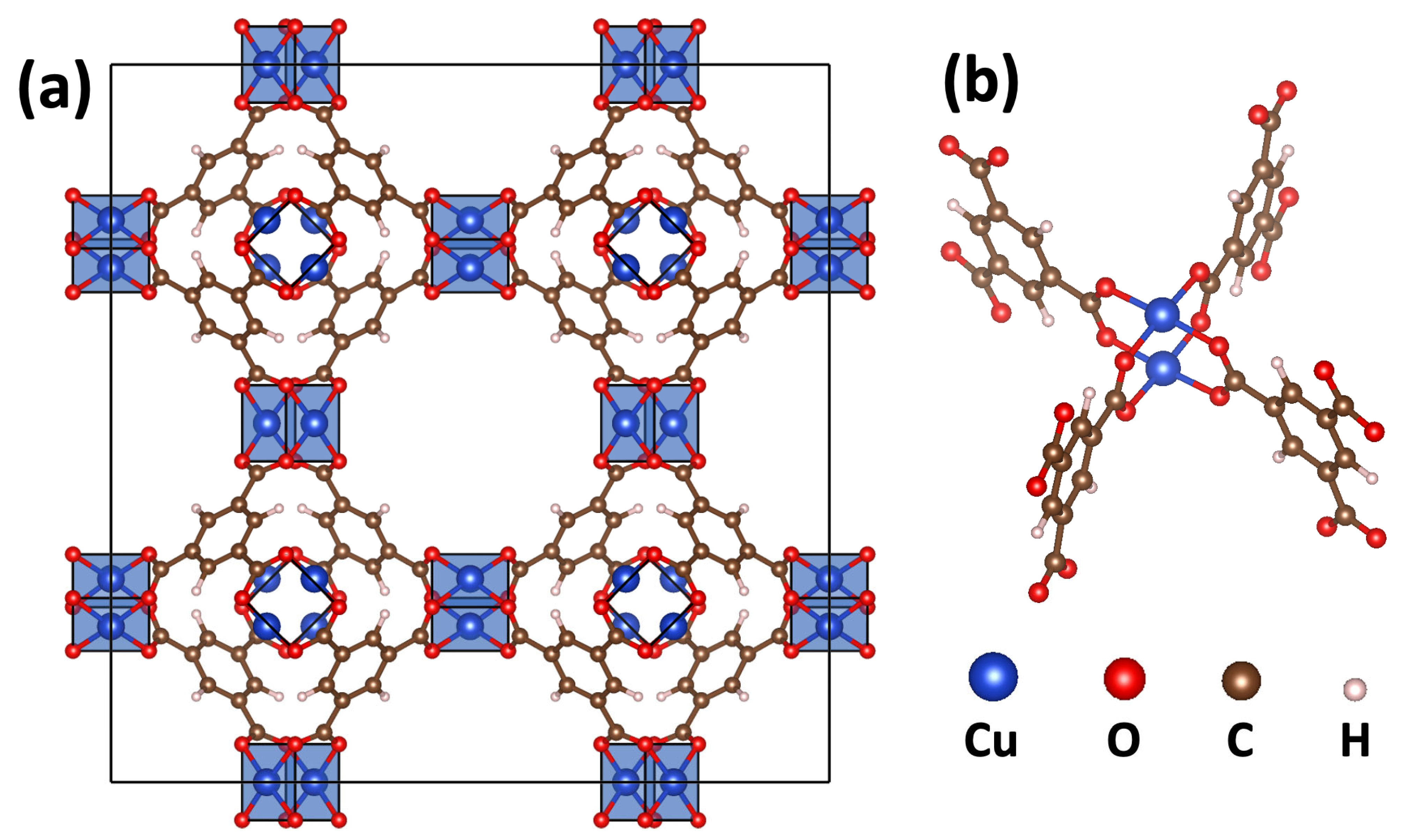
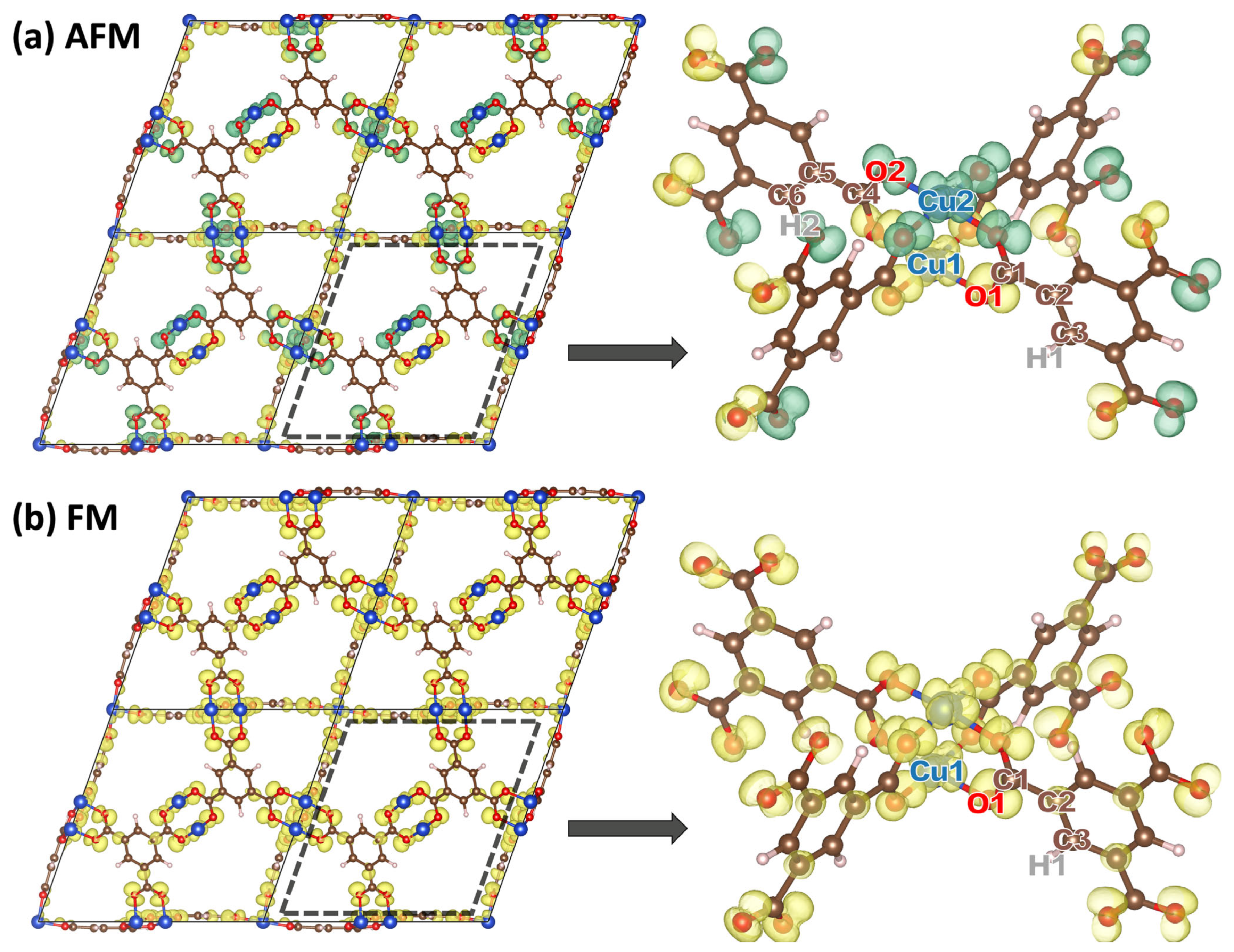
2.1. DFT-Calculated Energetics and Spin Densities of HKUST-1 in the Non-, Ferro- and Antiferromagnetic States
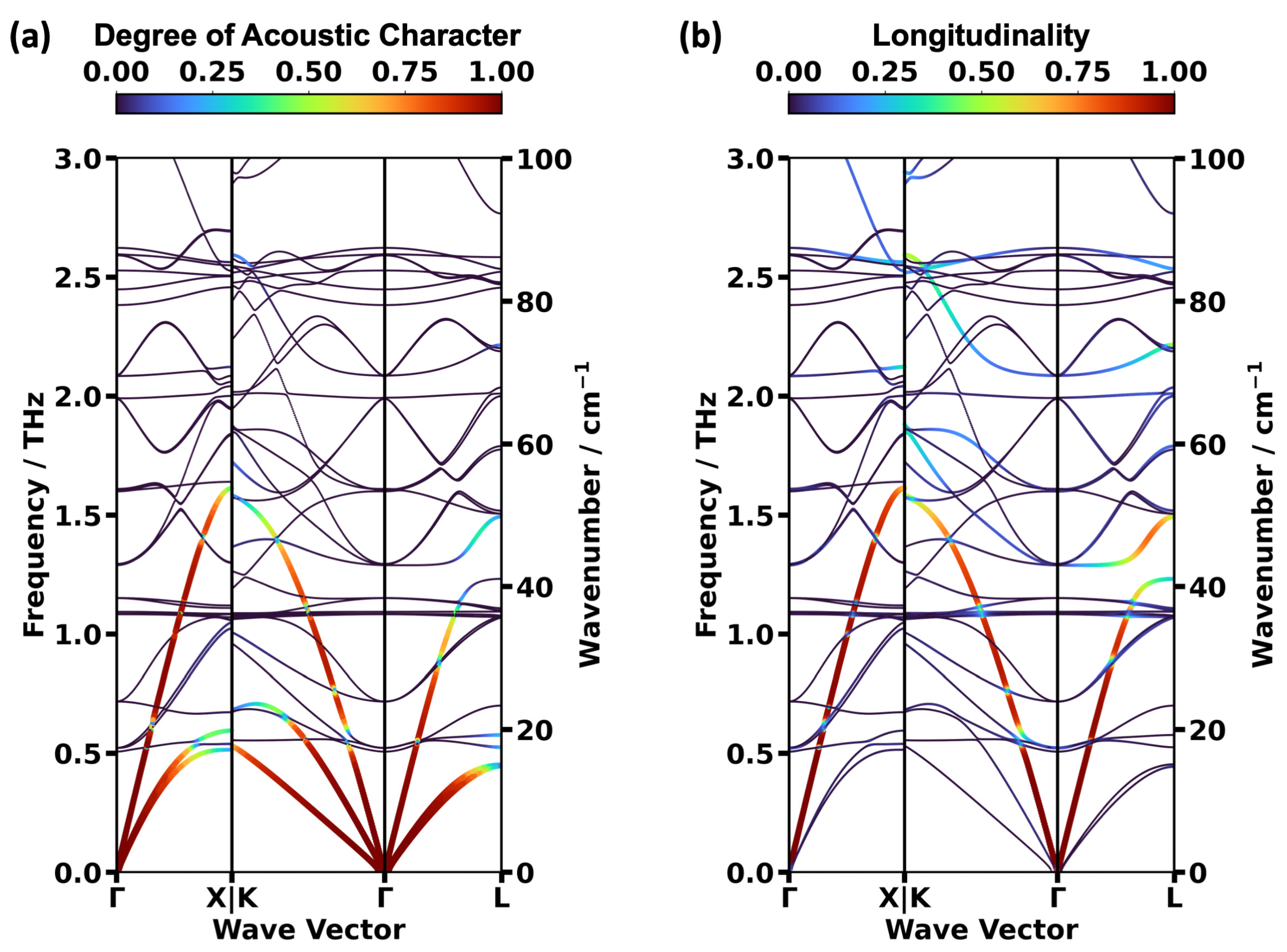
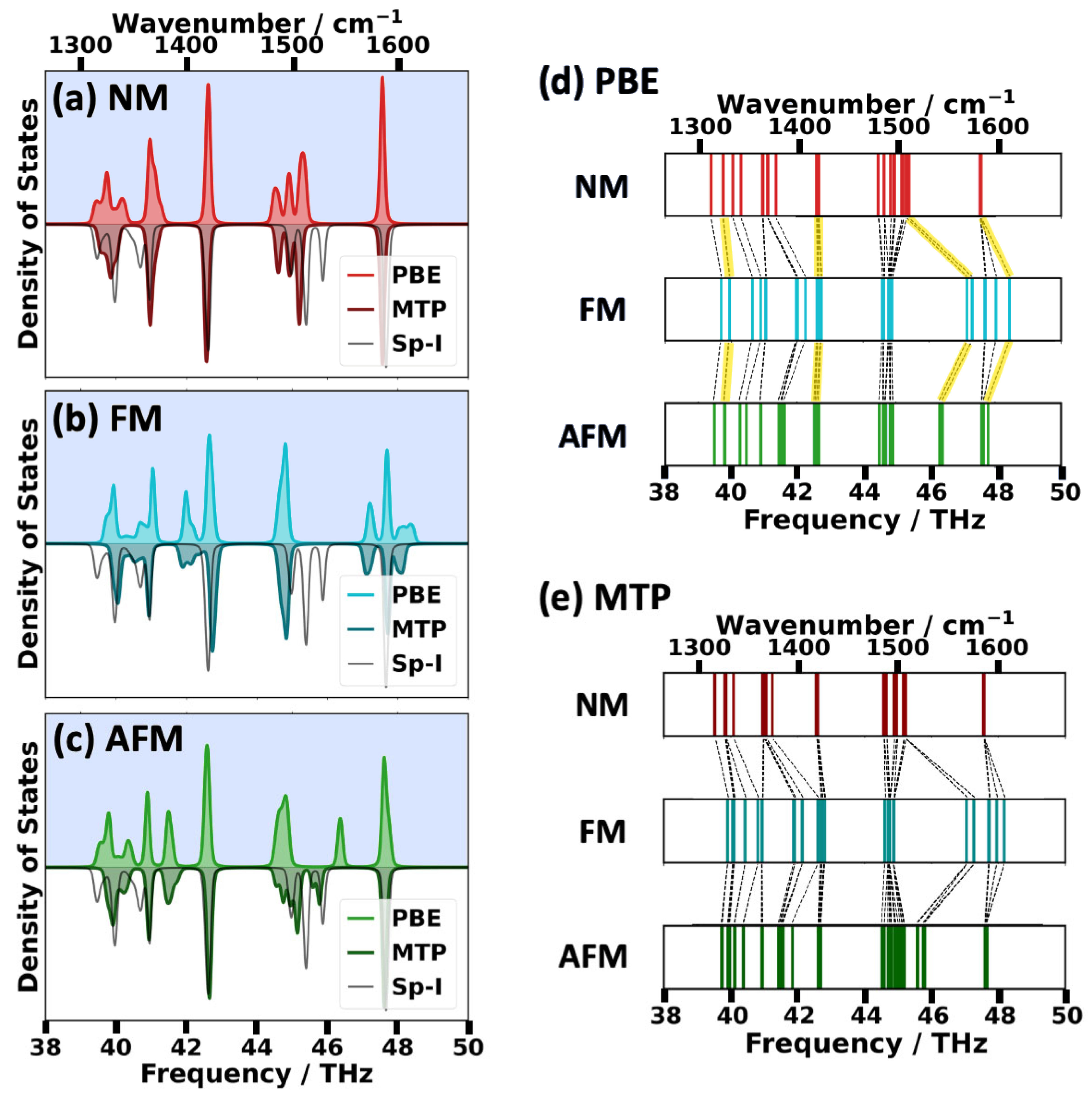
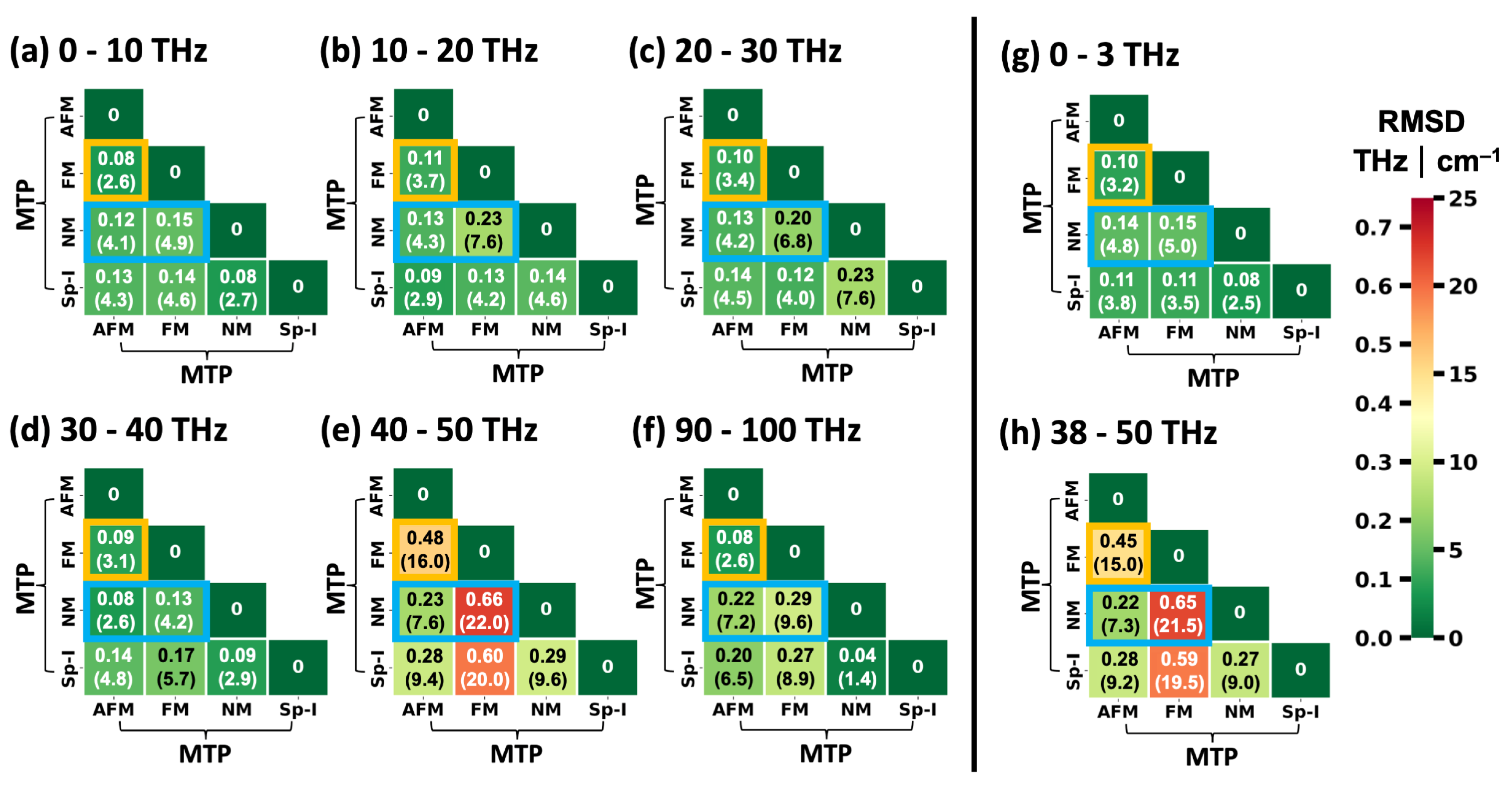
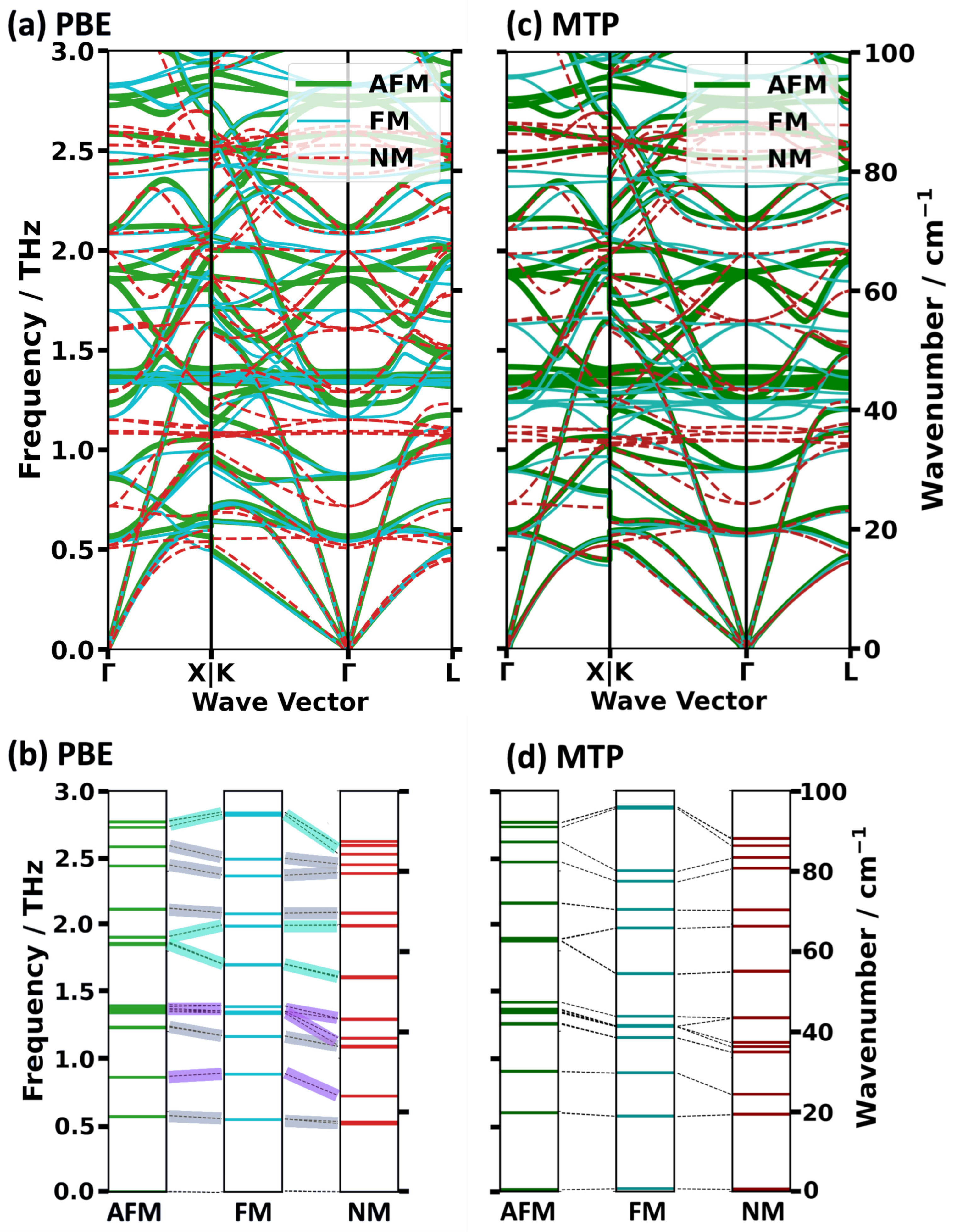
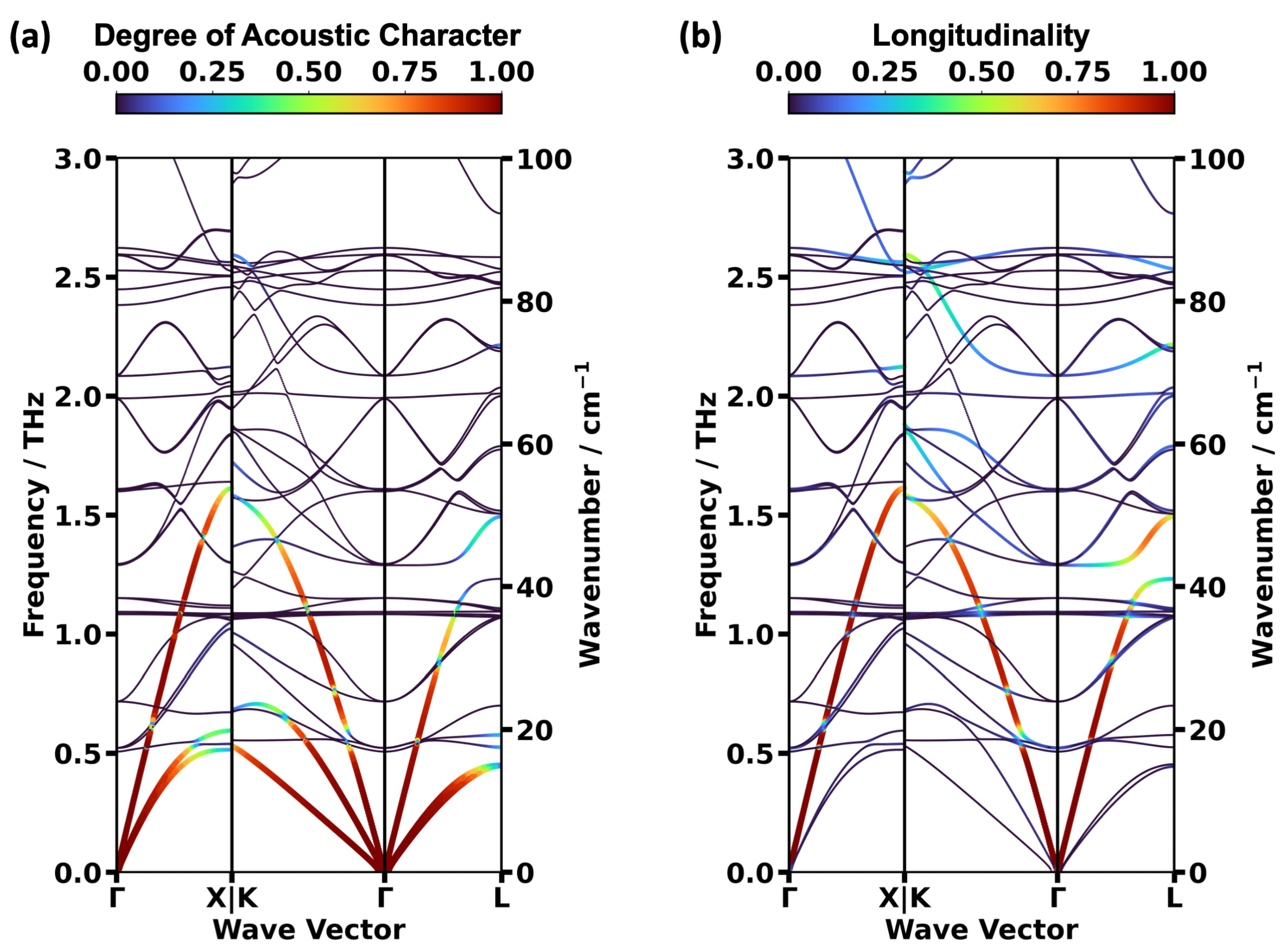
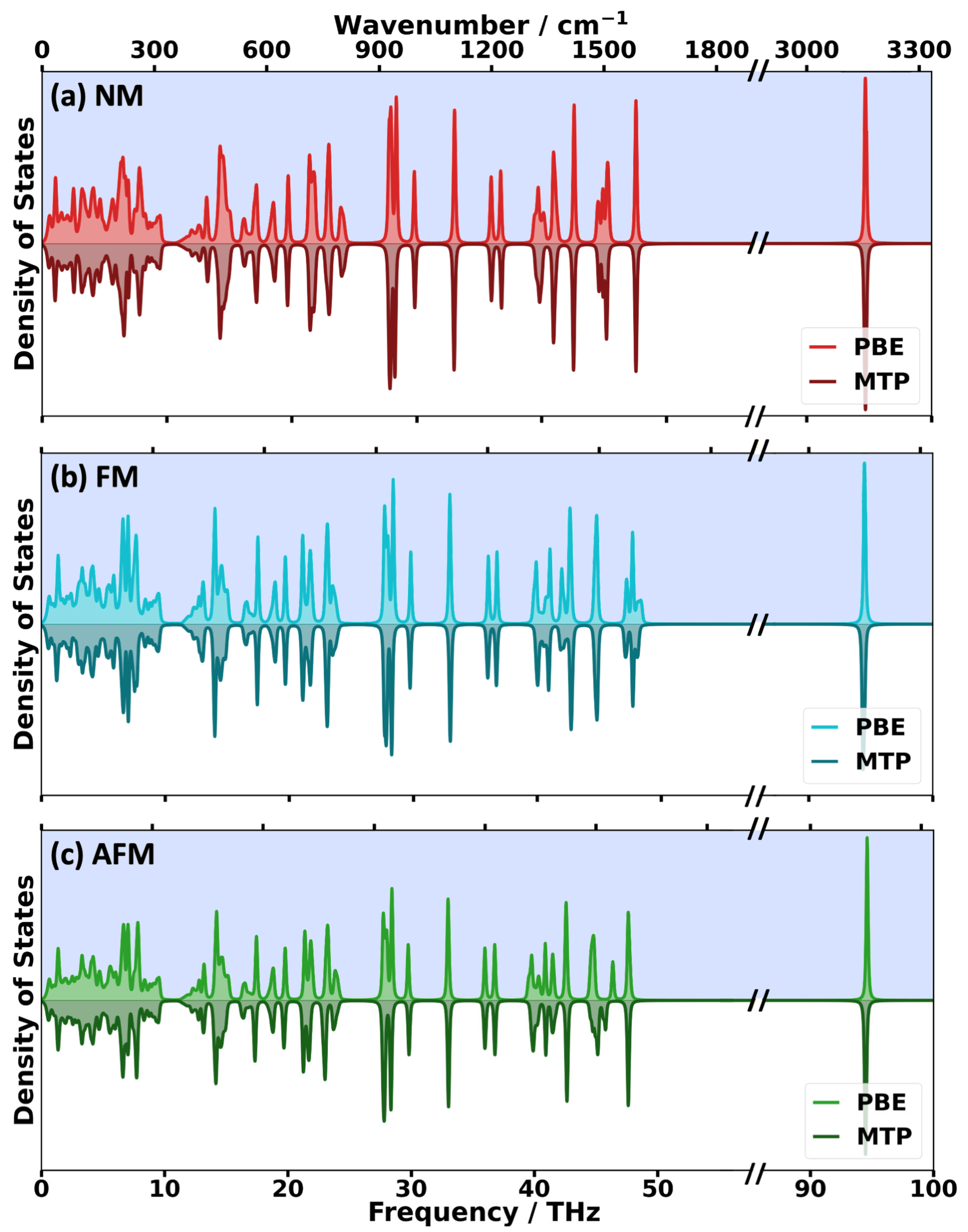
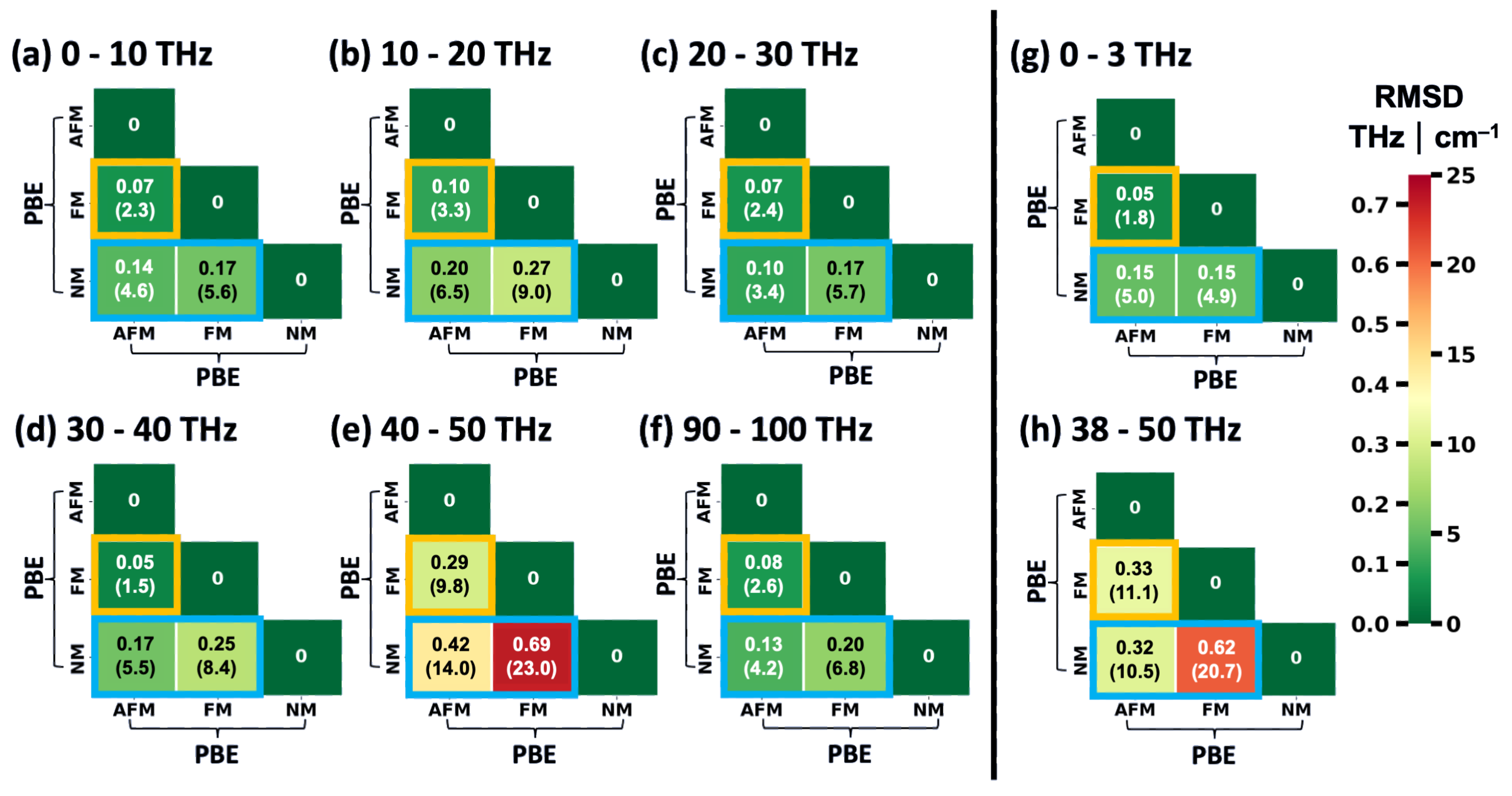
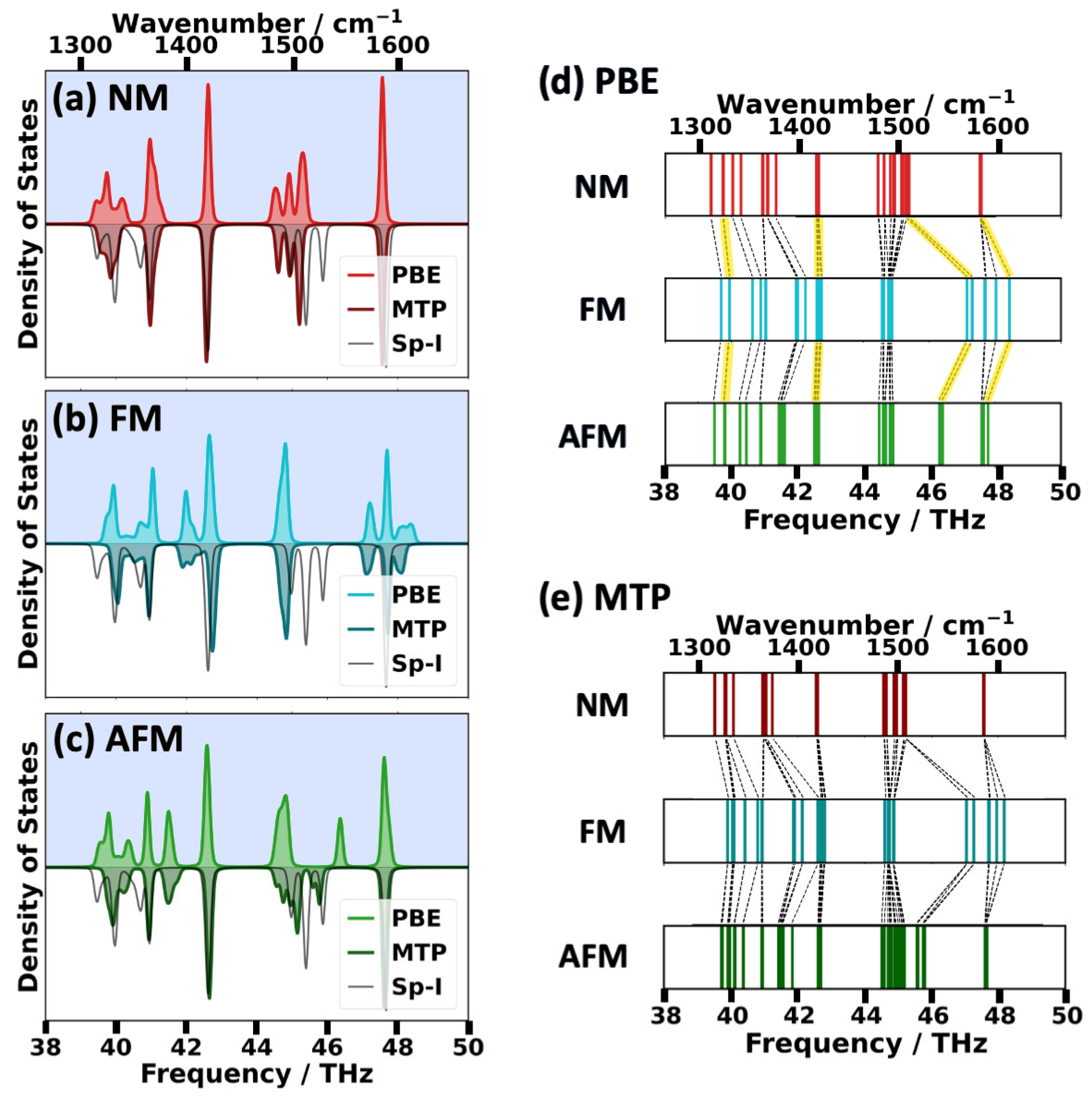
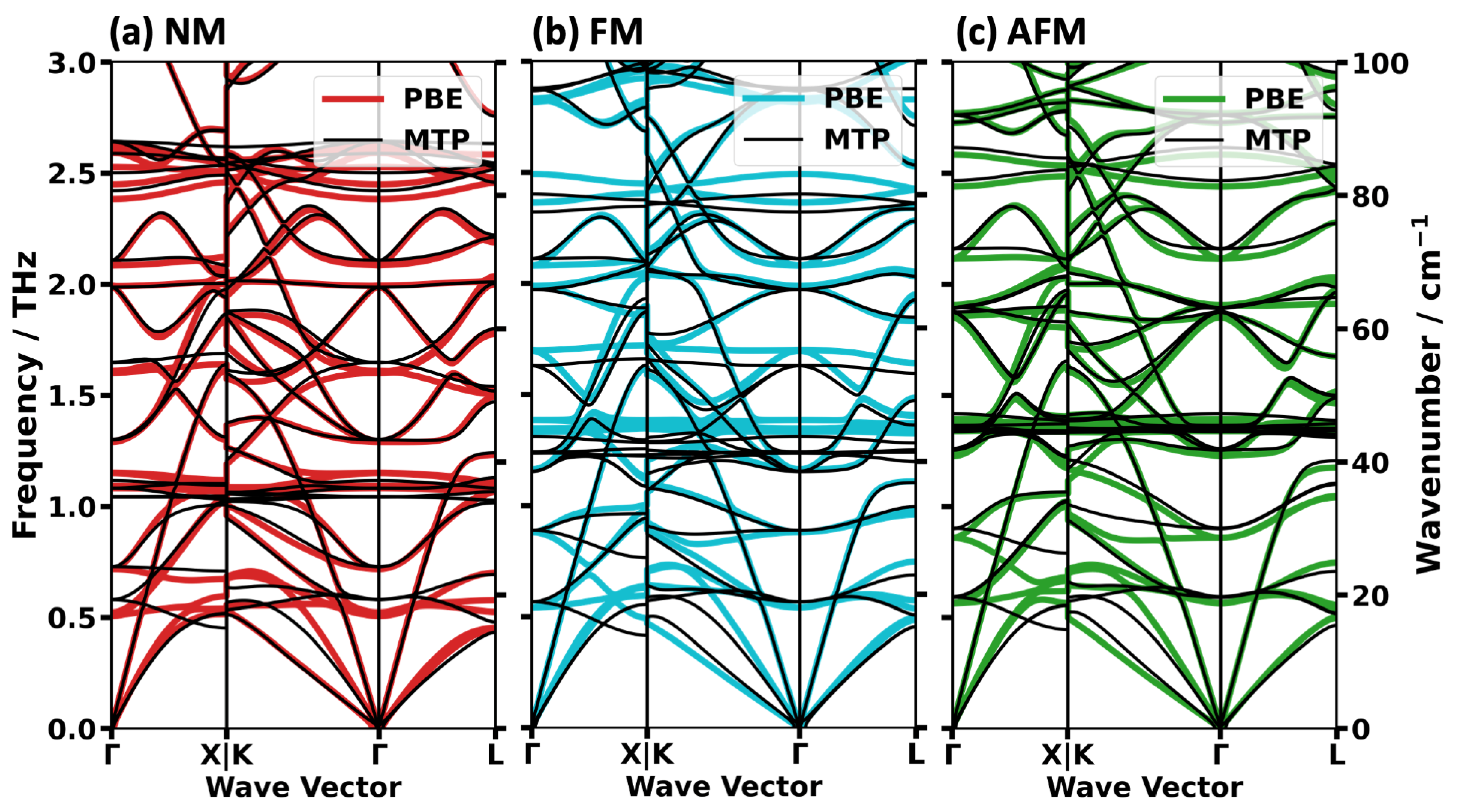
2.2. Impact of the Spin States of HKUST-1 on Phonon Properties
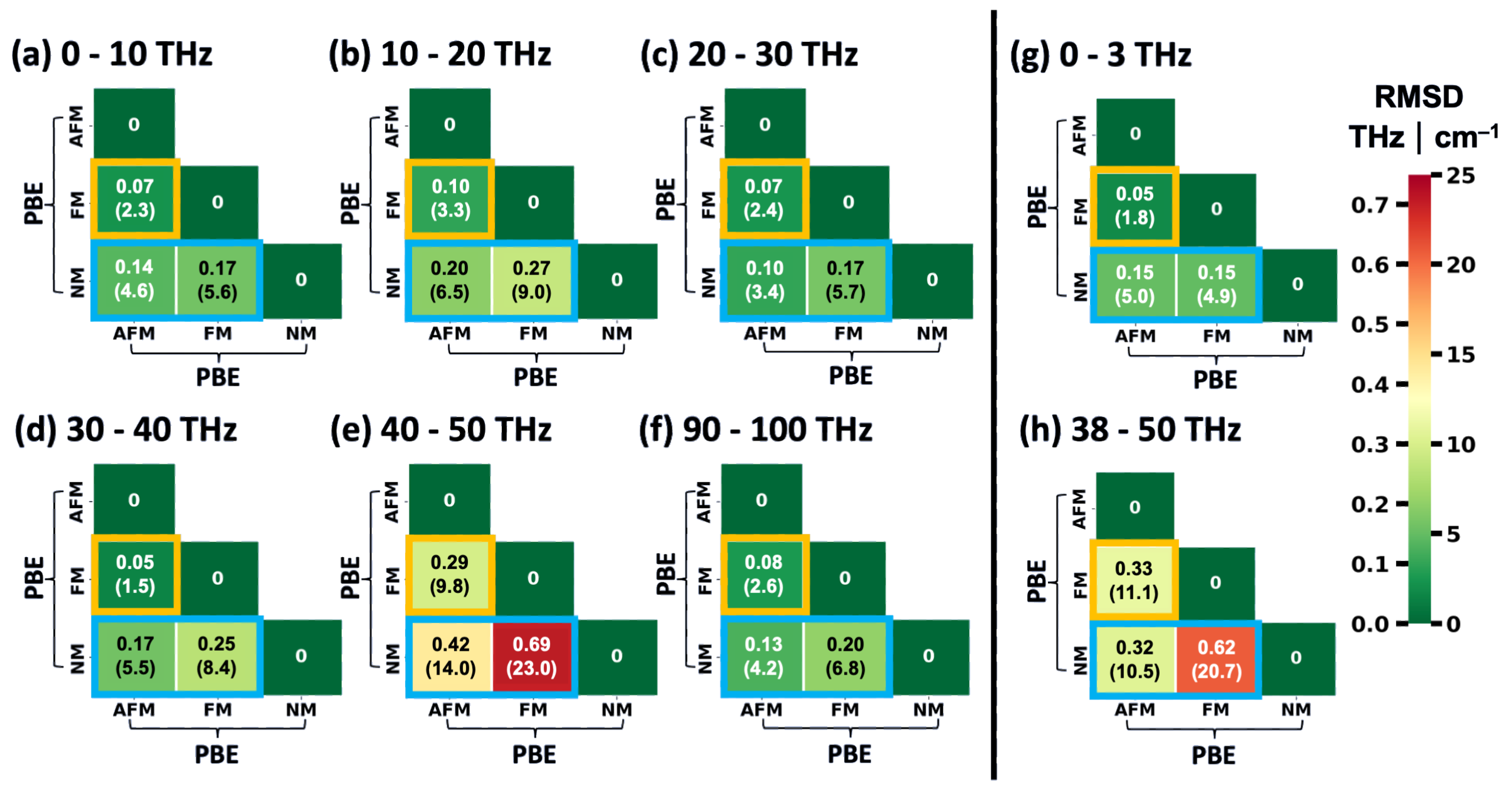
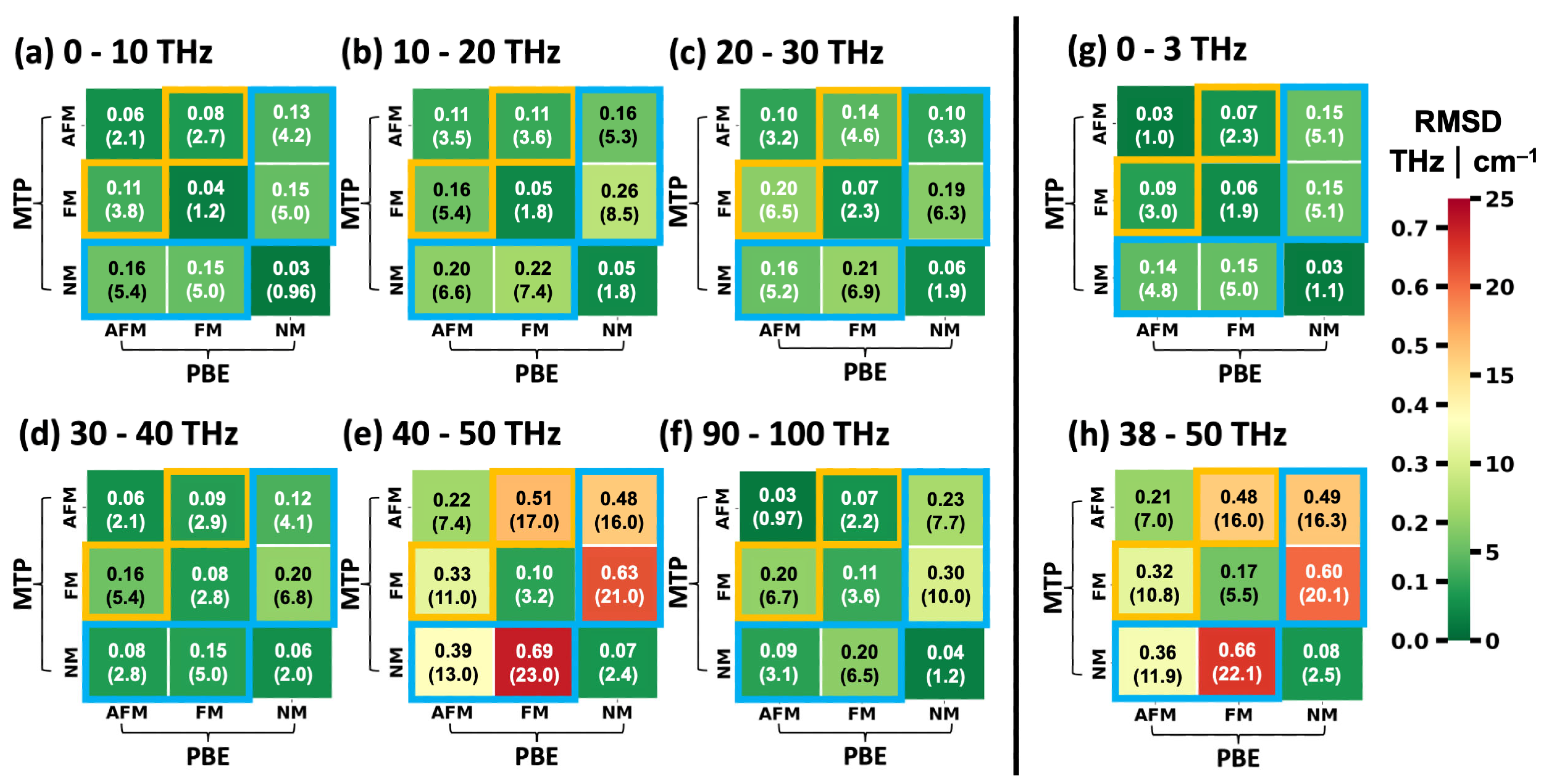
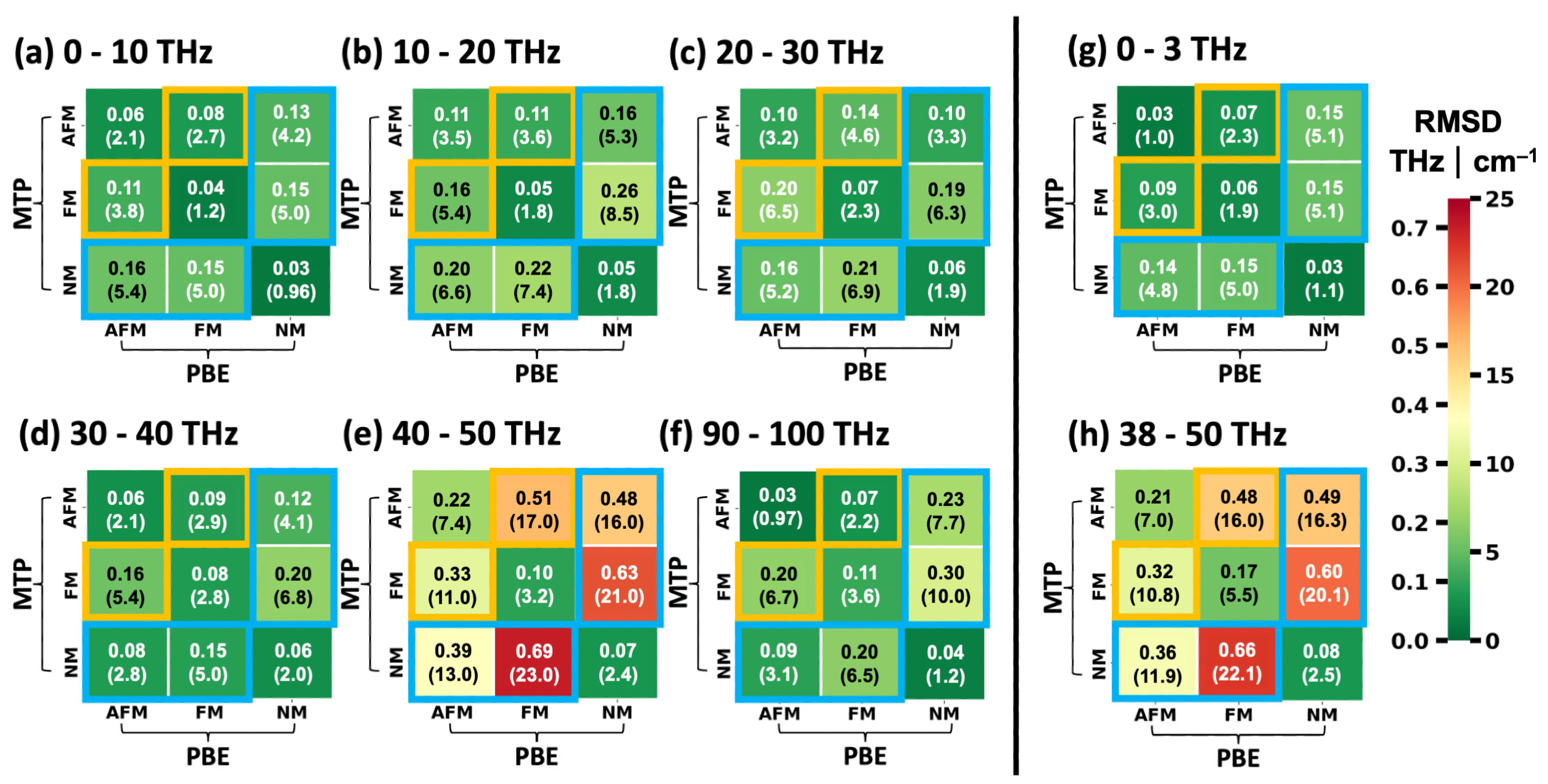
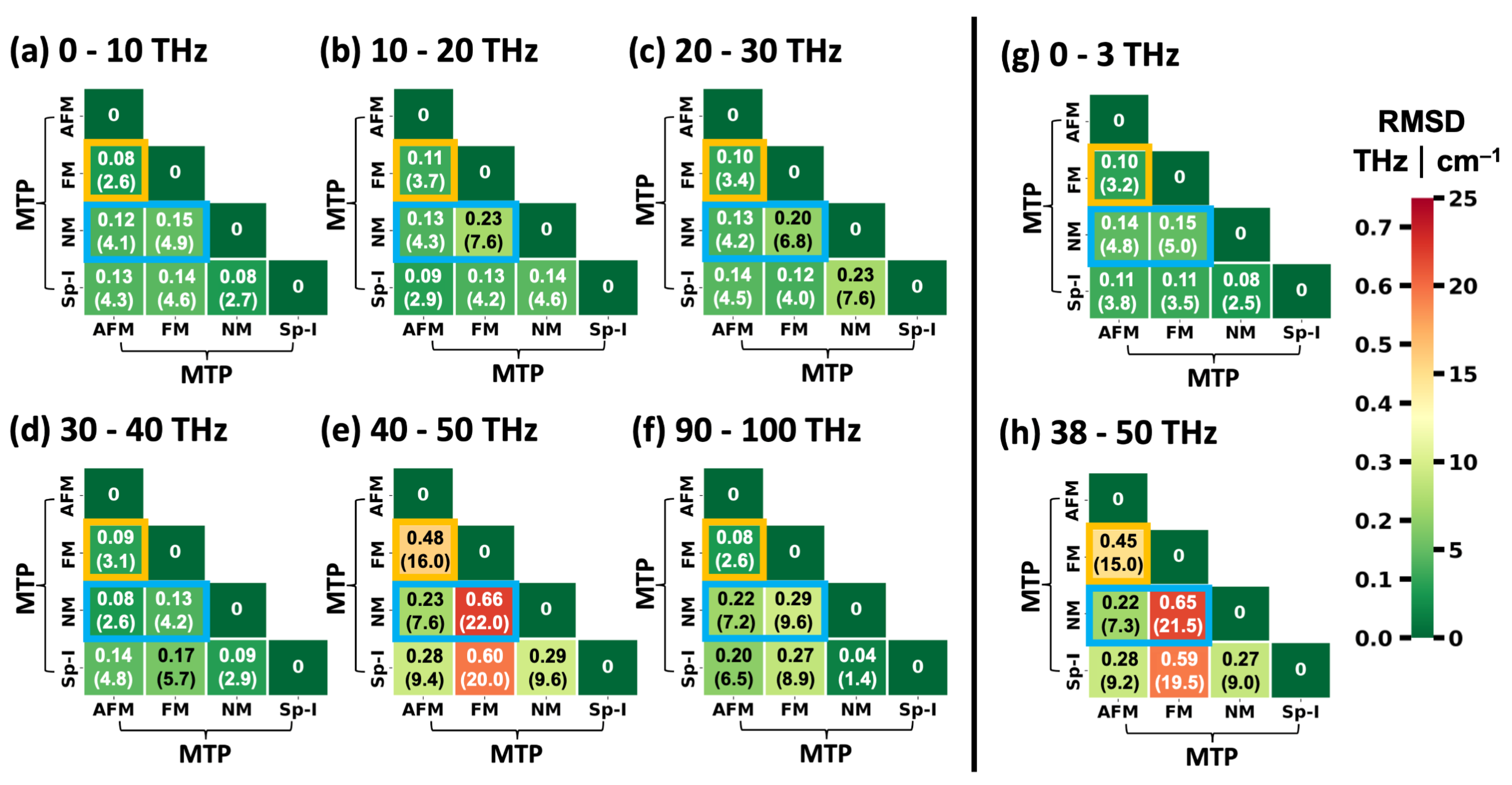
2.3. Performance of System-Specifically Trained MTPs Compared to DFT Reference Data
3. Materials and Methods
3.1. Density Functional Theory Calculations
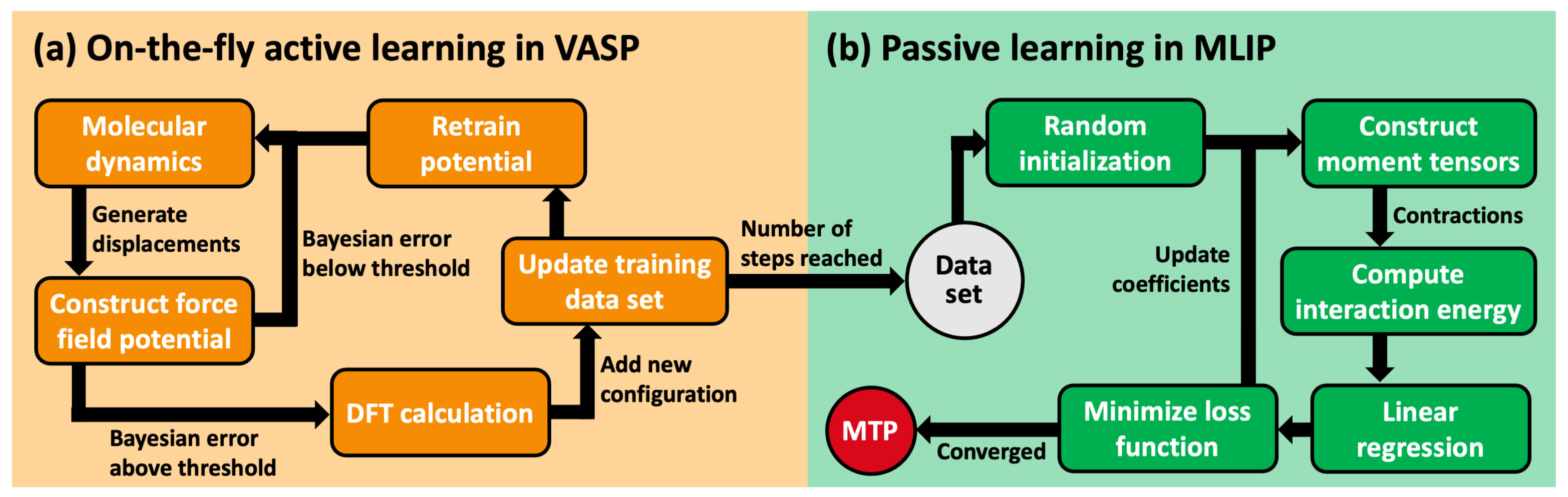
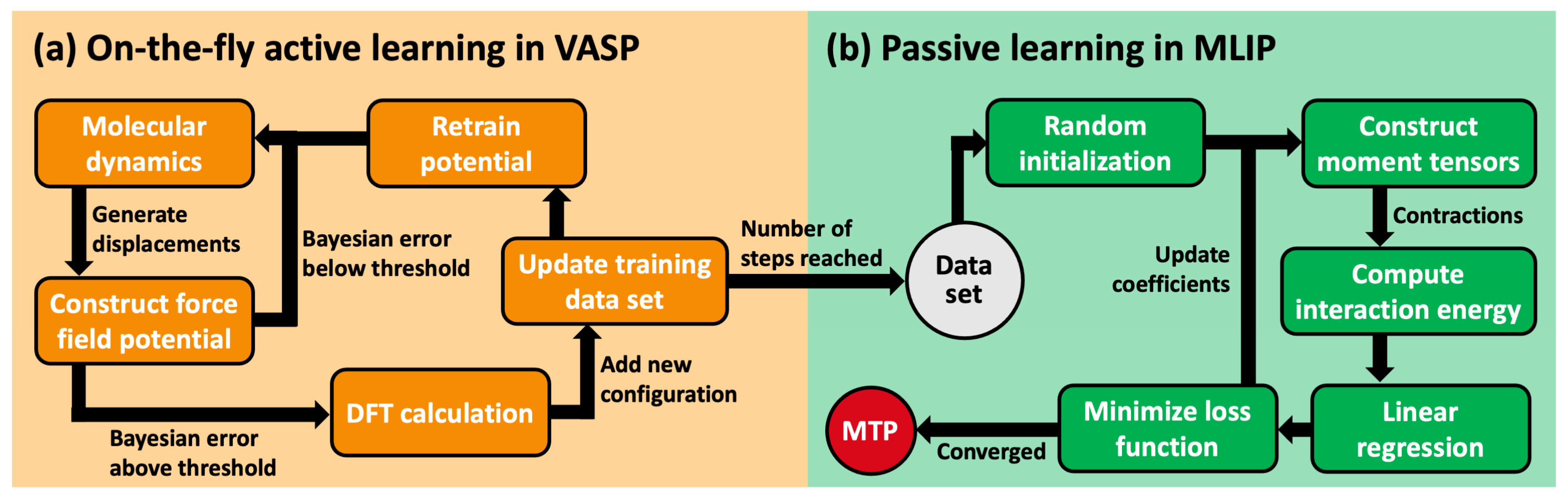
3.2. Generation of Training Data
3.3. Training of Moment Tensor Potentials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yaghi, O.M.; Li, G.; Li, H. Selective Binding and Removal of Guests in a Microporous Metal–Organic Framework. Nature 1995, 378, 703–706. [Google Scholar] [CrossRef]
- Farha, O.K.; Eryazici, I.; Jeong, N.C.; Hauser, B.G.; Wilmer, C.E.; Sarjeant, A.A.; Snurr, R.Q.; Nguyen, S.T.; Yazaydın, A.Ö.; Hupp, J.T. Metal–Organic Framework Materials with Ultrahigh Surface Areas: Is the Sky the Limit? J. Am. Chem. Soc. 2012, 134, 15016–15021. [Google Scholar] [CrossRef]
- Furukawa, H.; Ko, N.; Go, Y.B.; Aratani, N.; Choi, S.B.; Choi, E.; Yazaydin, A.O.; Snurr, R.Q.; O’Keeffe, M.; Kim, J.; et al. Ultrahigh Porosity in Metal-Organic Frameworks. Science 2010, 329, 424–428. [Google Scholar] [CrossRef]
- Pascanu, V.; González Miera, G.; Inge, A.K.; Martín-Matute, B. Metal–Organic Frameworks as Catalysts for Organic Synthesis: A Critical Perspective. J. Am. Chem. Soc. 2019, 141, 7223–7234. [Google Scholar] [CrossRef]
- Mason, J.A.; Veenstra, M.; Long, J.R. Evaluating Metal–Organic Frameworks for Natural Gas Storage. Chem. Sci. 2014, 5, 32–51. [Google Scholar] [CrossRef]
- Li, J.-R.; Kuppler, R.J.; Zhou, H.-C. Selective Gas Adsorption and Separation in Metal–Organic Frameworks. Chem. Soc. Rev. 2009, 38, 1477. [Google Scholar] [CrossRef]
- Khadiv-Parsi, P.; Moradi, H.; Ardestani, M.S.; Taheri, P. Preparation and Drug-Delivery Properties of Metal-Organic Framework HKUST-1. Iran. J. Chem. Eng. 2019, 16, 8. [Google Scholar]
- Velásquez-Hernández, M.d.J.; Linares-Moreau, M.; Astria, E.; Carraro, F.; Alyami, M.Z.; Khashab, N.M.; Sumby, C.J.; Doonan, C.J.; Falcaro, P. Towards Applications of bioentities@MOFs in Biomedicine. Coord. Chem. Rev. 2021, 429, 213651. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; Wiley: Hoboken, NJ, USA, 2005; ISBN 978-0-471-41526-8. [Google Scholar]
- Ziman, J.M. Electrons and Phonons: The Theory of Transport Phenomena in Solids; Oxford University Press: Oxford, UK, 2001; Volume 540. [Google Scholar]
- Pallikara, I.; Kayastha, P.; Skelton, J.M.; Whalley, L.D. The Physical Significance of Imaginary Phonon Modes in Crystals. Electron. Struct. 2022, 4, 033002. [Google Scholar] [CrossRef]
- Chui, S.S.-Y.; Lo, S.M.-F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef]
- Bureekaew, S.; Amirjalayer, S.; Schmid, R. Orbital Directing Effects in Copper and Zinc Based Paddle-Wheel Metal Organic Frameworks: The Origin of Flexibility. J. Mater. Chem. 2012, 22, 10249. [Google Scholar] [CrossRef]
- Scatena, R.; Guntern, Y.T.; Macchi, P. Electron Density and Dielectric Properties of Highly Porous MOFs: Binding and Mobility of Guest Molecules in Cu3(BTC)2 and Zn3(BTC)2. J. Am. Chem. Soc. 2019, 141, 9382–9390. [Google Scholar] [CrossRef]
- Butler, K.T.; Hendon, C.H.; Walsh, A. Electronic Chemical Potentials of Porous Metal–Organic Frameworks. J. Am. Chem. Soc. 2014, 136, 2703–2706. [Google Scholar] [CrossRef]
- Zhang, X.X.; Chui, S.S.-Y.; Williams, I.D. Cooperative Magnetic Behavior in the Coordination Polymers [Cu3(TMA)2L3] (L=H2O, Pyridine). J. Appl. Phys. 2000, 87, 6007–6009. [Google Scholar] [CrossRef]
- Pöppl, A.; Kunz, S.; Himsl, D.; Hartmann, M. CW and Pulsed ESR Spectroscopy of Cupric Ions in the Metal−Organic Framework Compound Cu3(BTC)2. J. Phys. Chem. C 2008, 112, 2678–2684. [Google Scholar] [CrossRef]
- Tiana, D.; Hendon, C.H.; Walsh, A. Ligand Design for Long-Range Magnetic Order in Metal–Organic Frameworks. Chem. Commun. 2014, 50, 13990–13993. [Google Scholar] [CrossRef]
- Tafipolsky, M.; Amirjalayer, S.; Schmid, R. First-Principles-Derived Force Field for Copper Paddle-Wheel-Based Metal−Organic Frameworks. J. Phys. Chem. C 2010, 114, 14402–14409. [Google Scholar] [CrossRef]
- Hendon, C.H.; Walsh, A. Chemical Principles Underpinning the Performance of the Metal–Organic Framework HKUST-1. Chem. Sci. 2015, 6, 3674–3683. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Constantin, L.A.; Sun, J.; Csonka, G.I. Some Fundamental Issues in Ground-State Density Functional Theory: A Guide for the Perplexed. J. Chem. Theory Comput. 2009, 5, 902–908. [Google Scholar] [CrossRef]
- Tada, K.; Mori, M.; Tanaka, S. Spin Contamination Errors in DFT+U/Plane-Wave Calculations for LixFeF3 Systems (x = 0–1). Chem. Lett. 2021, 50, 1057–1061. [Google Scholar] [CrossRef]
- Mohr, S.; Ratcliff, L.E.; Genovese, L.; Caliste, D.; Boulanger, P.; Goedecker, S.; Deutsch, T. Accurate and Efficient Linear Scaling DFT Calculations with Universal Applicability. Phys. Chem. Chem. Phys. 2015, 17, 31360–31370. [Google Scholar] [CrossRef]
- Kamencek, T.; Schrode, B.; Resel, R.; Ricco, R.; Zojer, E. Understanding the Origin of the Particularly Small and Anisotropic Thermal Expansion of MOF-74. Adv. Theory Simul. 2022, 5, 2200031. [Google Scholar] [CrossRef]
- Rimmer, L.H.N.; Dove, M.T.; Goodwin, A.L.; Palmer, D.C. Acoustic Phonons and Negative Thermal Expansion in MOF-5. Phys. Chem. Chem. Phys. 2014, 16, 21144–21152. [Google Scholar] [CrossRef]
- Wieser, S.; Kamencek, T.; Schmid, R.; Bedoya-Martínez, N.; Zojer, E. Exploring the Impact of the Linker Length on Heat Transport in Metal-Organic Frameworks. Nanomaterials 2022, 12, 2142. [Google Scholar] [CrossRef]
- Behler, J.; Parrinello, M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 2007, 98, 146401. [Google Scholar] [CrossRef]
- Eyert, V.; Wormald, J.; Curtin, W.A.; Wimmer, E. Machine-Learned Interatomic Potentials: Recent Developments and Prospective Applications. J. Mater. Res. 2023, 38, 5079–5094. [Google Scholar] [CrossRef]
- Zuo, Y.; Chen, C.; Li, X.; Deng, Z.; Chen, Y.; Behler, J.; Csányi, G.; Shapeev, A.V.; Thompson, A.P.; Wood, M.A.; et al. Performance and Cost Assessment of Machine Learning Interatomic Potentials. J. Phys. Chem. A 2020, 124, 731–745. [Google Scholar] [CrossRef]
- Friederich, P.; Häse, F.; Proppe, J.; Aspuru-Guzik, A. Machine-Learned Potentials for next-Generation Matter Simulations. Nat. Mater. 2021, 20, 750–761. [Google Scholar] [CrossRef]
- Castel, N.; André, D.; Edwards, C.; Evans, J.D.; Coudert, F.-X. Machine Learning Interatomic Potentials for Amorphous Zeolitic Imidazolate Frameworks. Digit. Discov. 2024, 3, 355–368. [Google Scholar] [CrossRef]
- Goeminne, R.; Vanduyfhuys, L.; Van Speybroeck, V.; Verstraelen, T. DFT-Quality Adsorption Simulations in Metal–Organic Frameworks Enabled by Machine Learning Potentials. J. Chem. Theory Comput. 2023, 19, 6313–6325. [Google Scholar] [CrossRef]
- Mortazavi, B.; Rajabpour, A.; Zhuang, X.; Rabczuk, T.; Shapeev, A.V. Exploring Thermal Expansion of Carbon-Based Nanosheets by Machine-Learning Interatomic Potentials. Carbon 2022, 186, 501–508. [Google Scholar] [CrossRef]
- Mortazavi, B.; Zhuang, X. Ultrahigh Strength and Negative Thermal Expansion and Low Thermal Conductivity in Graphyne Nanosheets Confirmed by Machine-Learning Interatomic Potentials. FlatChem 2022, 36, 100446. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shojaei, F.; Shapeev, A.V.; Zhuang, X. A Combined First-Principles and Machine-Learning Investigation on the Stability, Electronic, Optical, and Mechanical Properties of Novel C6N7-Based Nanoporous Carbon Nitrides. Carbon 2022, 194, 230–239. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shapeev, A.V. Anisotropic Mechanical Response, High Negative Thermal Expansion, and Outstanding Dynamical Stability of Biphenylene Monolayer Revealed by Machine-Learning Interatomic Potentials. FlatChem 2022, 32, 100347. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shojaei, F.; Zhuang, X. A Novel Two-Dimensional C36 Fullerene Network; an Isotropic, Auxetic Semiconductor with Low Thermal Conductivity and Remarkable Stiffness. Mater. Today Nano 2023, 21, 100280. [Google Scholar] [CrossRef]
- Wieser, S.; Zojer, E. Machine Learned Force-Fields for an Ab-Initio Quality Description of Metal-Organic Frameworks. npj Comput. Mater. 2024, 10, 18. [Google Scholar] [CrossRef]
- Ying, P.; Liang, T.; Xu, K.; Zhang, J.; Xu, J.; Zhong, Z.; Fan, Z. Sub-Micrometer Phonon Mean Free Paths in Metal–Organic Frameworks Revealed by Machine Learning Molecular Dynamics Simulations. ACS Appl. Mater. Interfaces 2023, 15, 36412–36422. [Google Scholar] [CrossRef]
- Novikov, I.S.; Gubaev, K.; Podryabinkin, E.V.; Shapeev, A.V. The MLIP Package: Moment Tensor Potentials with MPI and Active Learning. Mach. Learn. Sci. Technol. 2021, 2, 025002. [Google Scholar] [CrossRef]
- Shapeev, A.V. Moment Tensor Potentials: A Class of Systematically Improvable Interatomic Potentials. Multiscale Model. Simul. 2016, 14, 1153–1173. [Google Scholar] [CrossRef]
- Novikov, I.; Grabowski, B.; Körmann, F.; Shapeev, A. Magnetic Moment Tensor Potentials for Collinear Spin-Polarized Materials Reproduce Different Magnetic States of Bcc Fe. npj Comput. Mater. 2022, 8, 13. [Google Scholar] [CrossRef]
- Salem, L. Electrons in Chemical Reactions: First Principles; John Wiley & Sons: Toronto, ON, Canada, 1982. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized Gradient Approximation for the Exchange-Correlation Hole of a Many-Electron System. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef]
- Bleaney, B.; Bowers, K.D. Anomalous Paramagnetism of Copper Acetate. Proc. R. Soc. Lond. 1952, 214, 415. [Google Scholar] [CrossRef]
- Melnik, M.; Dunaj-Jurco, M.; Handlovic, M. Crystal Structure, Spectral and Magnetic Characterization of Bis(p-Benzoato-0, 0′) (dimethylsuIphoxide)Copper(II). Inorg. Chim. Acta 1984, 86, 185–190. [Google Scholar] [CrossRef]
- Jacob, C.R.; Reiher, M. Spin in Density-Functional Theory. Int. J. Quantum Chem. 2012, 112, 3661–3684. [Google Scholar] [CrossRef]
- Schüler, M.; Peil, O.E.; Kraberger, G.J.; Pordzik, R.; Marsman, M.; Kresse, G.; Wehling, T.O.; Aichhorn, M. Charge Self-Consistent Many-Body Corrections Using Optimized Projected Localized Orbitals. J. Phys. Condens. Matter 2018, 30, 475901. [Google Scholar] [CrossRef]
- Lindsay, L.; Katre, A.; Cepellotti, A.; Mingo, N. Perspective on Ab Initio Phonon Thermal Transport. J. Appl. Phys. 2019, 126, 050902. [Google Scholar] [CrossRef]
- Jaeken, J.W.; Cottenier, S. Solving the Christoffel Equation: Phase and Group Velocities. Comput. Phys. Commun. 2016, 207, 445–451. [Google Scholar] [CrossRef]
- Kamencek, T.; Wieser, S.; Kojima, H.; Bedoya-Martínez, N.; Dürholt, J.P.; Schmid, R.; Zojer, E. Evaluating Computational Shortcuts in Supercell-Based Phonon Calculations of Molecular Crystals: The Instructive Case of Naphthalene. J. Chem. Theory Comput. 2020, 16, 2716–2735. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tanaka, I. Distributions of Phonon Lifetimes in Brillouin Zones. Phys. Rev. B 2015, 91, 094306. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Miwa, K.; Karsai, F.; Kresse, G.; Asahi, R. On-the-Fly Active Learning of Interatomic Potentials for Large-Scale Atomistic Simulations. J. Phys. Chem. Lett. 2020, 11, 6946–6955. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Fuchs, F.; Furthmüller, J.; Bechstedt, F.; Shishkin, M.; Kresse, G. Quasiparticle Band Structure Based on a Generalized Kohn-Sham Scheme. Phys. Rev. B 2007, 76, 115109. [Google Scholar] [CrossRef]
- Lebernegg, S.; Schmitt, M.; Tsirlin, A.A.; Janson, O.; Rosner, H. Magnetism of Cu X 2 Frustrated Chains (X = F, Cl, Br): Role of Covalency. Phys. Rev. B 2013, 87, 155111. [Google Scholar] [CrossRef]
- Kvashnin, Y.O.; Rudenko, A.N.; Thunström, P.; Rösner, M.; Katsnelson, M.I. Dynamical Correlations in Single-Layer CrI 3. Phys. Rev. B 2022, 105, 205124. [Google Scholar] [CrossRef]
- Ren, X.; Rinke, P.; Blum, V.; Wieferink, J.; Tkatchenko, A.; Sanfilippo, A.; Reuter, K.; Scheffler, M. Resolution-of-Identity Approach to Hartree–Fock, Hybrid Density Functionals, RPA, MP2 and GW with Numeric Atom-Centered Orbital Basis Functions. New J. Phys. 2012, 14, 053020. [Google Scholar] [CrossRef]
- Blum, V.; Gehrke, R.; Hanke, F.; Havu, P.; Havu, V.; Ren, X.; Reuter, K.; Scheffler, M. Ab Initio Molecular Simulations with Numeric Atom-Centered Orbitals. Comput. Phys. Commun. 2009, 180, 2175–2196. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A Generally Applicable Atomic-Charge Dependent London Dispersion Correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 Dispersion Coefficient Model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Mewes, J.-M.; Ehlert, S.; Grimme, S. Extension and Evaluation of the D4 London-Dispersion Model for Periodic Systems. Phys. Chem. Chem. Phys. 2020, 22, 8499–8512. [Google Scholar] [CrossRef]
- Ambrosetti, A.; Reilly, A.M.; DiStasio, R.A.; Tkatchenko, A. Long-Range Correlation Energy Calculated from Coupled Atomic Response Functions. J. Chem. Phys. 2014, 140, 18A508. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- PREC. Available online: https://www.vasp.at/wiki/index.php/PREC (accessed on 14 August 2023).
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Togo, A. First-Principles Phonon Calculations with Phonopy and Phono3py. J. Phys. Soc. Jpn. 2023, 92, 012001. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First Principles Phonon Calculations in Materials Science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Kamencek, T. Understanding Phonon-Related Properties in Metal-Organic Frameworks for Controlling Their Mechanical and Thermal Characteristics. Ph.D. Thesis, Graz University of Technology, Graz, Austria, 2022. [Google Scholar]
- Legenstein, L.; Reicht, L.; Kamencek, T.; Zojer, E. Anisotropic Phonon Bands in H-Bonded Molecular Crystals: The Instructive Case of α-Quinacridone. ACS Mater. Au 2023, 3, 371–385. [Google Scholar] [CrossRef]
- Verdi, C.; Karsai, F.; Liu, P.; Jinnouchi, R.; Kresse, G. Thermal Transport and Phase Transitions of Zirconia by On-the-Fly Machine-Learned Interatomic Potentials. npj Comput. Mater. 2021, 7, 156. [Google Scholar] [CrossRef]
- Liu, P.; Verdi, C.; Karsai, F.; Kresse, G. α—β Phase Transition of Zirconium Predicted by On-the-Fly Machine-Learned Force Field. Phys. Rev. Mater. 2021, 5, 053804. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Karsai, F.; Kresse, G. On-the-Fly Machine Learning Force Field Generation: Application to Melting Points. Phys. Rev. B 2019, 100, 014105. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 2017; ISBN 978-0-19-252470-6. [Google Scholar]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; In ’T Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Golub, G.H.; Van Loan, C.F. Matrix Computations; Johns Hopkins Studies in the Mathematical Sciences, 4th ed.; The Johns Hopkins University Press: Baltimore, MD, USA, 2013; ISBN 978-1-4214-0794-4. [Google Scholar]
- Lampin, E.; Palla, P.L.; Francioso, P.-A.; Cleri, F. Thermal Conductivity from Approach-to-Equilibrium Molecular Dynamics. J. Appl. Phys. 2013, 114, 033525. [Google Scholar] [CrossRef]
- Evans, D.J.; Morriss, G.P. Statistical Mechanics of Nonequilibrium Liquids, 2nd ed.; ANU E Press: Canberra, Australia, 2007; ISBN 978-1-921313-22-6. [Google Scholar]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and Analysis of Atomistic Simulation Data with OVITO–the Open Visualization Tool. Modelling Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Lenthe, E.V.; Baerends, E.J.; Snijders, J.G. Relativistic Regular Two-Component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Gajdoš, M.; Hummer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear Optical Properties in the Projector-Augmented Wave Methodology. Phys. Rev. B 2006, 73, 045112. [Google Scholar] [CrossRef]
- Ambrosch-Draxl, C.; Sofo, J.O. Linear Optical Properties of Solids within the Full-Potential Linearized Augmented Planewave Method. Comput. Phys. Commun. 2006, 175, 1–14. [Google Scholar] [CrossRef]
- Ryder, M.R.; Civalleri, B.; Cinque, G.; Tan, J.-C. Discovering Connections between Terahertz Vibrations and Elasticity Underpinning the Collective Dynamics of the HKUST-1 Metal–Organic Framework. CrystEngComm 2016, 18, 4303–4312. [Google Scholar] [CrossRef]
- Mohammadnejad, M.; Fakhrefatemi, M. Synthesis of Magnetic HKUST-1 Metal-Organic Framework for Efficient Removal of Mefenamic Acid from Water. J. Mol. Struct. 2021, 1224, 129041. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E(AFM) [eV/UC] | 0.00 |
| E(FM) [eV/UC] | 0.32 |
| E(NM) [eV/UC] | 0.92 |
| J [cm−1/PW] | −215 |
| Atom Indices for AFM State | MAFM [μB] | Atom Indices for FM State | MFM [μB] |
|---|---|---|---|
| Cu1, Cu2 | ±0.51 | Cu1 | 0.55 |
| O1, O2 | ±0.08 | O1 | 0.09 |
| C1, C3, C4, C6 | 0.00 | C1, C3 | 0.00 |
| C2, C5 | 0.00 | C2 | 0.01 |
| H1, H2 | 0.00 | H1 | 0.00 |
| Mtotal [μB] | 0.00 | Mtotal [μB] | 11.6 |
| NM | FM | AFM | ||||
|---|---|---|---|---|---|---|
| vg(TA) [THzÅ] | vg(LA) [THzÅ] | vg(TA) [THzÅ] | vg(LA) [THzÅ] | vg(TA) [THzÅ] | vg(LA) [THzÅ] | |
| Γ-X | 25.1 (23.6) 27.0 (23.4) | 53.2 (53.1) | 25.0 (23.6) 27.2 (23.2) | 52.6 (53.4) | 26.9 (24.1) 28.0 (23.6) | 52.7 (53.6) |
| Γ-K | 13.3 (13.0) 28.4 (24.3) | 55.3 (54.9) | 11.6 (13.3) 27.4 (23.9) | 55.8 (55.5) | 12.0 (14.2) 27.8 (24.5) | 56.5 (55.6) |
| Γ-L | 19.7 (16.8) 21.7 (17.5) | 59.7 (58.2) | 18.3 (16.7) 19.5 (18.0) | 58.7 (57.9) | 19.1 (17.6) 19.3 (18.3) | 59.8 (58.3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strasser, N.; Wieser, S.; Zojer, E. Predicting Spin-Dependent Phonon Band Structures of HKUST-1 Using Density Functional Theory and Machine-Learned Interatomic Potentials. Int. J. Mol. Sci. 2024, 25, 3023. https://doi.org/10.3390/ijms25053023
Strasser N, Wieser S, Zojer E. Predicting Spin-Dependent Phonon Band Structures of HKUST-1 Using Density Functional Theory and Machine-Learned Interatomic Potentials. International Journal of Molecular Sciences. 2024; 25(5):3023. https://doi.org/10.3390/ijms25053023
Chicago/Turabian StyleStrasser, Nina, Sandro Wieser, and Egbert Zojer. 2024. "Predicting Spin-Dependent Phonon Band Structures of HKUST-1 Using Density Functional Theory and Machine-Learned Interatomic Potentials" International Journal of Molecular Sciences 25, no. 5: 3023. https://doi.org/10.3390/ijms25053023
APA StyleStrasser, N., Wieser, S., & Zojer, E. (2024). Predicting Spin-Dependent Phonon Band Structures of HKUST-1 Using Density Functional Theory and Machine-Learned Interatomic Potentials. International Journal of Molecular Sciences, 25(5), 3023. https://doi.org/10.3390/ijms25053023