

Targeted RNAseq Revealed the Gene Expression Signature of Ferroptosis-Related Processes Associated with Disease Severity in Patients with Multiple Sclerosis


 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Study Population
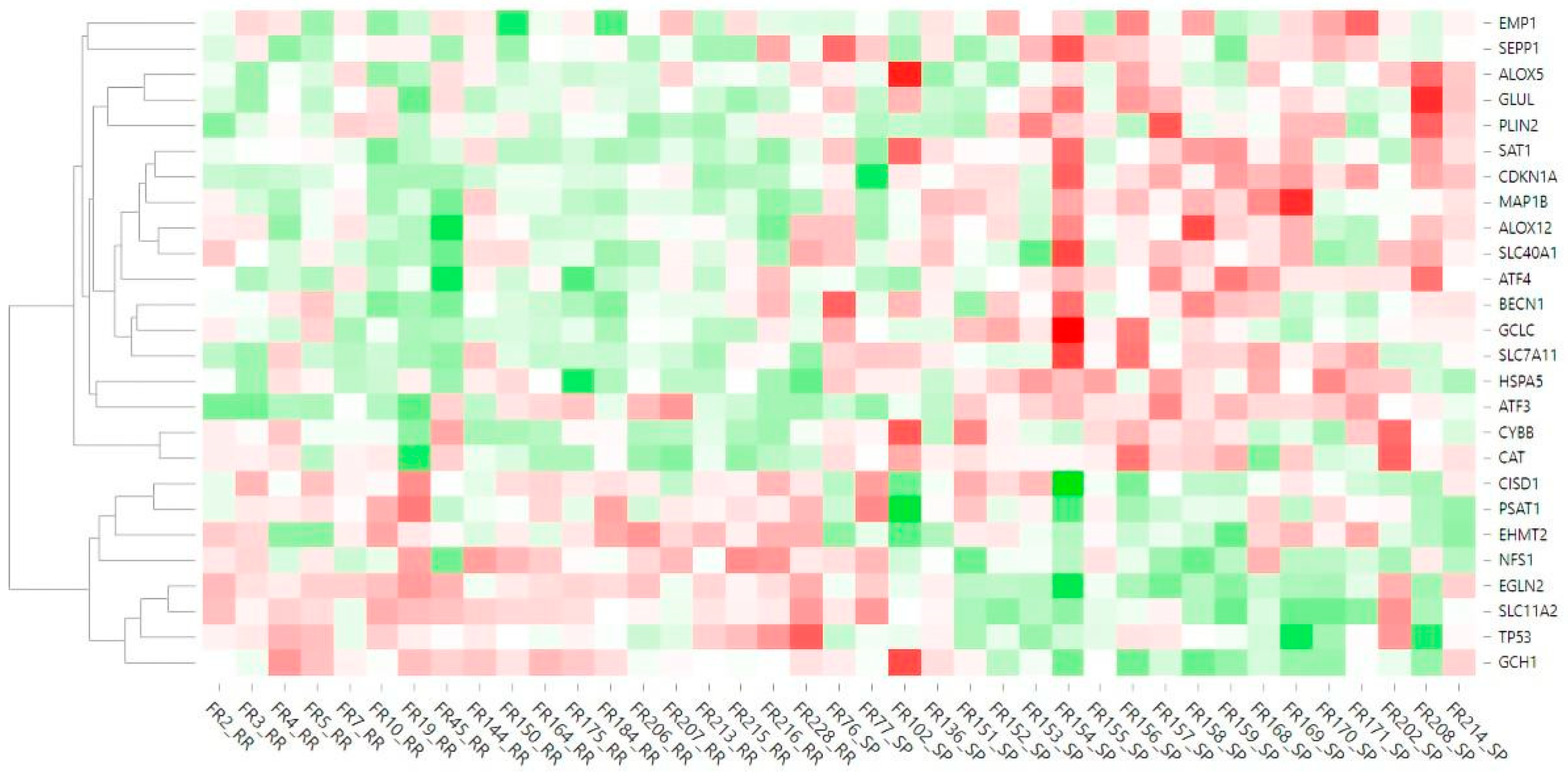
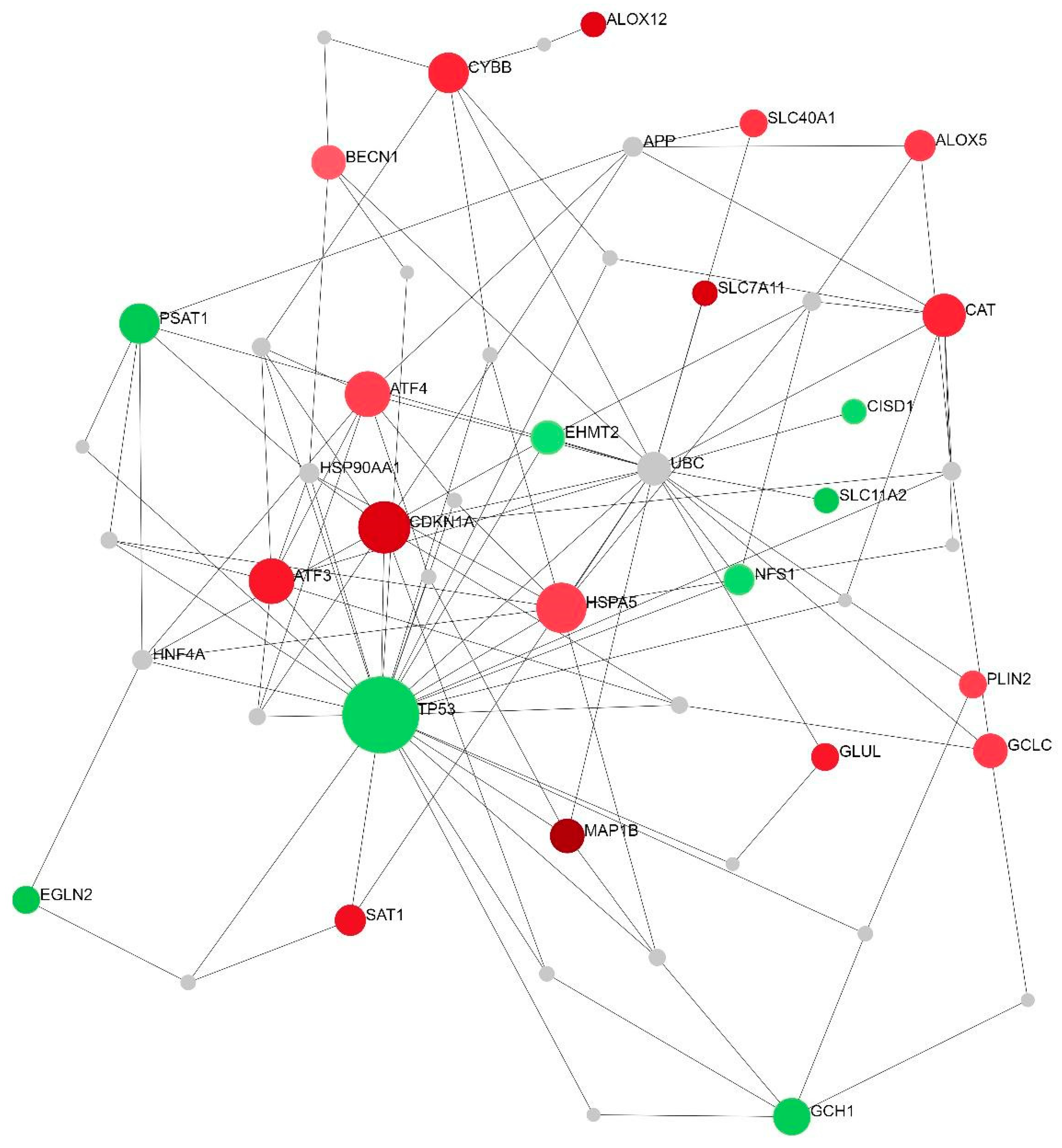
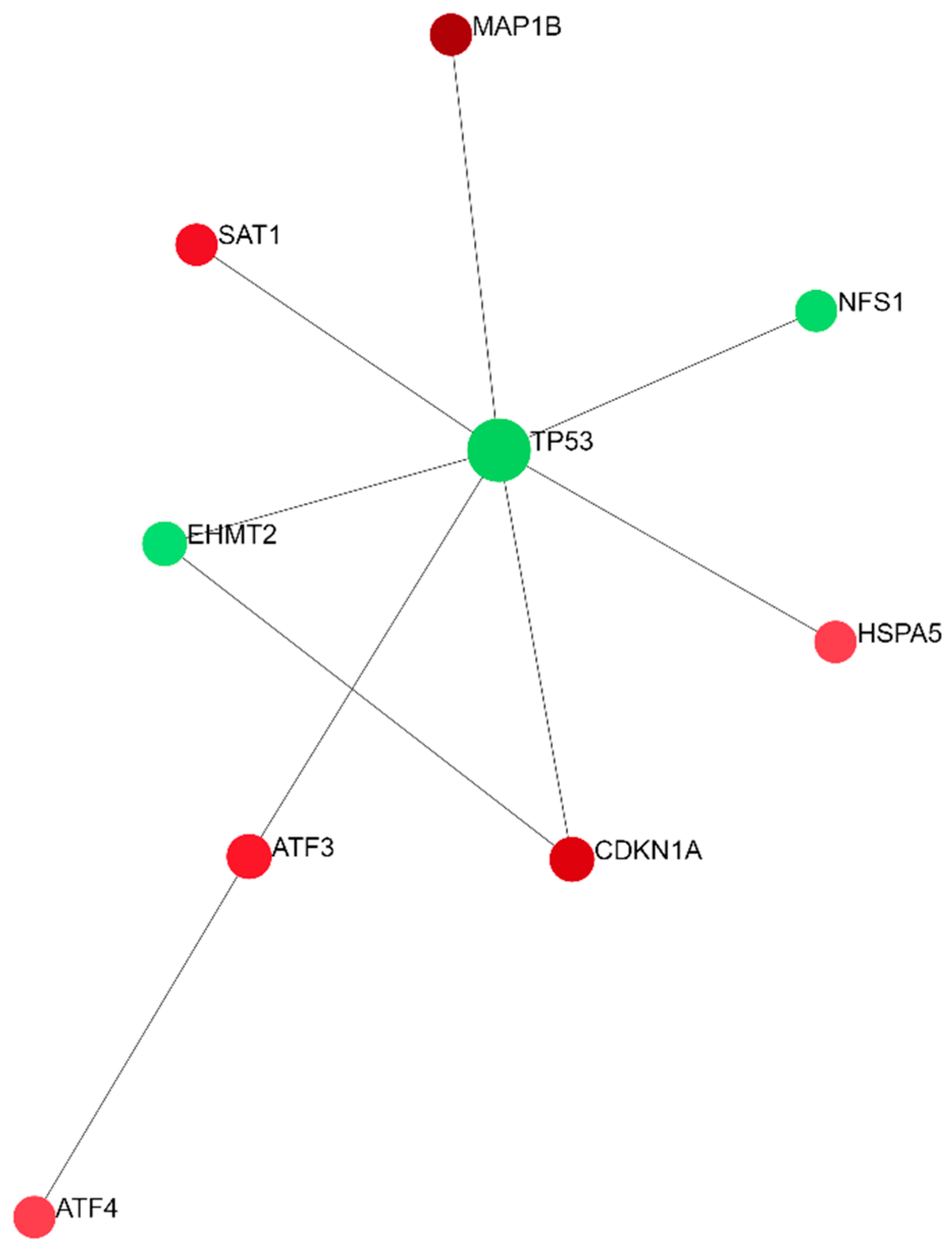
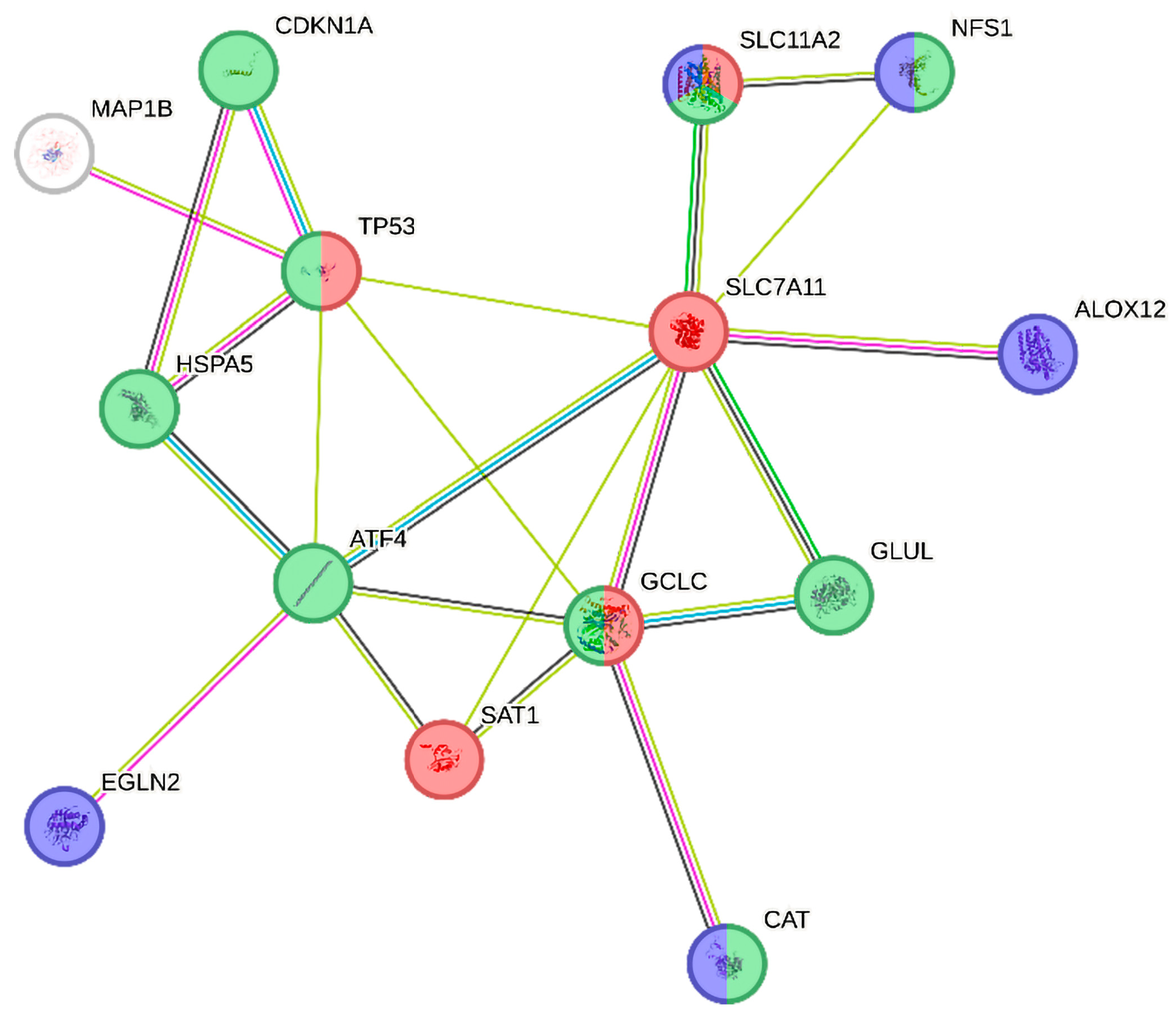
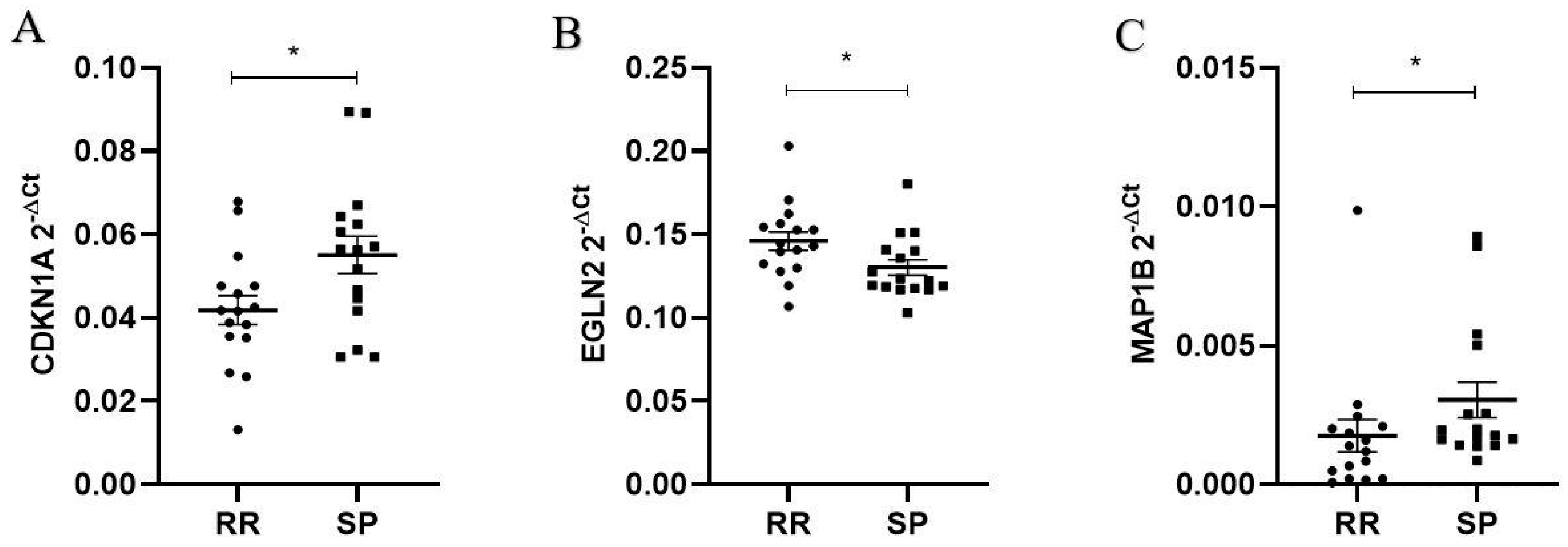
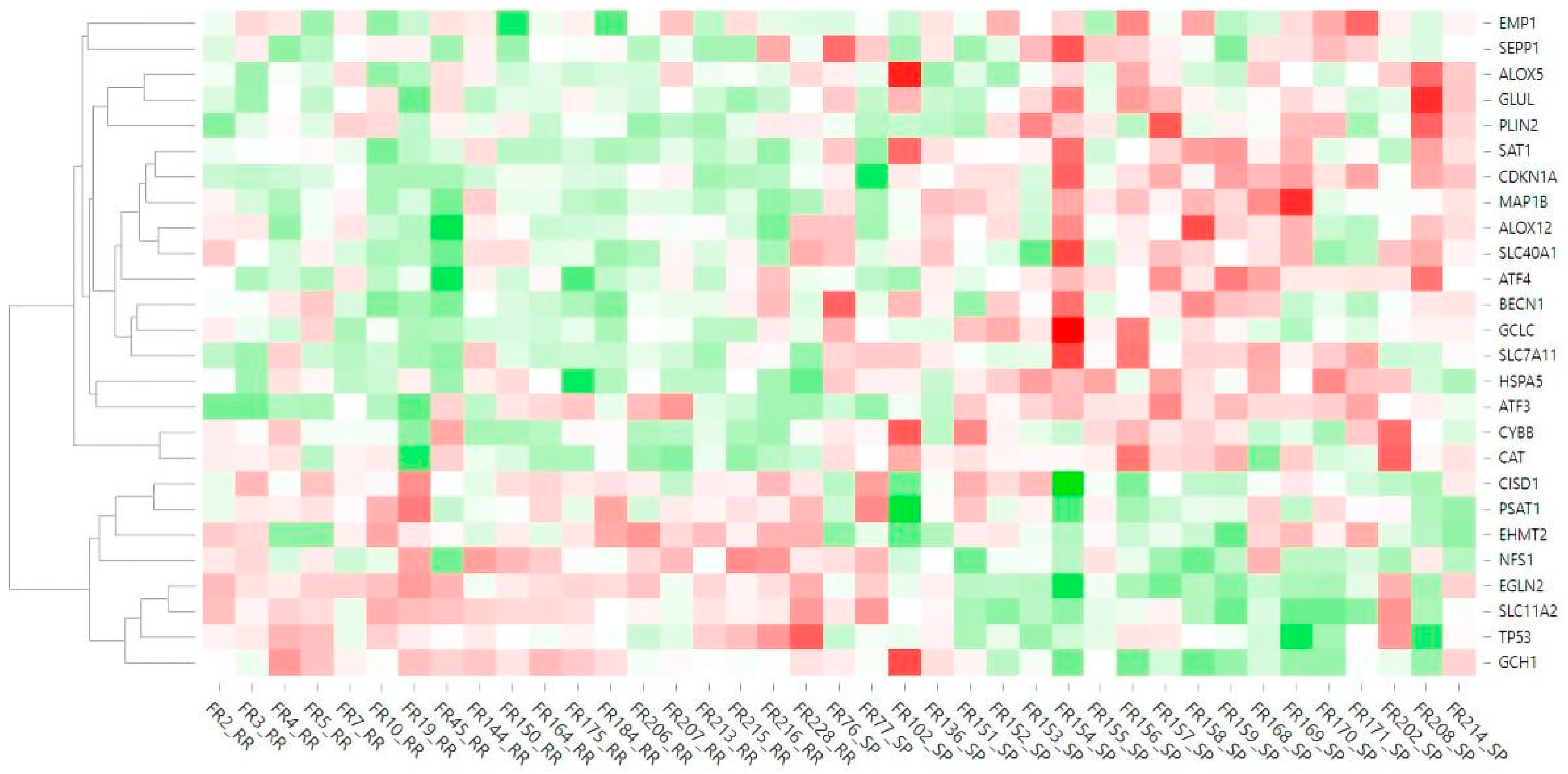
2.2. Differentially Expressed Ferroptosis-Related Genes in PBMCs between SP and RR MS Patients
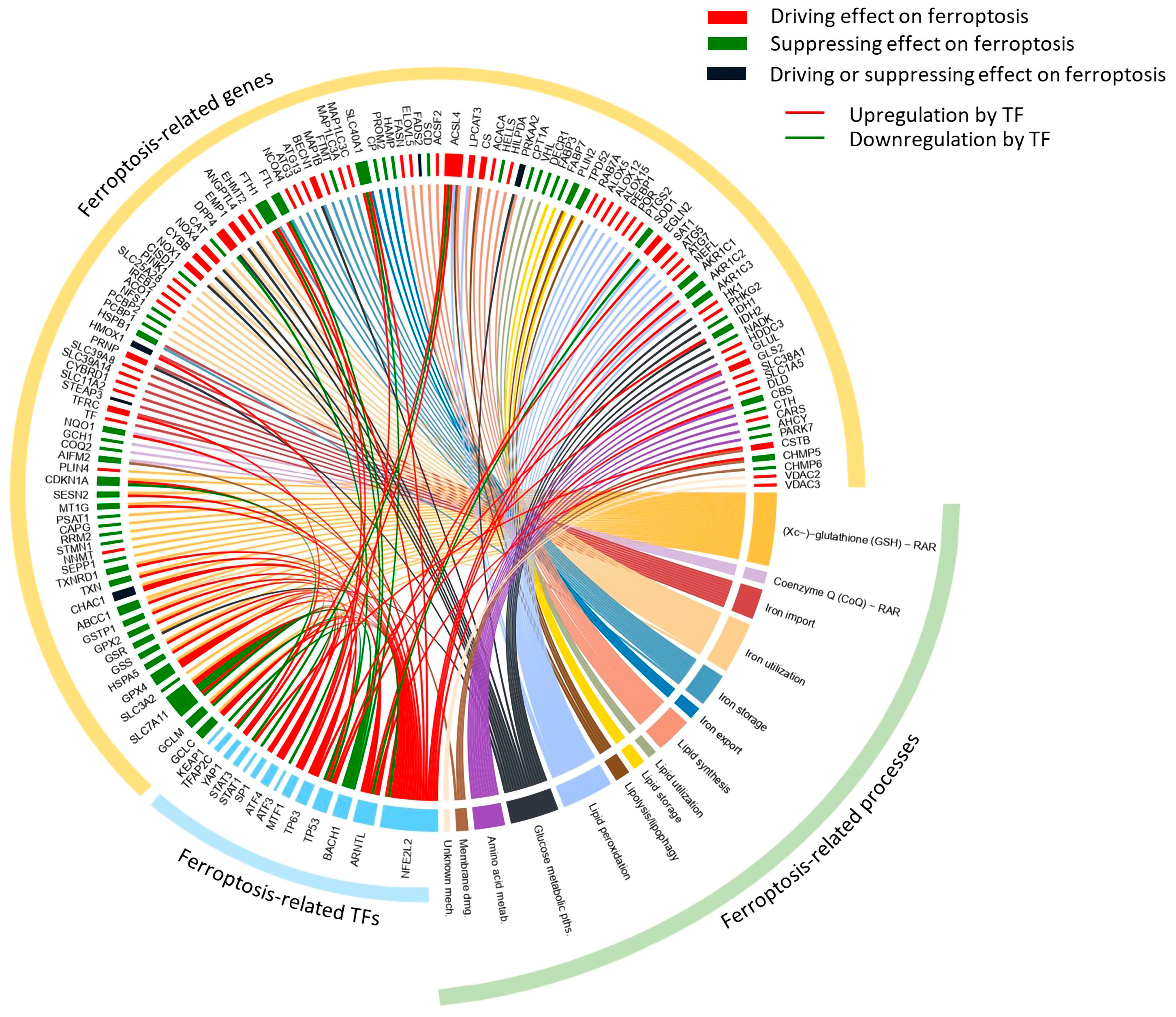
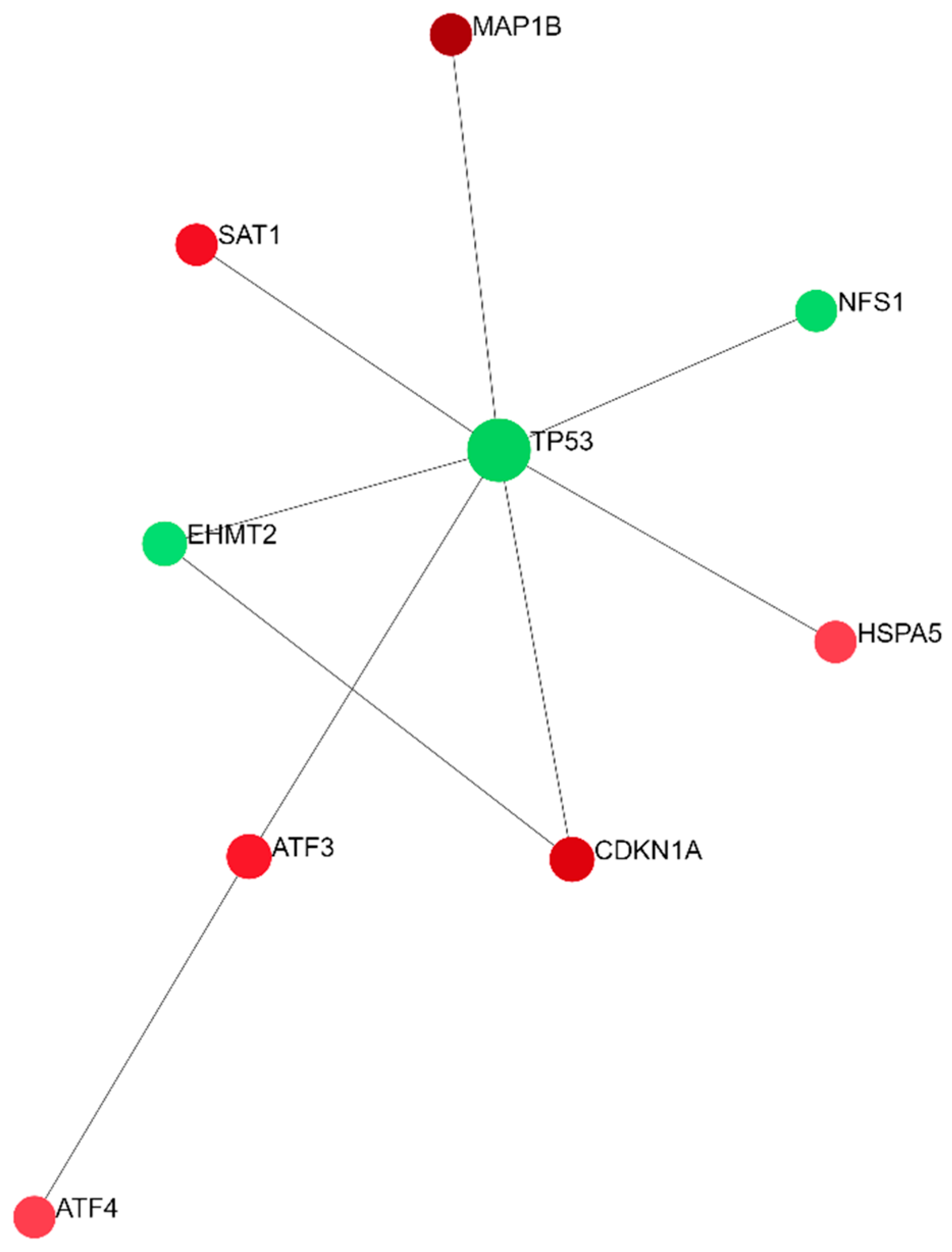
3. Discussion
4. Methods and Materials
4.1. Subjects
4.2. Curated Selection of the Ferroptosis-Related Genes
4.3. Design of the AmpliSeq Illumina Custom RNA Panel for the Profiling of Ferroptosis-Related Genes Using Targeted RNAseq
4.4. Isolation of PBMCs and Extraction of the Total RNA
4.5. Targeted RNAseq Library Synthesis
4.6. Targeted RNA Sequencing and Data Analysis
4.7. Bioinformatic Analysis
4.8. Relative Expression of the Candidate Target Genes by Quantitative Real-Time PCR in a Replication Group of MS Patients
4.9. Quantification of Iron, Transferrin and Ferritin in Serum
4.10. Quantification of GPX4, MDA and 4-HNE in Plasma
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cree, B.A.; Arnold, D.L.; Chataway, J.; Chitnis, T.; Fox, R.J.; Ramajo, A.P.; Murphy, N.; Lassmann, H. Secondary Progressive Multiple Sclerosis: New Insights. Neurology 2021, 97, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X. The Chemistry and Biology of Ferroptosis. Cell Chem. Biol. 2020, 27, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Viktorinova, A. Future Perspectives of Oxytosis/Ferroptosis Research in Neurodegeneration Diseases. Cell Mol. Neurobiol. 2023, 43, 2761–2768. [Google Scholar] [CrossRef]
- Ryan, S.K.; Zelic, M.; Han, Y.; Teeple, E.; Chen, L.; Sadeghi, M.; Shankara, S.; Guo, L.; Li, C.; Pontarelli, F.; et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci. 2023, 26, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Nydes, M.; Shanley, K.L.; Morales Pantoja, I.E.; Howard, T.A.; Bizzozero, O.A. Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurochem. 2019, 148, 426–439. [Google Scholar] [CrossRef]
- Jhelum, P.; Santos-Nogueira, E.; Teo, W.; Haumont, A.; Lenoël, I.; Stys, P.K.; David, S. Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J. Neurosci. 2020, 40, 9327–9341. [Google Scholar] [CrossRef]
- Jhelum, P.; Zandee, S.; Ryan, F.; Zarruk, J.G.; Michalke, B.; Venkataramani, V.; Curran, L.; Klement, W.; Prat, A.; David, S. Ferroptosis induces detrimental effects in chronic EAE and its implications for progressive MS. Acta Neuropathol. Commun. 2023, 11, 121. [Google Scholar] [CrossRef]
- Luoqian, J.; Yang, W.; Ding, X.; Tuo, Q.-Z.; Xiang, Z.; Zheng, Z.; Guo, Y.-J.; Li, L.; Guan, P.; Ayton, S.; et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell Mol. Immunol. 2022, 19, 913–924. [Google Scholar] [CrossRef]
- Van San, E.; Debruyne, A.C.; Veeckmans, G.; Tyurina, Y.Y.; Tyurin, V.A.; Zheng, H.; Choi, S.M.; Augustyns, K.; van Loo, G.; Michalke, B.; et al. Ferroptosis contributes to multiple sclerosis and its pharmacological targeting suppresses experimental disease progression. Cell Death Differ. 2023, 30, 2092–2103. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Gaitán, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.-C.; Yuan, C.-X.; Gu, C. Identification of ferroptosis-related gene signatures associated with multiple sclerosis using weighted gene co-expression network analysis. Medicine 2022, 101, e31802. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, Z.; Tian, Z.; Wu, M.; Zhou, Y.; Zhang, J. Identification of Key Ferroptosis-Related Genes in the Peripheral Blood of Patients with Relapsing-Remitting Multiple Sclerosis and Its Diagnostic Value. Int. J. Mol. Sci. 2023, 24, 6399. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, A.M. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43, Erratum in Nat. Rev. Dis. Primers 2018, 4, 49. [Google Scholar] [CrossRef]
- Clark, M.B.; Mercer, T.R.; Bussotti, G.; Leonardi, T.; Haynes, K.R.; Crawford, J.; Brunck, M.E.; Le Cao, K.-A.; Thomas, G.P.; Chen, W.Y.; et al. Quantitative gene profiling of long noncoding RNAs with targeted RNA sequencing. Nat. Methods 2015, 12, 339–342. [Google Scholar] [CrossRef]
- Whitney, L.W.; Ludwin, S.K.; McFarland, H.F.; E Biddison, W. Microarray analysis of gene expression in multiple sclerosis and EAE identifies 5-lipoxygenase as a component of inflammatory lesions. J. Neuroimmunol. 2001, 121, 40–48. [Google Scholar] [CrossRef]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Dobrian, A.D.; Lieb, D.C.; Cole, B.K.; Taylor-Fishwick, D.A.; Chakrabarti, S.K.; Nadler, J.L. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog. Lipid Res. 2011, 50, 115–131. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Li, J.; Jimenez, D.A.; Levitan, E.S.; Aizenman, E.; Rosenberg, P.A. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J. Neurosci. 2004, 24, 10616–10627. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, J.; Follett, P.L.; Zhang, Y.; Cotanche, D.A.; Jensen, F.E.; Volpe, J.J.; Rosenberg, P.A. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur. J. Neurosci. 2004, 20, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, M.; Huang, Z.-H.; Wang, M.; Sun, W.-Y.; Kurihara, H.; Huang, R.-T.; Wang, R.; Huang, F.; Liang, L.; et al. ALOX5 inhibition protects against dopaminergic neurons undergoing ferroptosis. Pharmacol. Res. 2023, 193, 106779. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Kim, J.Y.; Yenari, M.A.; Lee, J.E. The role of NOX inhibitors in neurodegenerative diseases. IBRO Rep. 2019, 7, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-F.; Wu, S.-P.; Lin, G.-J.; Shieh, C.-C.; Hsu, C.-S.; Chen, J.-W.; Chen, S.-H.; Hong, J.-S.; Chen, S.-J. Microglial Nox2 Plays a Key Role in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2021, 12, 638381. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-H.; Huang, Z.; Wu, J.; Ding, C.-K.C.; Murphy, S.K.; Chi, J.-T. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol. Cancer Res. 2020, 18, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Aksu, N.; Samadi, A.; Yalçınkaya, A.; Çetin, T.; Eser, B.; Lay, I.; Öziş, T.N.; Öztaş, Y.; Sabuncuoğlu, S. Evaluation of oxysterol levels of patients with silicosis by LC-MS/MS method. Mol. Cell Biochem. 2020, 467, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Merckx, E.; Albertini, G.; Paterka, M.; Jensen, C.; Albrecht, P.; Dietrich, M.; Van Liefferinge, J.; Bentea, E.; Verbruggen, L.; Demuyser, T.; et al. Absence of system xc- on immune cells invading the central nervous system alleviates experimental autoimmune encephalitis. J. Neuroinflammation 2017, 14, 9. [Google Scholar] [CrossRef]
- Pampliega, O.; Domercq, M.; Soria, F.N.; Villoslada, P.; Rodríguez-Antigüedad, A.; Matute, C. Increased expression of cystine/glutamate antiporter in multiple sclerosis. J. Neuroinflammation 2011, 8, 63. [Google Scholar] [CrossRef]
- Evonuk, K.S.; Baker, B.J.; Doyle, R.E.; Moseley, C.E.; Sestero, C.M.; Johnston, B.P.; De Sarno, P.; Tang, A.; Gembitsky, I.; Hewett, S.J.; et al. Inhibition of System Xc(-) Transporter Attenuates Autoimmune Inflammatory Demyelination. J. Immunol. 2015, 195, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Ma, P.; Yang, P.; Zhai, S.; He, M.; Zhang, X.; Tu, Q.; Jiao, L.; Ye, L.; Feng, Z.; et al. Alpha lipoic acid ameliorates motor deficits by inhibiting ferroptosis in Parkinson’s disease. Neurosci. Lett. 2023, 810, 137346. [Google Scholar] [CrossRef]
- Fan, J.; Han, Y.; Sun, H.; Sun, S.; Wang, Y.; Guo, R.; Guo, J.; Tian, X.; Wang, J.; Wang, J. Mesenchymal stem cell-derived exosomal microRNA-367-3p alleviates experimental autoimmune encephalomyelitis via inhibition of microglial ferroptosis by targeting EZH2. Biomed. Pharmacother. 2023, 162, 114593. [Google Scholar] [CrossRef]
- Yardim, A.; Gur, C.; Comakli, S.; Ozdemir, S.; Kucukler, S.; Celik, H.; Kandemir, F.M. Investigation of the effects of berberine on bortezomib-induced sciatic nerve and spinal cord damage in rats through pathways involved in oxidative stress and neuro-inflammation. Neurotoxicology 2022, 89, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Matsuba, Y.; Takahashi, M.; Kamano, N.; Watamura, N.; Sasaguri, H.; Takado, Y.; Yoshihara, Y.; Saito, T.; Saido, T.C. Neuronal glutathione loss leads to neurodegeneration involving gasdermin activation. Sci. Rep. 2023, 13, 1109. [Google Scholar] [CrossRef]
- Welker, A.F.; Moreira, D.C.; Hermes-Lima, M. Roles of catalase and glutathione peroxidase in the tolerance of a pulmonate gastropod to anoxia and reoxygenation. J. Comp. Physiol. B 2016, 186, 553–568. [Google Scholar] [CrossRef]
- van Horssen, J.; Schreibelt, G.; Drexhage, J.; Hazes, T.; Dijkstra, C.; van der Valk, P.; de Vries, H. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic. Biol. Med. 2008, 45, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Wu, M.; Tsirka, S.E. Endothelial NOS-deficient mice reveal dual roles for nitric oxide during experimental autoimmune encephalomyelitis. Glia 2009, 57, 1204–1215. [Google Scholar] [CrossRef]
- Luchetti, S.; Fransen, N.L.; van Eden, C.G.; Ramaglia, V.; Mason, M.; Huitinga, I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: A retrospective autopsy cohort analysis. Acta Neuropathol. 2018, 135, 511–528. [Google Scholar] [CrossRef]
- Fernandes, M.G.F.; Mohammadnia, A.; Pernin, F.; Schmitz-Gielsdorf, L.E.; Hodgins, C.; Cui, Q.-L.; Yaqubi, M.; Blain, M.; Hall, J.; Dudley, R.; et al. Mechanisms of metabolic stress induced cell death of human oligodendrocytes: Relevance for progressive multiple sclerosis. Acta Neuropathol. Commun. 2023, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Wang, S.-J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Meira, M.; Sievers, C.; Hoffmann, F.; Rasenack, M.; Kuhle, J.; Derfuss, T.; Kappos, L.; Lindberg, R.L. Unraveling natalizumab effects on deregulated miR-17 expression in CD4+ T cells of patients with relapsing-remitting multiple sclerosis. J. Immunol. Res. 2014, 2014, 897249. [Google Scholar] [CrossRef]
- Ku, C.-C.; Wuputra, K.; Kato, K.; Lin, W.-H.; Pan, J.-B.; Tsai, S.-C.; Kuo, C.-J.; Lee, K.-H.; Lee, Y.-L.; Lin, Y.-C.; et al. Jdp2-deficient granule cell progenitors in the cerebellum are resistant to ROS-mediated apoptosis through xCT/Slc7a11 activation. Sci. Rep. 2020, 10, 4933. [Google Scholar] [CrossRef] [PubMed]
- Kennel, K.B.; Burmeister, J.; Schneider, M.; Taylor, C.T. The PHD1 oxygen sensor in health and disease. J. Physiol. 2018, 596, 3899–3913. [Google Scholar] [CrossRef]
- Li, S.; Fu, J.; Wang, Y.; Hu, C.; Xu, F. LncRNA MIAT enhances cerebral ischaemia/reperfusion injury in rat model via interacting with EGLN2 and reduces its ubiquitin-mediated degradation. J. Cell Mol. Med. 2021, 25, 10140–10151. [Google Scholar] [CrossRef]
- Strocchi, S.; Reggiani, F.; Gobbi, G.; Ciarrocchi, A.; Sancisi, V. The multifaceted role of EGLN family prolyl hydroxylases in cancer: Going beyond HIF regulation. Oncogene 2022, 41, 3665–3679. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.; Chen, X.; Takada, M.; Fan, C.; Zheng, X.; Wen, H.; Liu, Y.; Wang, C.; Pestell, R.G.; et al. EglN2 associates with the NRF1-PGC1α complex and controls mitochondrial function in breast cancer. EMBO J. 2015, 34, 2953–2970. [Google Scholar] [CrossRef]
- Weinberg, W.C.; Denning, M.F. P21Waf1 control of epithelial cell cycle and cell fate. Crit. Rev. Oral. Biol. Med. 2002, 13, 453–464. [Google Scholar] [CrossRef]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P.M. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004, 304, 596. [Google Scholar] [CrossRef] [PubMed]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef]
- Tan, M.; Wang, S.; Song, J.; Jia, J. Combination of p53(ser15) and p21/p21(thr145) in peripheral blood lymphocytes as potential Alzheimer’s disease biomarkers. Neurosci. Lett. 2012, 516, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Kosik, K.S. Accelerated neuronal differentiation induced by p53 suppression. J. Cell Sci. 1996, 109 Pt 6, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Kim, J.-W.; Jeong, M.-H.; An, J.-H.; Jang, S.-M.; Song, K.-H.; Choi, K.-H. Microtubule-associated protein 1B light chain (MAP1B-LC1) negatively regulates the activity of tumor suppressor p53 in neuroblastoma cells. FEBS Lett. 2008, 582, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Weeks, P.R.; Fischer, I. MAP1B expression and microtubule stability in growing and regenerating axons. Microsc. Res. Tech. 2000, 48, 63–74. [Google Scholar] [CrossRef]
- Wu, H.Y.; Dawson, M.R.; Reynolds, R.; Hardy, R.J. Expression of QKI proteins and MAP1B identifies actively myelinating oligodendrocytes in adult rat brain. Mol. Cell Neurosci. 2001, 17, 292–302. [Google Scholar] [CrossRef]
- Bouquet, C.; Soares, S.; von Boxberg, Y.; Ravaille-Veron, M.; Propst, F.; Nothias, F. Microtubule-associated protein 1B controls directionality of growth cone migration and axonal branching in regeneration of adult dorsal root ganglia neurons. J. Neurosci. 2004, 24, 7204–7213. [Google Scholar] [CrossRef]
- Meixner, A.; Haverkamp, S.; Wässle, H.; Führer, S.; Thalhammer, J.; Kropf, N.; Bittner, R.E.; Lassmann, H.; Wiche, G.; Propst, F. MAP1B is required for axon guidance and Is involved in the development of the central and peripheral nervous system. J. Cell Biol. 2000, 151, 1169–1178. [Google Scholar] [CrossRef]
- Allen, E.; Ding, J.; Wang, W.; Pramanik, S.; Chou, J.; Yau, V.; Yang, Y. Gigaxonin-controlled degradation of MAP1B light chain is critical to neuronal survival. Nature 2005, 438, 224–228. [Google Scholar] [CrossRef]
- Uchida, Y. Overexpression of full-length but not N-terminal truncated isoform of microtubule-associated protein (MAP) 1B accelerates apoptosis of cultured cortical neurons. J. Biol. Chem. 2003, 278, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Edan, G.; Filippi, M.; Hartung, H.; Kappos, L.; Lublin, F.D.; Metz, L.M.; McFarland, H.F.; O’Connor, P.W.; et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann. Neurol. 2005, 58, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Classification of demyelinating diseases at the interface between etiology and pathogenesis. Curr. Opin. Neurol. 2001, 14, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathophysiology of inflammation and tissue injury in multiple sclerosis: What are the targets for therapy. J. Neurol. Sci. 2011, 306, 167–169. [Google Scholar] [CrossRef]
- Lorscheider, J.; Buzzard, K.; Jokubaitis, V.; Spelman, T.; Havrdova, E.; Horakova, D.; Trojano, M.; Izquierdo, G.; Girard, M.; Duquette, P.; et al. Defining secondary progressive multiple sclerosis. Brain 2016, 139 Pt 9, 2395–2405. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983, 33, 1444–1452. [Google Scholar] [CrossRef]
- Roxburgh, R.H.; Seaman, S.R.; Masterman, T.; Hensiek, A.E.; Sawcer, S.J.; Vukusic, S.; Achiti, I.; Confavreux, C.; Coustans, M.; Le Page, E.; et al. Multiple Sclerosis Severity Score: Using disability and disease duration to rate disease severity. Neurology 2005, 64, 1144–1151. [Google Scholar] [CrossRef]
- Manouchehrinia, A.; Westerlind, H.; Kingwell, E.; Zhu, F.; Carruthers, R.; Ramanujam, R.; Ban, M.; Glaser, A.; Sawcer, S.; Tremlett, H.; et al. Age Related Multiple Sclerosis Severity Score: Disability ranked by age. Mult. Scler. 2017, 23, 1938–1946. [Google Scholar] [CrossRef]
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.genome.jp/pathway/hsa04216 (accessed on 30 May 2023).
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- FerrDb V2. Regulator & Disease Association. Available online: http://www.zhounan.org/ferrdb/current (accessed on 1 June 2023).
- Rothammer, N.; Woo, M.S.; Bauer, S.; Binkle-Ladisch, L.; Di Liberto, G.; Egervari, K.; Wagner, I.; Haferkamp, U.; Pless, O.; Merkler, D.; et al. G9a dictates neuronal vulnerability to inflammatory stress via transcriptional control of ferroptosis. Sci. Adv. 2022, 8, eabm5500. [Google Scholar] [CrossRef] [PubMed]
- GTEx Portal. Available online: https://www.gtexportal.org/home/gene (accessed on 5 June 2023).
- Gene Cards®: The Human Gene Database. Available online: https://www.genecards.org/ (accessed on 5 June 2023).
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Chen, X.; Li, J.; Comish, P.; Kang, R.; Tang, D. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020, 27, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Yuan, X.; Du, Q.; Zhang, Z.; Shi, X.; Bao, J.; Ning, Y.; Peng, L. FerrDb V2: Update of the manually curated database of ferroptosis regulators and ferroptosis-disease associations. Nucleic Acids Res. 2023, 51, D571–D582. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Galaxy. Available online: https://usegalaxy.eu (accessed on 3 July 2023).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Gill, E.E.; Hancock, R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef]
- NetworkAnalyst—Comprehensive Gene Expression Profiling via Network Visual Analytics. Available online: https://www.networkanalyst.ca (accessed on 15 June 2023).
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.W.; Brinkman, F.S.L.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef] [PubMed]
- String. Available online: https://string-db.org (accessed on 22 June 2023).
- von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- MyAssays Online. Available online: https://www.myassays.com/four-parameter-logistic-curve.assay (accessed on 1 August 2023).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical and Biochemical Parameters | Discovery Group | Replication Group | ||||
|---|---|---|---|---|---|---|
| RR (N = 24) | SP (N = 24) | p | RR (N = 16) | SP (N = 16) | p | |
| Age at sampling, y | 48.0 ± 6.2 | 49.8 ± 9.0 | 0.43 | 42.8 ± 8.2 | 48.5 ± 7.6 | 0.04 |
| Sex, n (m/f) | 16/8 | 7/17 | 0.004 | 8/8 | 8/8 | 1.00 |
| BMI (kg/m2) | 23.6 ± 2.4 | 23.0 ± 3.6 | 0.51 | 25.6 ± 5.0 | 23.9 ± 4.7 | 0.34 |
| Age at onset, y | 31.52 ± 6.26 | 31.16 ± 9.77 | 0.89 | 31.2 ± 8.3 | 34.3 ± 7.2 | 0.27 |
| Disease duration, y | 16.48 ± 4.64 | 18.64 ± 7.99 | 0.33# | 11.5 ± 3.4 | 13.6 ± 7.0 | 0.40# |
| EDSS | 0.98 ± 0.65 | 6.60 ± 0.48 | 0.00# | 0.97 ± 0.67 | 5.56 ± 0.79 | 0.00# |
| MSSS | 0.47 ± 0.30 | 7.16 ± 1.10 | 0.00# | 0.94 ± 0.67 | 6.17 ± 1.56 | 0.00# |
| Total number of relapses | 1.17 ± 1.34 | 7.54 ± 3.80 | 0.00# | 2.14 ± 3.41 | 7.00 ± 4.77 | 0.00# |
| Number of relapses in last 2 y | 0.13± 0.34 | 1.28± 1.06 | 0.0001# | 0.12± 0.33 | 1.31± 0.79 | 0.00# |
| GPX4 (pg/mL) | 2738.39 ± 1872.95 | 2862.81 ± 1005.21 | 0.56# | 1970.32 ± 1191.13 | 1955.91 ± 1387.92 | 0.78# |
| MDA (ng/mL) | 96.55 ± 25.47 | 99.91 ± 64.54 | 0.36# | 108.12 ± 47.28 | 92.88 ± 31.07 | 0.38# |
| 4-HNE (pg/mL) | 1490.10 ± 337.04 | 1171.19 ± 325.46 | 0.003 | 1561.37 ± 279.62 | 1512.06 ± 278.80 | 0.58# |
| Fe (µmol/L) | 16.00 ± 3.75 | 14.60 ± 4.80 | 0.28 | 13.65 ± 5.78 | 14.84 ± 3.39 | 0.47 |
| Transferrin (g/L) | 2.34 ± 0.30 | 2.35 ± 0.39 | 0.93 | 2.61 ± 0.52 | 2.36 ± 0.25 | 0.21# |
| Ferritin (ng/mL) | 95.44 ± 100.98 | 59.3258.98 | 0.11# | 61.29 ± 65.45 | 85.47 ± 96.49 | 0.44# |
| SP vs. RR DEGs | baseMean | log2FoldChange | lfcSE | p Value | padj |
|---|---|---|---|---|---|
| CDKN1A | 9548.4098 | 0.591158406 | 0.114681 | 2.54 × 10−7 | 3.24 × 10−5 |
| EGLN2 | 5756.6618 | −0.338615318 | 0.067246 | 4.77 × 10−7 | 3.24 × 10−5 |
| MAP1B | 75.6888061 | 0.901324663 | 0.183941 | 9.58 × 10−7 | 4.34 × 10−5 |
| SLC7A11 | 126.622192 | 0.606526575 | 0.132314 | 4.56 × 10−6 | 0.000155 |
| SAT1 | 19,655.5985 | 0.4501215 | 0.099888 | 6.60 × 10−6 | 0.000179 |
| SLC11A2 | 1181.28541 | −0.307764602 | 0.082231 | 0.000182 | 0.004127 |
| CAT | 4651.63507 | 0.29174057 | 0.081601 | 0.00035 | 0.006799 |
| GLUL | 3109.18502 | 0.380189011 | 0.11015 | 0.000557 | 0.009475 |
| TP53 | 9275.04884 | −0.23651102 | 0.075289 | 0.001682 | 0.025412 |
| GCLC | 3856.85128 | 0.226106063 | 0.073082 | 0.001976 | 0.02687 |
| ALOX12 | 3420.79701 | 0.542998884 | 0.179849 | 0.002534 | 0.031335 |
| NFS1 | 1626.194 | −0.18163292 | 0.061148 | 0.002974 | 0.033708 |
| EHMT2 | 325.41458 | −0.146782661 | 0.053859 | 0.006424 | 0.067202 |
| CYBB | 22,370.9217 | 0.299323864 | 0.113882 | 0.00858 | 0.083345 |
| SEPP1 | 55.4441827 | 0.391031784 | 0.153583 | 0.010895 | 0.087159 |
| GCH1 | 1363.03231 | −0.285756188 | 0.111031 | 0.010063 | 0.087159 |
| BECN1 | 7986.9329 | 0.110282249 | 0.043312 | 0.01089 | 0.087159 |
| ATF3 | 438.943629 | 0.386548037 | 0.177873 | 0.029768 | 0.202423 |
| CISD1 | 1126.71629 | −0.193534746 | 0.088844 | 0.029378 | 0.202423 |
| PLIN2 | 4488.7674 | 0.216134924 | 0.101398 | 0.033044 | 0.208698 |
| ALOX5 | 7082.81376 | 0.221153845 | 0.104174 | 0.03376 | 0.208698 |
| SLC40A1 | 13,636.9118 | 0.250180235 | 0.119032 | 0.035571 | 0.210334 |
| PSAT1 | 455.492259 | −0.292751394 | 0.142061 | 0.039327 | 0.222852 |
| EMP1 | 168.560458 | 0.275828894 | 0.13874 | 0.046801 | 0.2546 |
| HSPA5 # | 3974.35589 | 0.198734257 | 0.063625 | 0.001787 | 0.010721 |
| ATF4 # | 3565.62181 | 0.199836813 | 0.063727 | 0.001714 | 0.010721 |
| Pathway | Total | Hits | p Value | FDR |
|---|---|---|---|---|
| Ferroptosis | 40 | 7 | 2.23 × 10−9 | 5.10 × 10−7 |
| Prostate cancer | 97 | 9 | 3.21 × 10−9 | 5.10 × 10−7 |
| Mitophagy—animal | 65 | 7 | 7.49 × 10−8 | 7.94 × 10−6 |
| Hepatitis B | 163 | 9 | 3.03 × 10−7 | 2.41 × 10−5 |
| IL-17 signaling pathway | 93 | 7 | 9.03 × 10−7 | 5.74 × 10−5 |
| Endocrine resistance | 98 | 7 | 1.29 × 10−6 | 6.84 × 10−5 |
| Pathways in cancer | 530 | 13 | 6.02 × 10−6 | 0.000273 |
| Kaposi’s sarcoma-associated herpesvirus infection | 186 | 8 | 9.66 × 10−6 | 0.000384 |
| Viral carcinogenesis | 201 | 8 | 1.70 × 10−5 | 0.000602 |
| Choline metabolism in cancer | 99 | 6 | 2.05 × 10−5 | 0.000652 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stojkovic, L.; Jovanovic, I.; Dincic, E.; Djordjevic, A.; Kuveljic, J.; Djuric, T.; Stankovic, A.; Vojinovic, S.; Zivkovic, M. Targeted RNAseq Revealed the Gene Expression Signature of Ferroptosis-Related Processes Associated with Disease Severity in Patients with Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 3016. https://doi.org/10.3390/ijms25053016
Stojkovic L, Jovanovic I, Dincic E, Djordjevic A, Kuveljic J, Djuric T, Stankovic A, Vojinovic S, Zivkovic M. Targeted RNAseq Revealed the Gene Expression Signature of Ferroptosis-Related Processes Associated with Disease Severity in Patients with Multiple Sclerosis. International Journal of Molecular Sciences. 2024; 25(5):3016. https://doi.org/10.3390/ijms25053016
Chicago/Turabian StyleStojkovic, Ljiljana, Ivan Jovanovic, Evica Dincic, Ana Djordjevic, Jovana Kuveljic, Tamara Djuric, Aleksandra Stankovic, Slobodan Vojinovic, and Maja Zivkovic. 2024. "Targeted RNAseq Revealed the Gene Expression Signature of Ferroptosis-Related Processes Associated with Disease Severity in Patients with Multiple Sclerosis" International Journal of Molecular Sciences 25, no. 5: 3016. https://doi.org/10.3390/ijms25053016
APA StyleStojkovic, L., Jovanovic, I., Dincic, E., Djordjevic, A., Kuveljic, J., Djuric, T., Stankovic, A., Vojinovic, S., & Zivkovic, M. (2024). Targeted RNAseq Revealed the Gene Expression Signature of Ferroptosis-Related Processes Associated with Disease Severity in Patients with Multiple Sclerosis. International Journal of Molecular Sciences, 25(5), 3016. https://doi.org/10.3390/ijms25053016