
Involvement of Ferroptosis Induction and Oxidative Phosphorylation Inhibition in the Anticancer-Drug-Induced Myocardial Injury: Ameliorative Role of Pterostilbene

Abstract
1. Introduction
2. Results
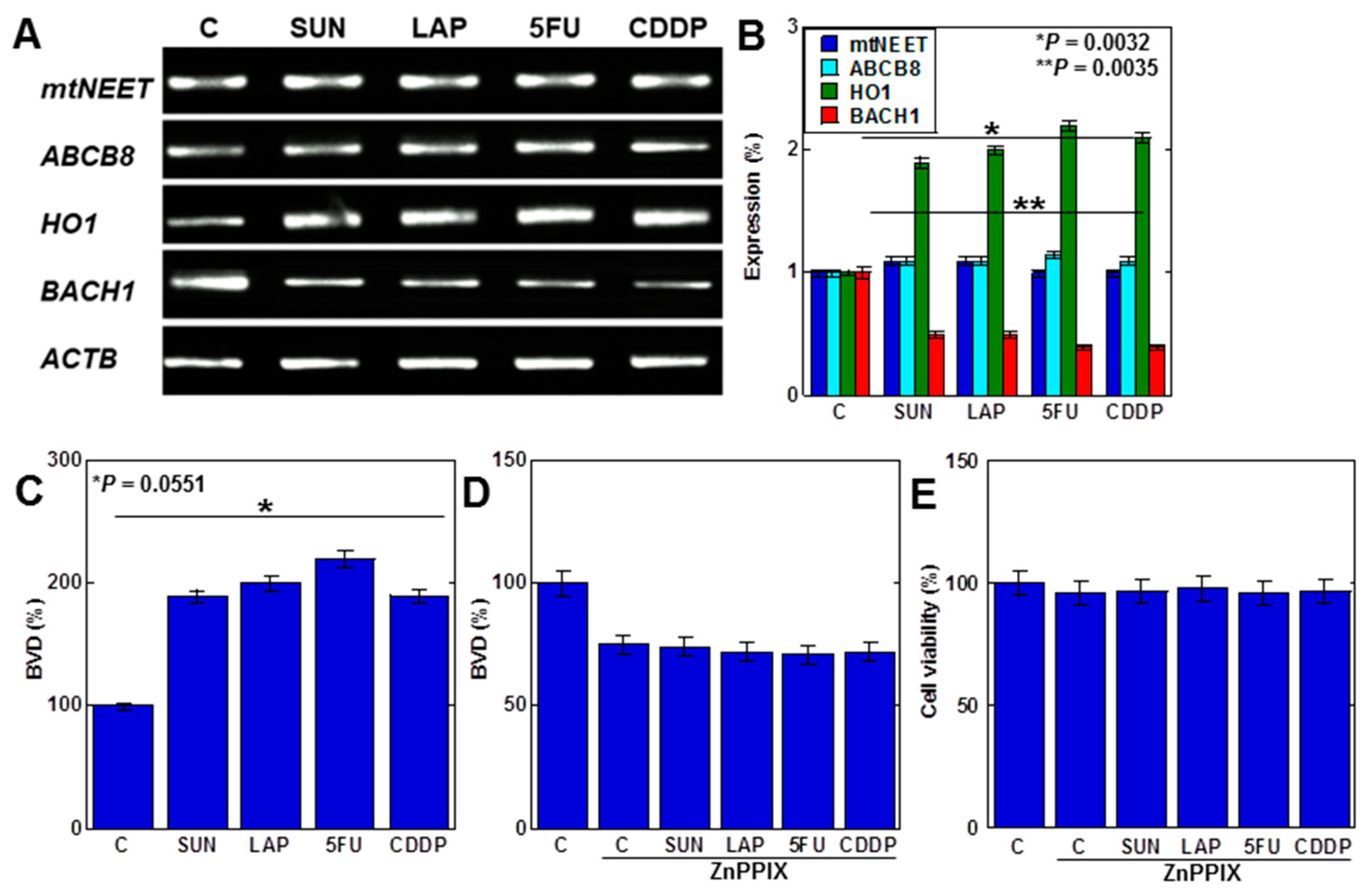
2.1. Effects of Anticancer Drugs on H9c2 Cells
2.2. Redox Alteration in H9c2 Cells Induced by Anticancer Drugs
2.3. Induction of Cell Death by Anticancer Drugs in H9c2 Cells
2.4. MtFe Deposition by Anticancer Drugs in H9c2 Cells
2.5. Anticancer Drugs Inhibit Mitochondrial DNA (mtDNA) Damage and Oxidative Phosphorylation (OXPHOS)
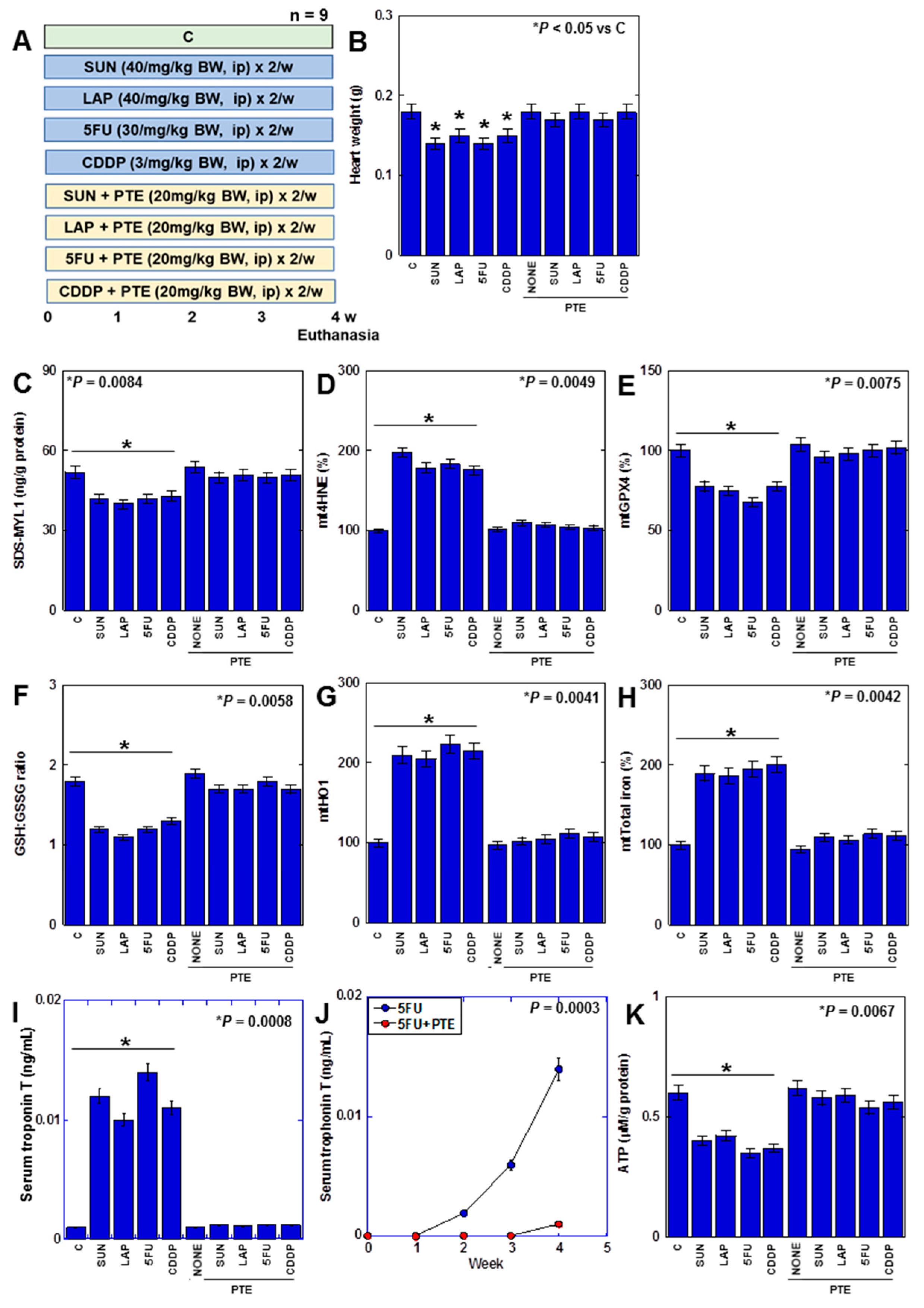
2.6. Effects of PTE on Cell Protection in H9c2 Cells
2.7. Effects of PTE on Anticancer-Drug-Induced Myocardial Damage in Rats
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Mitochondrial Imaging
4.3. Protein Extraction
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Cell Death Inhibitor Assay
4.6. RNA Isolation
4.7. Reverse Transcription–Polymerase Chain Reaction (RT-PCR)
4.8. Quantification of BVD
4.9. Mitochondrial Stress Test (Seahorse Assay)
4.10. Glycolytic Stress Test
4.11. Animals
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Paillaud, E.; Caillet, P.; Campillo, B.; Bories, P.N. Increased risk of alteration of nutritional status in hospitalized elderly patients with advanced cancer. J. Nutr. Health Aging 2006, 10, 91–95. [Google Scholar]
- Ausoni, S.; Calamelli, S.; Saccà, S.; Azzarello, G. How progressive cancer endangers the heart: An intriguing and underestimated problem. Cancer Metastasis Rev. 2020, 39, 535–552. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Arteaga, C.L.; Force, T.; Humphreys, B.D.; Demetri, G.D.; Druker, B.J.; Moslehi, J.J. Cardio-Oncology: How New Targeted Cancer Therapies and Precision Medicine Can Inform Cardiovascular Discovery. Circulation 2015, 132, 2248–2258. [Google Scholar] [CrossRef]
- Asnani, A.; Moslehi, J.J.; Adhikari, B.B.; Baik, A.H.; Beyer, A.M.; de Boer, R.A.; Ghigo, A.; Grumbach, I.M.; Jain, S.; Zhu, H. Preclinical Models of Cancer Therapy-Associated Cardiovascular Toxicity: A Scientific Statement From the American Heart Association. Circ. Res. 2021, 129, e21–e34. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Ewer, M.S. Cardiac and cardiovascular toxicity of nonanthracycline anticancer drugs. Expert. Rev. Anticancer Ther. 2006, 6, 1249–1269. [Google Scholar] [CrossRef] [PubMed]
- Kitakata, H.; Endo, J.; Ikura, H.; Moriyama, H.; Shirakawa, K.; Katsumata, Y.; Sano, M. Therapeutic Targets for DOX-Induced Cardiomyopathy: Role of Apoptosis vs. Ferroptosis. Int. J. Mol. Sci. 2022, 23, 1414. [Google Scholar] [CrossRef]
- Hasegawa, K. Clinical Practice Guideline. Available online: http://www.jsco-cpg.jp/ (accessed on 23 February 2024).
- Polk, A.; Vistisen, K.; Vaage-Nilsen, M.; Nielsen, D.L. A systematic review of the pathophysiology of 5-fluorouracil-induced cardiotoxicity. BMC Pharmacol. Toxicol. 2014, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Demkow, U.; Stelmaszczyk-Emmel, A. Cardiotoxicity of cisplatin-based chemotherapy in advanced non-small cell lung cancer patients. Respir. Physiol. Neurobiol. 2013, 187, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Doherty, K.R.; Wappel, R.L.; Talbert, D.R.; Trusk, P.B.; Moran, D.M.; Kramer, J.W.; Brown, A.M.; Shell, S.A.; Bacus, S. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol. Appl. Pharmacol. 2013, 272, 245–255. [Google Scholar] [CrossRef]
- Roca-Alonso, L.; Pellegrino, L.; Castellano, L.; Stebbing, J. Breast cancer treatment and adverse cardiac events: What are the molecular mechanisms? Cardiology 2012, 122, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Coradduzza, D.; Congiargiu, A.; Chen, Z.; Zinellu, A.; Carru, C.; Medici, S. Ferroptosis and Senescence: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 3658. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Daniel, M.; Tollefsbol, T.O. Pterostilbene down-regulates hTERT at physiological concentrations in breast cancer cells: Potentially through the inhibition of cMyc. J. Cell Biochem. 2018, 119, 3326–3337. [Google Scholar] [CrossRef]
- Kosuru, R.; Rai, U.; Prakash, S.; Singh, A.; Singh, S. Promising therapeutic potential of pterostilbene and its mechanistic insight based on preclinical evidence. Eur. J. Pharmacol. 2016, 789, 229–243. [Google Scholar] [CrossRef]
- Kang, L.L.; Zhang, D.M.; Jiao, R.Q.; Pan, S.M.; Zhao, X.J.; Zheng, Y.J.; Chen, T.Y.; Kong, L.D. Pterostilbene Attenuates Fructose-Induced Myocardial Fibrosis by Inhibiting ROS-Driven Pitx2c/miR-15b Pathway. Oxid. Med. Cell Longev. 2019, 2019, 1243215. [Google Scholar] [CrossRef]
- Lv, M.; Liu, K.; Fu, S.; Li, Z.; Yu, X. Pterostilbene attenuates the inflammatory reaction induced by ischemia/reperfusion in rat heart. Mol. Med. Rep. 2015, 11, 724–728. [Google Scholar] [CrossRef]
- Lee, D.I.; Acosta, C.; Anderson, C.M.; Anderson, H.D. Peripheral and Cerebral Resistance Arteries in the Spontaneously Hypertensive Heart Failure Rat: Effects of Stilbenoid Polyphenols. Molecules 2017, 22, 380. [Google Scholar] [CrossRef]
- Akinwumi, B.C.; Raj, P.; Lee, D.I.; Acosta, C.; Yu, L.; Thomas, S.M.; Nagabhushanam, K.; Majeed, M.; Davies, N.M.; Netticadan, T.; et al. Disparate Effects of Stilbenoid Polyphenols on Hypertrophic Cardiomyocytes In Vitro vs. in the Spontaneously Hypertensive Heart Failure Rat. Molecules 2017, 22, 204. [Google Scholar] [CrossRef] [PubMed]
- Couto, G.K.; Fernandes, R.O.; Lacerda, D.; Campos-Carraro, C.; Turck, P.; Bianchi, S.E.; Ferreira, G.D.; Brum, I.S.; Bassani, V.L.; Bello-Klein, A.; et al. Profile of pterostilbene-induced redox homeostasis modulation in cardiac myoblasts and heart tissue. J. Biosci. 2018, 43, 931–940. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, S.; Zhang, X.; Li, Y.; Zhao, Q.; Liu, T. Pterostilbene protects against myocardial ischemia/reperfusion injury via suppressing oxidative/nitrative stress and inflammatory response. Int. Immunopharmacol. 2017, 43, 7–15. [Google Scholar] [CrossRef]
- Lacerda, D.; Ortiz, V.; Türck, P.; Campos-Carraro, C.; Zimmer, A.; Teixeira, R.; Bianchi, S.; de Castro, A.L.; Schenkel, P.C.; Belló-Klein, A.; et al. Stilbenoid pterostilbene complexed with cyclodextrin preserves left ventricular function after myocardial infarction in rats: Possible involvement of thiol proteins and modulation of phosphorylated GSK-3β. Free Radic. Res. 2018, 52, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, Z.; Fang, M.; Zou, T.; Zhang, Z.; Meng, X.; Wang, T.; Meng, H.; Chen, Y.; Duan, Y.; et al. Novel pterostilbene derivatives ameliorate heart failure by reducing oxidative stress and inflammation through regulating Nrf2/NF-κB signaling pathway. Eur. J. Med. Chem. 2023, 258, 115602. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ma, Z.; Xu, L.; Zhang, X.; Qiao, S.; Yuan, J. PGC1α activation by pterostilbene ameliorates acute doxorubicin cardiotoxicity by reducing oxidative stress via enhancing AMPK and SIRT1 cascades. Aging 2019, 11, 10061–10073. [Google Scholar] [CrossRef]
- Nishiguchi, Y.; Fujiwara-Tani, R.; Nukaga, S.; Nishida, R.; Ikemoto, A.; Sasaki, R.; Mori, S.; Ogata, R.; Kishi, S.; Hojo, Y.; et al. Pterostilbene induces apoptosis from endoplasmic reticulum stress synergistically with anticancer drugs that deposit iron in mitochondria. Int. J. Mol. Sci. 2024; in print. [Google Scholar]
- Hojo, Y.; Kishi, S.; Mori, S.; Fujiwara-Tani, R.; Sasaki, T.; Fujii, K.; Nishiguchi, Y.; Nakashima, C.; Luo, Y.; Shinohara, H.; et al. Sunitinib and Pterostilbene Combination Treatment Exerts Antitumor Effects in Gastric Cancer via Suppression of PDZD8. Int. J. Mol. Sci. 2022, 23, 4002. [Google Scholar] [CrossRef]
- Vela, D. Keeping heart homeostasis in check through the balance of iron metabolism. Acta Physiol. 2020, 228, e13324. [Google Scholar] [CrossRef]
- Hewer, R.L. Study of fatal cases of Friedreich’s ataxia. Br. Med. J. 1968, 3, 649–652. [Google Scholar] [CrossRef]
- Tsou, A.Y.; Paulsen, E.K.; Lagedrost, S.J.; Perlman, S.L.; Mathews, K.D.; Wilmot, G.R.; Ravina, B.; Koeppen, A.H.; Lynch, D.R. Mortality in Friedreich ataxia. J. Neurol. Sci. 2011, 307, 46–49. [Google Scholar] [CrossRef]
- Zhao, W.K.; Zhou, Y.; Xu, T.T.; Wu, Q. Ferroptosis: Opportunities and Challenges in Myocardial Ischemia-Reperfusion Injury. Oxid. Med. Cell Longev. 2021, 2021, 9929687. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, W.; Zhang, C.; Yin, Y.; Mu, N.; Wang, Y.; Yu, L.; Ma, H. Frataxin inhibits the sensitivity of the myocardium to ferroptosis by regulating iron homeostasis. Free Radic. Biol. Med. 2023, 205, 305–317. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Hassannia, B.; Van Coillie, S.; Vanden Berghe, T. Ferroptosis: Biological Rust of Lipid Membranes. Antioxid. Redox Signal. 2021, 35, 487–509. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Gianazza, E.; Brioschi, M.; Martinez Fernandez, A.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid Peroxidation in Atherosclerotic Cardiovascular Diseases. Antioxid. Redox Signal. 2021, 34, 49–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, X.; Zeng, Y.; Mo, X.; Hong, S.; He, H.; Li, J.; Fatima, S.; Liu, Q. Oxidative stress induces mitochondrial iron overload and ferroptotic cell death. Sci. Rep. 2023, 13, 15515. [Google Scholar] [CrossRef] [PubMed]
- Horii, S.; Mori, S.; Ogata, R.; Nukaga, S.; Nishida, R.; Kishi, S.; Sasaki, R.; Ikemoto, A.; Owari, T.; Maesaka, F.; et al. 5-Aminolevrinic Acid Exhibits Dual Effects on Stemness in Human Sarcoma Cell Lines under Dark Conditions. Int. J. Mol. Sci. 2023, 24, 6189. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Santhosh Kumar, T.R.; Kartha, C.C. Mitochondrial membrane transporters and metabolic switch in heart failure. Heart Fail. Rev. 2019, 24, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Renal. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef]
- Adamcova, M.; Skarkova, V.; Seifertova, J.; Rudolf, E. Cardiac Troponins are Among Targets of Doxorubicin-Induced Cardiotoxicity in hiPCS-CMs. Int. J. Mol. Sci. 2019, 20, 2638. [Google Scholar] [CrossRef]
- Herman, E.H.; Lipshultz, S.E.; Rifai, N.; Zhang, J.; Papoian, T.; Yu, Z.X.; Takeda, K.; Ferrans, V.J. Use of cardiac troponin T levels as an indicator of doxorubicin-induced cardiotoxicity. Cancer Res. 1998, 58, 195–197. [Google Scholar]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Bernuzzi, F.; Recalcati, S.; Alberghini, A.; Cairo, G. Reactive oxygen species-independent apoptosis in doxorubicin-treated H9c2 cardiomyocytes: Role for heme oxygenase-1 down-modulation. Chem. Biol. Interact 2009, 177, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Pan, C.; Guo, H.; Chang, J.; Gao, X.; Wu, X.; Zhi, X.; Ren, C.; Chen, Q.; Jiang, H.; et al. Early diagnostic value of high-sensitivity cardiac troponin T for cancer treatment-related cardiac dysfunction: A meta-analysis. ESC Heart Fail. 2023, 10, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Reply to ‘Metabolic remodelling in heart failure revisited’. Nat. Rev. Cardiol. 2018, 15, 780–781. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, S.K.; Dhalla, N.S. Status of Mitochondrial Oxidative Phosphorylation during the Development of Heart Failure. Antioxidants 2023, 12, 1941. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Quan, Y.; Xin, Y.; Tian, G.; Zhou, J.; Liu, X. Mitochondrial ROS-Modulated mtDNA: A Potential Target for Cardiac Aging. Oxid. Med. Cell Longev. 2020, 2020, 9423593. [Google Scholar] [CrossRef]
- Armoundas, A.A.; Hobai, I.A.; Tomaselli, G.F.; Winslow, R.L.; O’Rourke, B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ. Res. 2003, 93, 46–53. [Google Scholar] [CrossRef]
- Takagi, T.; Fujiwara-Tani, R.; Mori, S.; Kishi, S.; Nishiguchi, Y.; Sasaki, T.; Ogata, R.; Ikemoto, A.; Sasaki, R.; Ohmori, H.; et al. Lauric Acid Overcomes Hypoxia-Induced Gemcitabine Chemoresistance in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 7506. [Google Scholar] [CrossRef]
- Fujiwara-Tani, R.; Sasaki, T.; Takagi, T.; Mori, S.; Kishi, S.; Nishiguchi, Y.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Gemcitabine Resistance in Pancreatic Ductal Carcinoma Cell Lines Stems from Reprogramming of Energy Metabolism. Int. J. Mol. Sci. 2022, 23, 7824. [Google Scholar] [CrossRef]
- Xu, W.; Tang, J.; Zhao, L. DNA-protein cross-links between abasic DNA damage and mitochondrial transcription factor A (TFAM). Nucleic Acids Res. 2023, 51, 41–53. [Google Scholar] [CrossRef]
- Song, Y.; Wang, W.; Wang, B.; Shi, Q. The Protective Mechanism of TFAM on Mitochondrial DNA and its Role in Neurodegenerative Diseases. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef]
- Branda, R.F.; Brooks, E.M.; Chen, Z.; Naud, S.J.; Nicklas, J.A. Dietary modulation of mitochondrial DNA deletions and copy number after chemotherapy in rats. Mutat. Res. 2002, 501, 29–36. [Google Scholar] [CrossRef]
- Ashley, N.; Poulton, J. Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs. Biochem. Biophys. Res. Commun. 2009, 378, 450–455. [Google Scholar] [CrossRef]
- Forgie, B.N.; Prakash, R.; Telleria, C.M. Revisiting the Anti-Cancer Toxicity of Clinically Approved Platinating Derivatives. Int. J. Mol. Sci. 2022, 23, 15410. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.K.; Gao, J.; Chen, Z.; Shi, H.; Yuan, J.; Cui, H.L.; Yeh, C.N.; Bränström, R.; Larsson, C.; Li, S.; et al. Heterogeneity of Metabolic Vulnerability in Imatinib -Resistant Gastrointestinal Stromal Tumor. Cells 2020, 9, 1333. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, J. Mitochondrial Dynamics in Rat Heart Induced by 5-Fluorouracil. Med. Sci. Monit. 2018, 24, 6666–6672. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Zhou, X.; Lei, X.; Li, X.; Wei, L. Dynamic observation of 5-fluorouracil-induced myocardial injury and mitochondrial autophagy in aging rats. Exp. Ther. Med. 2021, 22, 1451. [Google Scholar] [CrossRef]
- Tasca, S.; Campos, C.; Lacerda, D.; Ortiz, V.D.; Turck, P.; Bianchi, S.E.; Castro, A.L.; Belló-Klein, A.; Bassani, V.; Araújo, A. Pterostilbene Reduces Experimental Myocardial Infarction-Induced Oxidative Stress in Lung and Right Ventricle. Arq. Bras. Cardiol. 2022, 118, 435–445. [Google Scholar] [CrossRef]
- Xu, C.; Song, Y.; Wang, Z.; Jiang, J.; Piao, Y.; Li, L.; Jin, S.; Li, L.; Zhu, L.; Yan, G. Pterostilbene suppresses oxidative stress and allergic airway inflammation through AMPK/Sirt1 and Nrf2/HO-1 pathways. Immun. Inflamm. Dis. 2021, 9, 1406–1417. [Google Scholar] [CrossRef]
- Ganesh, G.V.; Ramkumar, K.M. Pterostilbene accelerates wound healing response in diabetic mice through Nrf2 regulation. Mol. Immunol. 2023, 164, 17–27. [Google Scholar] [CrossRef]
- Nagarajan, S.; Mohandas, S.; Ganesan, K.; Xu, B.; Ramkumar, K.M. New Insights into Dietary Pterostilbene: Sources, Metabolism, and Health Promotion Effects. Molecules 2022, 27, 6316. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Oue, N.; Wakikawa, A.; Shigeishi, H.; Matsutani, N.; Kuraoka, K.; Ito, R.; Yokozaki, H.; Yasui, W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J. Pathol. 2002, 196, 163–170. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sakagishi, Y. Diazo coupling reaction for the determination of biliverdin. Bunseki Kagaku 1991, 40, 377–381. [Google Scholar] [CrossRef][Green Version]
- Karbownik, A.; Szałek, E.; Sobańska, K.; Grabowski, T.; Klupczynska, A.; Plewa, S.; Wolc, A.; Magiera, M.; Porażka, J.; Kokot, Z.J.; et al. The concomitant use of lapatinib and paracetamol—The risk of interaction. Investig. New Drugs 2018, 36, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, J.L.; Berger, M.R.; Frank, N. Locoregional application of 5-fluorouracil to the liver: Alteration of pharmacokinetics and bone marrow toxicity in rats with thioacetamide-induced cirrhosis. Toxicol. Lett. 1987, 36, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Prihatno, S.A.; Padeta, I.; Larasati, A.D.; Sundari, B.; Hidayati, A.; Fibrianto, Y.H.; Budipitojo, T. Effects of secretome on cisplatin-induced testicular dysfunction in rats. Vet. World 2018, 11, 1349–1356. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Set | |||||
|---|---|---|---|---|---|
| Gene symbol | Gene bank ID | Forward primer (5′–3′) | Reverse primer (5′–3′) | ||
| HO1 | NM_002133.2 | taagctggtgatggcttcct | atgatttcctgccagtgagg | ||
| FCH | KR712044.1 | gatgaattgtcccccaacac | gcttccgtcccacttgatta | ||
| mitoNEET | BC007043.1 | tccagaaagacaaccccaag | gcccacattgtctccagtct | ||
| ABCB8 | NM_001282291.2 | cgtggggtctcgctttaact | cctgacactggcgagacaat | ||
| ACTB | NM_001101.3 | ggacttcgagcaagagatgg | agcactgtgttggcgtacag | ||
| ELISA | |||||
| Protein | Catalog number | Range | Company | ||
| 4HNE | ab238538 | 1.2–200 μg/mL | Abcam, Cambridge, MA, USA | ||
| GPX4 | ARP-E4145 | 78.125–5000 pg/mL | Biocompare. South San Francisco, CA, USA | ||
| Troponin T | CSB-E16443r | 12.5–800 pg/mL | Cusabio Biotech Co., Ltd., Houston, TX, USA | ||
| HO1 | ab279414 | 15.63–1000 pg/mL | Abcam, Cambridge, MA, USA | ||
| GSH/GSSG | ab138881 | 10 nM | Abcam, Cambridge, MA, USA | ||
| Myogenin | LS-F16037 | 0.625–40 ng/mL | LS Bio, Shirley, MA, USA | ||
| MYL1 | MBS261960 | 0.312–20 ng/mL | MyBioSource, San Diego, CA, USA | ||
| TFAM | OKCA01941 | 20–1800 pg/mL | Aviva System Biology, San Diego, CA, USA | ||
| ETC Complex I | KLR2154 | 0.1–20 ng/mL | Krishgen Biosystems, Cerritos, CA, USA | ||
| ETC Complex III | MBS2606461 | 0.1–20 ng/mL | Biocompare. South San Francisco, CA, USA | ||
| ATP | LS-F24998 | 12.35–1000 ng/mL | LS Bio, Shirley, MA, USA | ||
| Iron | MBS3801560 | 0.1 μmol/L | MyBioSource, San Diego, CA, USA | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujii, K.; Fujiwara-Tani, R.; Nukaga, S.; Ohmori, H.; Luo, Y.; Nishida, R.; Sasaki, T.; Miyagawa, Y.; Nakashima, C.; Kawahara, I.; et al. Involvement of Ferroptosis Induction and Oxidative Phosphorylation Inhibition in the Anticancer-Drug-Induced Myocardial Injury: Ameliorative Role of Pterostilbene. Int. J. Mol. Sci. 2024, 25, 3015. https://doi.org/10.3390/ijms25053015
Fujii K, Fujiwara-Tani R, Nukaga S, Ohmori H, Luo Y, Nishida R, Sasaki T, Miyagawa Y, Nakashima C, Kawahara I, et al. Involvement of Ferroptosis Induction and Oxidative Phosphorylation Inhibition in the Anticancer-Drug-Induced Myocardial Injury: Ameliorative Role of Pterostilbene. International Journal of Molecular Sciences. 2024; 25(5):3015. https://doi.org/10.3390/ijms25053015
Chicago/Turabian StyleFujii, Kiyomu, Rina Fujiwara-Tani, Shota Nukaga, Hitoshi Ohmori, Yi Luo, Ryoichi Nishida, Takamitsu Sasaki, Yoshihiro Miyagawa, Chie Nakashima, Isao Kawahara, and et al. 2024. "Involvement of Ferroptosis Induction and Oxidative Phosphorylation Inhibition in the Anticancer-Drug-Induced Myocardial Injury: Ameliorative Role of Pterostilbene" International Journal of Molecular Sciences 25, no. 5: 3015. https://doi.org/10.3390/ijms25053015
APA StyleFujii, K., Fujiwara-Tani, R., Nukaga, S., Ohmori, H., Luo, Y., Nishida, R., Sasaki, T., Miyagawa, Y., Nakashima, C., Kawahara, I., Ogata, R., Ikemoto, A., Sasaki, R., & Kuniyasu, H. (2024). Involvement of Ferroptosis Induction and Oxidative Phosphorylation Inhibition in the Anticancer-Drug-Induced Myocardial Injury: Ameliorative Role of Pterostilbene. International Journal of Molecular Sciences, 25(5), 3015. https://doi.org/10.3390/ijms25053015