
Oxidative Stress and Cerebral Vascular Tone: The Role of Reactive Oxygen and Nitrogen Species

 ,
, 
 ,
,  and
and 
Abstract
1. Introduction
2. Experimental Approach and Methodology
3. Cerebral Vascular Tone
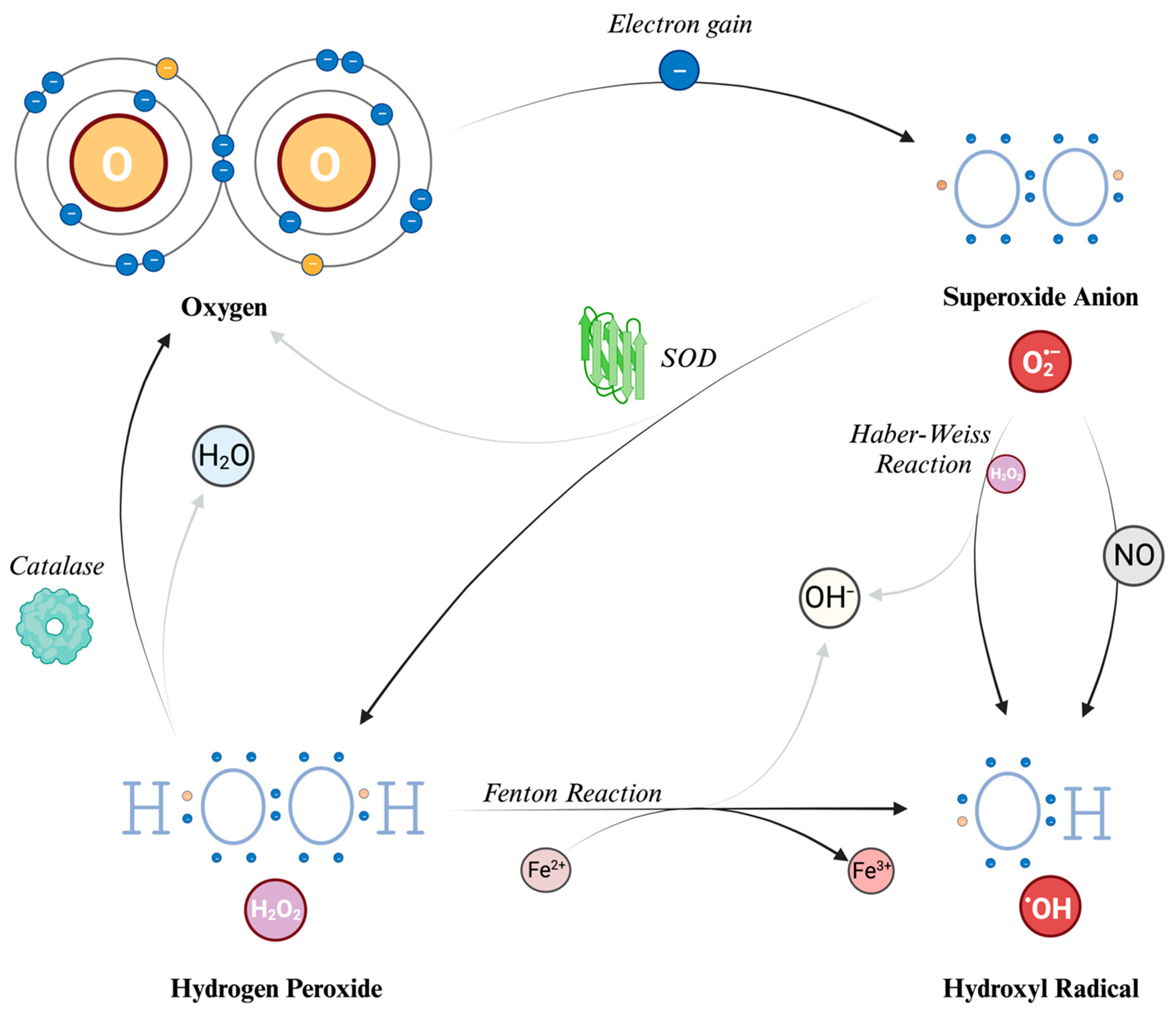
4. Reactive Oxygen Species
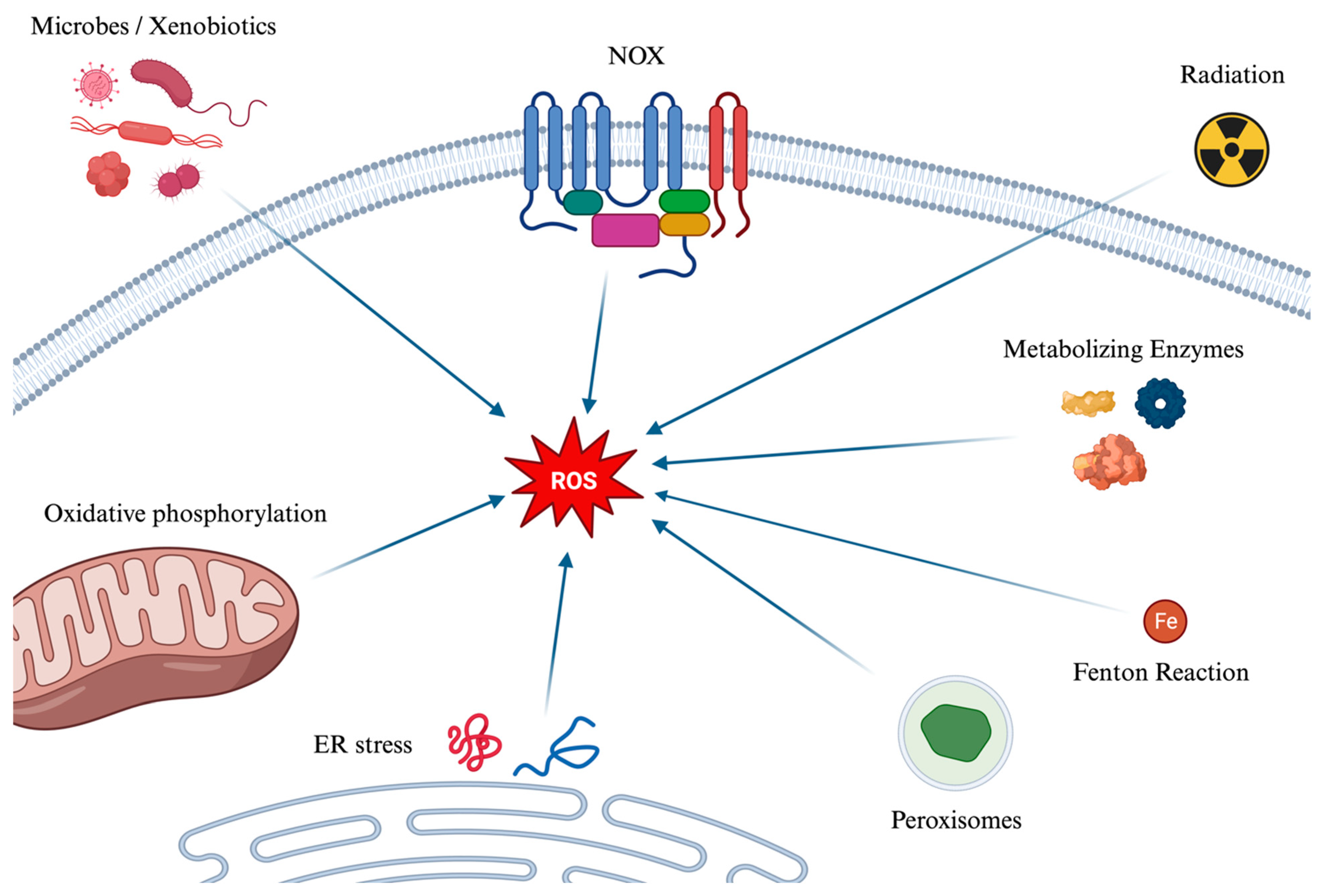
4.1. Generation Pathways of Reactive Oxygen Species
4.2. The Effects of ROS on Cerebral Vascular Tone
4.3. NADPH Oxidases
4.4. ROS as Signaling Molecules in HIF-1α Regulation
4.5. Regional Variability in Oxidative Stress Responses within Brain Vasculature
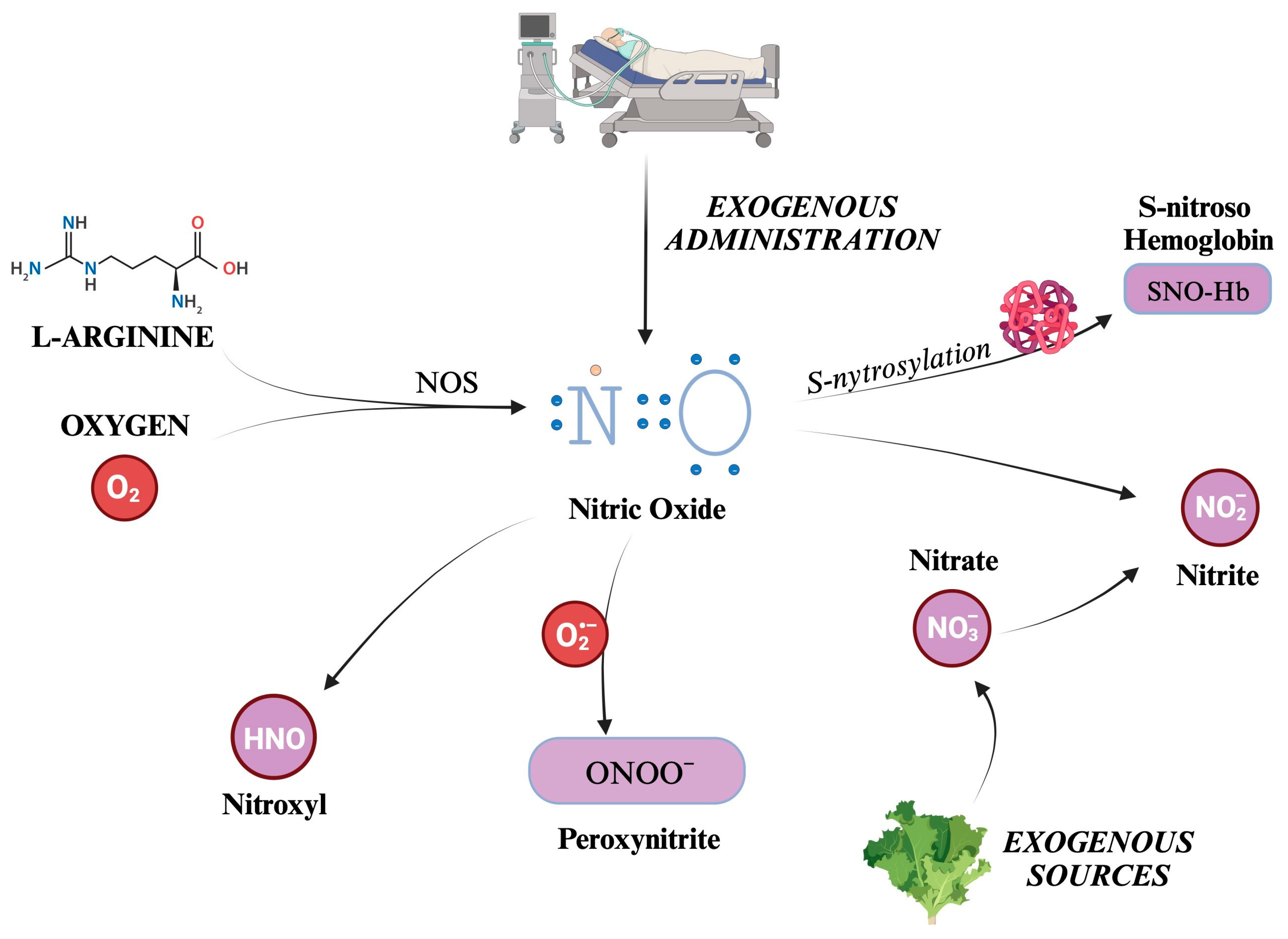
5. Reactive Nitrogen Species
5.1. Generation Pathways of the Main Reactive Nitrogen Species
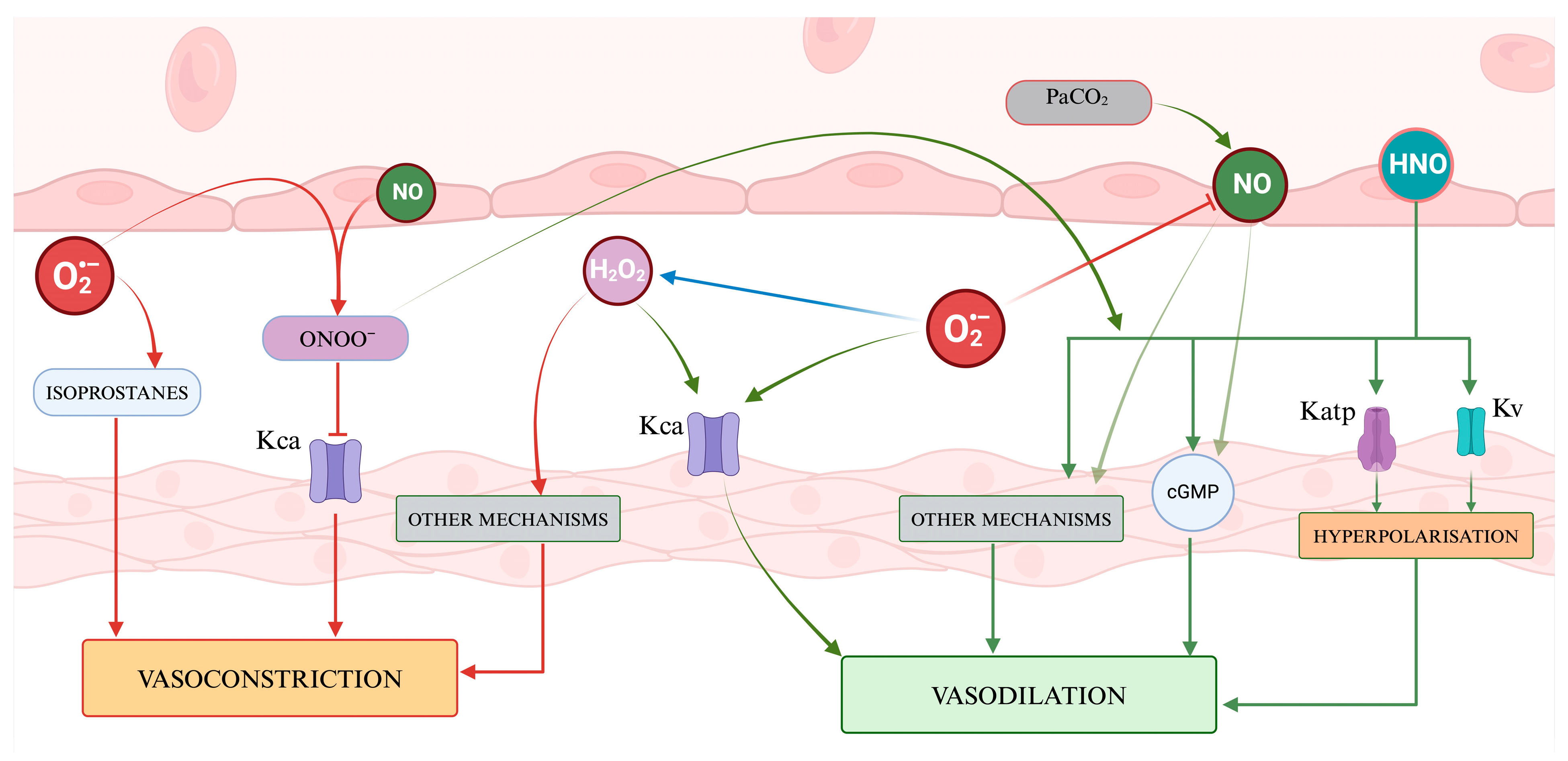
5.2. The Effects of RNS on Cerebral Vascular Tone
5.2.1. Nitric Oxide
5.2.2. Peroxynitrite
5.2.3. Nitroxyl
6. Antioxidants
6.1. Superoxide Dismutase
6.2. Glutathione Redox System
6.3. Vitamin C (Ascorbic Acid)
6.4. Vitamin E (Tocopherols and Tocotrienols)
6.5. Other Antioxidants
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Ascorbic Acid |
| AIS | Acute Ischemic Stroke |
| ATP | Adenosine Triphosphate |
| CAR | Cerebral Autoregulation |
| CBF | Cerebral Blood Flow |
| CGRP | Calcitonin Gene-Related Peptide |
| CMRO2 | Cerebral Metabolic Rate of Oxygen |
| cGMP | cyclic Guanosine Monophosphate |
| Cu-Zn SOD | Copper Zinc Superoxide Dismutase |
| EDRF | Endothelium-Derived Relaxing Factor |
| ER | Endoplasmic Reticulum |
| G6PD | Glucose-6-Phosphate Dehydrogenase |
| GGS | Glutathione Disulfide |
| GSH | Glutathione |
| GPx | Glutathione Peroxidase |
| Hb | Hemoglobin |
| HIF-1α | Hypoxia Inducible Factor 1 alpha |
| HNO | Nitroxyl |
| HRE | Hypoxia Responsive Elements |
| IH | Isocapnic Hyperoxia |
| iNOS | inducible Nitric Oxide Synthase |
| ICP | Intracranial Pressure |
| KATP | ATP–Sensitive Potassium |
| Kca | Calcium–Dependent Potassium Channels |
| Kv | Voltage–Dependent Potassium |
| MnSOD | Manganese Superoxide Dismutase |
| NaNO2 | Sodium Nitrite |
| NAC | N-acetylcysteine |
| NADH | Nicotinamide Adenine Dinucleotide |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NO | Nitric Oxide |
| NO2− | Nitrite |
| NO3− | Nitrate |
| NOX | NADPH Oxidase |
| NOS | Nitric Oxide Synthase |
| ONOO− | Peroxynitrite |
| Oxy-Hb | Oxyhemoglobin |
| PaCO2 | Partial Pressure of Arterial Carbon Dioxide |
| ROS | Reactive Oxygen Species |
| RNS | Reactive Nitrogen Species |
| SNO-Hb | S-nitroso Hemoglobin |
| SOD | Superoxide Dismutase |
| SOD1 | Copper-Zinc Superoxide Dismutase |
| SOD2 | Manganese Superoxide Dismutase |
| SOD3 | Extracellular Superoxide Dismutase |
| TBI | Traumatic Brain Injury |
| TRPA1 | Transient Receptor Potential Channel A1 |
References
- Rink, C.; Khanna, S. Significance of Brain Tissue Oxygenation and the Arachidonic Acid Cascade in Stroke. Antioxid. Redox Signal. 2011, 14, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.D.; Sokoloff, L. Regulation of Cerebral Metabolic Rate. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; American Society for Neurochemistry: Windermere, FL, USA, 1999. [Google Scholar]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, M.J. Regulation of Cerebrovascular Tone. In The Cerebral Circulation; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2009. [Google Scholar]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H Oxidase: Role in Cardiovascular Biology and Disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Staiculescu, M.C.; Foote, C.; Meininger, G.A.; Martinez-Lemus, L.A. The Role of Reactive Oxygen Species in Microvascular Remodeling. Int. J. Mol. Sci. 2014, 15, 23792–23835. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, S.R.; Park, S.J.; Park, H.S.; Min, K.H.; Lee, M.H.; Jin, S.M.; Jin, G.Y.; Yoo, W.H.; Lee, Y.C. Hydrogen Peroxide Induces Vascular Permeability via Regulation of Vascular Endothelial Growth Factor. Am. J. Respir. Cell Mol. Biol. 2006, 35, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Oxidative Stress in Vascular Disease and Its Pharmacological Prevention. Trends Pharmacol. Sci. 2013, 34, 313–319. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Reactive Oxygen Species, Vascular Noxs, and Hypertension: Focus on Translational and Clinical Research. Antioxid. Redox Signal. 2014, 20, 164–182. [Google Scholar] [CrossRef]
- Miller, A.A.; Drummond, G.R.; De Silva, T.M.; Mast, A.E.; Hickey, H.; Williams, J.P.; Broughton, B.R.S.; Sobey, C.G. NADPH Oxidase Activity Is Higher in Cerebral versus Systemic Arteries of Four Animal Species: Role of Nox2. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, 220–225. [Google Scholar] [CrossRef]
- Miller, A.A.; Drummond, G.R.; Schmidt, H.H.H.W.; Sobey, C.G. NADPH Oxidase Activity and Function Are Profoundly Greater in Cerebral versus Systemic Arteries. Circ. Res. 2005, 97, 1055–1062. [Google Scholar] [CrossRef]
- Terashvili, M.; Pratt, P.F.; Gebremedhin, D.; Narayanan, J.; Harder, D.R. Reactive Oxygen Species Cerebral Autoregulation in Health and Disease. Pediatr. Clin. N. Am. 2006, 53, 1029–1037. [Google Scholar] [CrossRef]
- Kitazono, T.; Faraci, F.M.; Taguchi, H.; Heistad, D.D. Role of Potassium Channels in Cerebral Blood Vessels. Stroke 1995, 26, 1713–1723. [Google Scholar] [CrossRef]
- Gutterman, D.D.; Miura, H.; Liu, Y. Redox Modulation of Vascular Tone: Focus of Potassium Channel Mechanisms of Dilation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 671–678. [Google Scholar] [CrossRef]
- Yang, J. The Role of Reactive Oxygen Species in Angiogenesis and Preventing Tissue Injury after Brain Ischemia. Microvasc. Res. 2019, 123, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef]
- Jackson, W.F. Ion Channels and Vascular Tone. Hypertension 2000, 35, 173–178. [Google Scholar] [CrossRef]
- Claassen, J.A.H.R.; Thijssen, D.H.J.; Panerai, R.B.; Faraci, F.M. Regulation of Cerebral Blood Flow in Humans: Physiology and Clinical Implications of Autoregulation. Physiol. Rev. 2021, 101, 1487–1559. [Google Scholar] [CrossRef]
- Weyland, A.; Buhre, W.; Grund, S.; Ludwig, H.; Kazmaier, S.; Weyland, W.; Sonntag, H. Cerebrovascular Tone Rather than Intracranial Pressure Determines the Effective Downstream Pressure of the Cerebral Circulation in the Absence of Intracranial Hypertension. J. Neurosurg. Anesthesiol. 2000, 12, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Michael De Silva, T.; Chen, J.; Faraci, F.M. Cerebral Vascular Disease and Neurovascular Injury in Ischemic Stroke. Circ. Res. 2017, 120, 449–471. [Google Scholar] [CrossRef]
- Battisti-Charbonney, A.; Fisher, J.; Duffin, J. The Cerebrovascular Response to Carbon Dioxide in Humans. J. Physiol. 2011, 589, 3039–3048. [Google Scholar] [CrossRef] [PubMed]
- Hoiland, R.L.; Bain, A.R.; Rieger, M.G.; Bailey, D.M.; Ainslie, P.N. Oxygen as a Regulator of Biological Systems: Hypoxemia, Oxygen Content, and the Regulation of Cerebral Blood Flow. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R398–R413. [Google Scholar] [CrossRef]
- Pearce, W.J. Mechanisms of Hypoxic Cerebral Vasodilatation. Pharmacol. Ther. 1995, 65, 75–91. [Google Scholar] [CrossRef]
- Xu, Y.; Liachenko, S.M.; Tang, P.; Chan, P.H. Faster Recovery of Cerebral Perfusion in SOD1-Overexpressed Rats after Cardiac Arrest and Resuscitation. Stroke 2009, 40, 2512–2518. [Google Scholar] [CrossRef][Green Version]
- Didion, S.P.; Hathaway, C.A.; Faraci, F.M. Superoxide Levels and Function of Cerebral Blood Vessels after Inhibition of CuZn-SOD. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1697–H1703. [Google Scholar] [CrossRef]
- Shibata, M.; Araki, N.; Hamada, J.; Sasaki, T.; Ohta, K.; Shimazu, K.; Fukuuchi, Y. Regional Difference in Nitric Oxide Production during Forebrain Ischemia. Microchem. J. 1997, 56, 188–191. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Altura, B.T.; Altura, B.M. Peroxynitrite-Induced Relaxation in Isolated Canine Cerebral Arteries and Mechanisms of Action. Toxicol. Appl. Pharmacol. 2004, 196, 176–182. [Google Scholar] [CrossRef]
- Brzezinska, A.K.; Gebremedhin, D.; Chilian, W.M.; Kalyanaraman, B.; Elliott, S.J. Peroxynitrite Reversibly Inhibits Ca(2+)-Activated K(+) Channels in Rat Cerebral Artery Smooth Muscle Cells. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1883–H1890. [Google Scholar] [CrossRef]
- Elliott, S.J.; Lacey, D.J.; Chilian, W.M.; Brzezinska, A.K. Peroxynitrite Is a Contractile Agonist of Cerebral Artery Smooth Muscle Cells. Am. J. Physiol. 1998, 275, H1585–H1591. [Google Scholar] [CrossRef] [PubMed]
- Aladag, M.A.; Turkoz, Y.; Sahna, E.; Parlakpinar, H.; Gul, M. The Attenuation of Vasospasm by Using a Sod Mimetic after Experimental Subarachnoidal Haemorrhage in Rats. Acta Neurochir. 2003, 145, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Güney, O.; Erdi, F.; Esen, H.; Kiyici, A.; Kocaogullar, Y. N-Acetylcysteine Prevents Vasospasm after Subarachnoid Hemorrhage. World Neurosurg. 2010, 73, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Mattos, J.D.; Campos, M.O.; Rocha, M.P.; Mansur, D.E.; Rocha, H.N.M.; Garcia, V.P.; Batista, G.; Alvares, T.S.; Oliveira, G.V.; Souza, M.V.; et al. Human Brain Blood Flow and Metabolism during Isocapnic Hyperoxia: The Role of Reactive Oxygen Species. J. Physiol. 2019, 597, 741–755. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Leveque, C.; Mrakic-Sposta, S.; Lafère, P.; Vezzoli, A.; Germonpré, P.; Beer, A.; Mievis, S.; Virgili, F.; Lambrechts, K.; Theunissen, S.; et al. Oxidative Stress Response’s Kinetics after 60 Minutes at Different (30% or 100%) Normobaric Hyperoxia Exposures. Int. J. Mol. Sci. 2023, 24, 664. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.P.; Kontos, H.A.; Beckman, J.S. Mechanisms of Cerebral Vasodilation by Superoxide, Hydrogen Peroxide, and Peroxynitrite. Am. J. Physiol. 1996, 271, H1262–H1266. [Google Scholar] [CrossRef] [PubMed]
- Marklund, S. Spectrophotometric Study of Spontaneous Disproportionation of Superoxide Anion Radical and Sensitive Direct Assay for Superoxide Dismutase. J. Biol. Chem. 1976, 251, 7504–7507. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent Hydroxyl Radical Production by Peroxynitrite: Implications for Endothelial Injury from Nitric Oxide and Superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Drummond, G.R.; Sobey, C.G. Reactive Oxygen Species in the Cerebral Circulation: Are They All Bad? Antioxid. Redox Signal. 2006, 8, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.S.; Sherrington, C.S. On the Regulation of the Blood-Supply of the Brain. J. Physiol. 1890, 11, 85–158. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef]
- Faraci, F.M. Oxidative Stress: The Curse That Underlies Cerebral Vascular Dysfunction? Stroke 2005, 36, 186–188. [Google Scholar] [CrossRef]
- De Silva, T.M.; Faraci, F.M. Reactive Oxygen Species and the Regulation of Cerebral Vascular Tone. In Oxidative Stress in Applied Basic Research and Clinical Practice; Humana Press: Boston, MA, USA, 2017; pp. 89–112. [Google Scholar] [CrossRef]
- Faraci, F.M. Reactive Oxygen Species: Influence on Cerebral Vascular Tone. J. Appl. Physiol. 2006, 100, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Kontos, H.A. Oxygen Radicals in Cerebral Ischemia. Stroke 2001, 32, 2712–2716. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Conway, M.A.; Knot, H.J.; Brayden, J.E. Chloride Channel Blockers Inhibit Myogenic Tone in Rat Cerebral Arteries. J. Physiol. 1997, 502 Pt 2, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Balestra, C.; Mrakic-Sposta, S.; Virgili, F. Oxygen Variations—Insights into Hypoxia, Hyperoxia and Hyperbaric Hyperoxia—Is the Dose the Clue? Int. J. Mol. Sci. 2023, 24, 13472. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Chrissobolis, S.; Drummond, G.R.; Sobey, C.G. Increased NADPH-Oxidase Activity and Nox4 Expression During Chronic Hypertension Is Associated With Enhanced Cerebral Vasodilatation to NADPH In Vivo. Stroke 2004, 35, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Clempus, R.E. Vascular NAD(P)H Oxidases: Specific Features, Expression, and Regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R277–R297. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed]
- Balestra, C.; Germonpré, P. Hypoxia, a Multifaceted Phenomenon: The Example of the Normobaric Oxygen Paradox. Eur. J. Appl. Physiol. 2012, 112, 4173–4175. [Google Scholar] [CrossRef]
- Salvagno, M.; Coppalini, G.; Taccone, F.S.; Strapazzon, G.; Mrakic-Sposta, S.; Rocco, M.; Khalife, M.; Balestra, C. The Normobaric Oxygen Paradox-Hyperoxic Hypoxic Paradox: A Novel Expedient Strategy in Hematopoiesis Clinical Issues. Int. J. Mol. Sci. 2022, 24, 82. [Google Scholar] [CrossRef]
- Nanduri, J.; Vaddi, D.R.; Khan, S.A.; Wang, N.; Makarenko, V.; Semenza, G.L.; Prabhakar, N.R. HIF-1α Activation by Intermittent Hypoxia Requires NADPH Oxidase Stimulation by Xanthine Oxidase. PLoS ONE 2015, 10, e0119762. [Google Scholar] [CrossRef]
- Mattos, J.D.; Campos, M.O.; Rocha, M.P.; Daniel, X.; Mansur, E.; Rocha, H.N.M.; Vinicius, X.; Garcia, P.; Rocha, N.G.; Thiago, X.; et al. Differential Vasomotor Responses to Isocapnic Hyperoxia: Cerebral versus Peripheral Circulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, 182–187. [Google Scholar] [CrossRef]
- Austin, S.A.; Santhanam, A.V.R.; D’Uscio, L.V.; Katusic, Z.S. Regional Heterogeneity of Cerebral Microvessels and Brain Susceptibility to Oxidative Stress. PLoS ONE 2015, 10, e0144062. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective Neuronal Vulnerability to Oxidative Stress in the Brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Specificity of a Third Kind: Reactive Oxygen and Nitrogen Intermediates in Cell Signaling. J. Clin. Investig. 2003, 111, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris Nikolaos Papageorgiou, C.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Raat, N.J.H.; Shiva, S.; Dezfulian, C.; Hogg, N.; Kim-Shapiro, D.B.; Patel, R.P. Nitrite as a Vascular Endocrine Nitric Oxide Reservoir That Contributes to Hypoxic Signaling, Cytoprotection, and Vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2026–H2035. [Google Scholar] [CrossRef]
- Premont, R.T.; Reynolds, J.D.; Zhang, R.; Stamler, J.S. Role of Nitric Oxide Carried by Hemoglobin in Cardiovascular Physiology: Developments on a Three-Gas Respiratory Cycle. Circ. Res. 2020, 126, 129–158. [Google Scholar] [CrossRef]
- Piacenza, L.; Zeida, A.; Trujillo, M.; Radi, R. The Superoxide Radical Switch in the Biology of Nitric Oxide and Peroxynitrite. Physiol. Rev. 2022, 102, 1881–1906. [Google Scholar] [CrossRef]
- Fukuda, S.; Koga, Y.; Fujita, M.; Suehiro, E.; Kaneda, K.; Oda, Y.; Ishihara, H.; Suzuki, M.; Tsuruta, R. Hyperoxemia during the Hyperacute Phase of Aneurysmal Subarachnoid Hemorrhage Is Associated with Delayed Cerebral Ischemia and Poor Outcome: A Retrospective Observational Study. J. Neurosurg. 2019, 134, 25–32. [Google Scholar] [CrossRef]
- Irvine, J.C.; Ritchie, R.H.; Favaloro, J.L.; Andrews, K.L.; Widdop, R.E.; Kemp-Harper, B.K. Nitroxyl (HNO): The Cinderella of the Nitric Oxide Story. Trends Pharmacol. Sci. 2008, 29, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; Gladwin, M.; Coleman, G.D.; Hord, N.; Howard, G.; Kim-Shapiro, D.B.; Lajous, M.; Larsen, F.J.; Lefer, D.J.; McClure, L.A.; et al. Dietary Nitrate and the Epidemiology of Cardiovascular Disease: Report from a National Heart, Lung, and Blood Institute Workshop. J. Am. Heart Assoc. 2016, 5, e003402. [Google Scholar] [CrossRef]
- DeMartino, A.W.; Kim-Shapiro, D.B.; Patel, R.P.; Gladwin, M.T. Nitrite and Nitrate Chemical Biology and Signalling. Br. J. Pharmacol. 2019, 176, 228–245. [Google Scholar] [CrossRef]
- Kapil, V.; Haydar, S.M.A.; Pearl, V.; Lundberg, J.O.; Weitzberg, E.; Ahluwalia, A. Physiological Role for Nitrate-Reducing Oral Bacteria in Blood Pressure Control. Free Radic. Biol. Med. 2013, 55, 93–100. [Google Scholar] [CrossRef]
- Cosby, K.; Partovi, K.S.; Crawford, J.H.; Patel, R.P.; Reiter, C.D.; Martyr, S.; Yang, B.K.; Waclawiw, M.A.; Zalos, G.; Xu, X.; et al. Nitrite Reduction to Nitric Oxide by Deoxyhemoglobin Vasodilates the Human Circulation. Nat. Med. 2003, 9, 1498–1505. [Google Scholar] [CrossRef]
- Crawford, J.H.; Isbell, T.S.; Huang, Z.; Shiva, S.; Chacko, B.K.; Schechter, A.N.; Darley-Usmar, V.M.; Kerby, J.D.; Lang, J.D.; Kraus, D.; et al. Hypoxia, Red Blood Cells, and Nitrite Regulate NO-Dependent Hypoxic Vasodilation. Blood 2006, 107, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Dalsgaard, T.; Simonsen, U.; Fago, A. Nitrite-Dependent Vasodilation Is Facilitated by Hypoxia and Is Independent of Known NO-Generating Nitrite Reductase Activities. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H3072–H3078. [Google Scholar] [CrossRef]
- Maher, A.R.; Milsom, A.B.; Gunaruwan, P.; Abozguia, K.; Ahmed, I.; Weaver, R.A.; Thomas, P.; Ashrafian, H.; Born, G.V.R.; James, P.E.; et al. Hypoxic Modulation of Exogenous Nitrite-Induced Vasodilation in Humans. Circulation 2008, 117, 670–677. [Google Scholar] [CrossRef]
- Pinder, A.G.; Pittaway, E.; Morris, K.; James, P.E. Nitrite Directly Vasodilates Hypoxic Vasculature via Nitric Oxide-Dependent and -Independent Pathways. Br. J. Pharmacol. 2009, 157, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Hataishi, R.; Rodrigues, A.C.; Neilan, T.G.; Morgan, J.G.; Buys, E.; Shiva, S.; Tambouret, R.; Jassal, D.S.; Raher, M.J.; Furutani, E.; et al. Inhaled Nitric Oxide Decreases Infarction Size and Improves Left Ventricular Function in a Murine Model of Myocardial Ischemia-Reperfusion Injury. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H379–H384. [Google Scholar] [CrossRef]
- Ma, L.; Hu, L.; Feng, X.; Wang, S. Nitrate and Nitrite in Health and Disease. Aging Dis. 2018, 9, 938–945. [Google Scholar] [CrossRef]
- Stamler, J.S.; Singel, D.J.; Loscalzo, J. Biochemistry of Nitric Oxide and Its Redox-Activated Forms. Science 1992, 258, 1898–1902. [Google Scholar] [CrossRef]
- Palmer, R.M.J.; Ferrige, A.G.; Moncada, S. Nitric Oxide Release Accounts for the Biological Activity of Endothelium-Derived Relaxing Factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.A.; Adachi, T. Nitric-Oxide-Induced Vasodilatation: Regulation by Physiologic s-Glutathiolation and Pathologic Oxidation of the Sarcoplasmic Endoplasmic Reticulum Calcium ATPase. Trends Cardiovasc. Med. 2006, 16, 109–114. [Google Scholar] [CrossRef]
- Sun, C.W.; Falck, J.R.; Okamoto, H.; Harder, D.R.; Roman, R.J. Role of CGMP versus 20-HETE in the Vasodilator Response to Nitric Oxide in Rat Cerebral Arteries. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H339–H350. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; MacVicar, B.A.; Newman, E.A. Glial and Neuronal Control of Brain Blood Flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef]
- Iadecola, C.; Pelligrino, D.A.; Moskowitz, M.A.; Lassen, N.A. Nitric Oxide Synthase Inhibition and Cerebrovascular Regulation. J. Cereb. Blood Flow Metab. 1994, 14, 175–192. [Google Scholar] [CrossRef]
- Matsuura, T.; Kanno, I. Effect of Nitric Oxide Synthase Inhibitor on the Local Cerebral Blood Flow Evoked by Rat Somatosensory Stimulation under Hyperoxia. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2002, 131, 267–274. [Google Scholar] [CrossRef]
- Takuwa, H.; Matsuura, T.; Bakalova, R.; Obata, T.; Kanno, I. Contribution of Nitric Oxide to Cerebral Blood Flow Regulation under Hypoxia in Rats. J. Physiol. Sci. 2010, 60, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, I.T.; Oury, T.D.; Crapo, J.D.; Piantadosi, C.A. Regulation of the Brain’s Vascular Responses to Oxygen. Circ. Res. 2002, 91, 1031–1037. [Google Scholar] [CrossRef]
- Iadecola, C. Does Nitric Oxide Mediate the Increases in Cerebral Blood Flow Elicited by Hypercapnia? Proc. Natl. Acad. Sci. USA 1992, 89, 3913–3916. [Google Scholar] [CrossRef]
- Hoiland, R.L.; Macleod, D.B.; Stacey, B.S.; Caldwell, H.G.; Howe, C.A.; Carr, J.M.J.R.; Tymko, M.M.; Nowak-flu, D.; Coombs, G.B.; Patrician, A.; et al. Hemoglobin and Cerebral Hypoxic Vasodilation in Humans: Evidence for Nitric Oxide-Dependent and S-Nitrosothiol Mediated Signal Transduction. J. Cereb. Blood Flow Metab. 2023, 43, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Lavi, S.; Gaitini, D.; Milloul, V.; Jacob, G. Impaired Cerebral CO2 Vasoreactivity: Association with Endothelial Dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1856–H1861. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.R.; Yang, C.; Bakhtian, K.D.; Qi, M.; Lonser, R.R.; Pluta, R.M. Carbon Dioxide Influence on Nitric Oxide Production in Endothelial Cells and Astrocytes: Cellular Mechanisms. Brain Res. 2011, 1386, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T.S. The Role of the Nitric Oxide Pathway in Brain Injury and Its Treatment--from Bench to Bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.K.; Schoenfeld, E.; Hou, W.; Singer, A.; Rakowski, E.; Ahmad, S.; Patel, R.; Parikh, P.B.; Smaldone, G. Inhaled Nitric Oxide in Adults with In-Hospital Cardiac Arrest: A Feasibility Study. Nitric Oxide 2021, 115, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Lenz, I.J.; Plesnila, N.; Terpolilli, N.A. Role of Endothelial Nitric Oxide Synthase for Early Brain Injury after Subarachnoid Hemorrhage in Mice. J. Cereb. Blood Flow Metab. 2021, 41, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Cheng, Y.; Peng, W.; Wu, Y.; Rui, T.; Luo, C.; Zhang, J. Targeting INOS Alleviates Early Brain Injury After Experimental Subarachnoid Hemorrhage via Promoting Ferroptosis of M1 Microglia and Reducing Neuroinflammation. Mol. Neurobiol. 2022, 59, 3124–3139. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, W.; Liu, M.; Zhang, X.; Zhang, Q.R.; Ni, L.; Hang, C.H. Increased Cerebrospinal Fluid Concentrations of Asymmetric Dimethylarginine Correlate with Adverse Clinical Outcome in Subarachnoid Hemorrhage Patients. J. Clin. Neurosci. 2014, 21, 1404–1408. [Google Scholar] [CrossRef]
- Ng, W.H.; Moochhala, S.; Yeo, T.T.; Ong, P.L.; Ng, P.Y. Nitric Oxide and Subarachnoid Hemorrhage: Elevated Level in Cerebrospinal Fluid and Their Implications. Neurosurgery 2001, 49, 622–627. [Google Scholar] [CrossRef]
- Fung, C.; Z’Graggen, W.J.; Jakob, S.M.; Gralla, J.; Haenggi, M.; Rothen, H.U.; Mordasini, P.; Lensch, M.; Söll, N.; Terpolilli, N.; et al. Inhaled Nitric Oxide Treatment for Aneurysmal SAH Patients With Delayed Cerebral Ischemia. Front. Neurol. 2022, 13, 817072. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.V.; Bahrami, S.; Redl, H.; Szabo, C. Alterations in Nitric Oxide Homeostasis during Traumatic Brain Injury. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2627–2632. [Google Scholar] [CrossRef] [PubMed]
- Angelis, D.; Savani, R.; Chalak, L. Nitric Oxide and the Brain. Part 2: Effects Following Neonatal Brain Injury-Friend or Foe? Pediatr. Res. 2021, 89, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Lauer, T.; Preik, M.; Rassaf, T.; Strauer, B.E.; Deussen, A.; Feelisch, M.; Kelm, M. Plasma Nitrite Rather than Nitrate Reflects Regional Endothelial Nitric Oxide Synthase Activity but Lacks Intrinsic Vasodilator Action. Proc. Natl. Acad. Sci. USA 2001, 98, 12814–12819. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Gruetter, C.S. Requirement of Thiols for Activation of Coronary Arterial Guanylate Cyclase by Glyceryl Trinitrate and Sodium Nitrite: Possible Involvement of S-Nitrosothiols. Biochim. Biophys. Acta 1980, 631, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Byrns, R.E.; Buga, G.M.; Wood, K.S. Endothelium-Derived Relaxing Factor from Pulmonary Artery and Vein Possesses Pharmacologic and Chemical Properties Identical to Those of Nitric Oxide Radical. Circ. Res. 1987, 61, 866–879. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Shelhamer, J.H.; Schechter, A.N.; Pease-Fye, M.E.; Waclawiw, M.A.; Panza, J.A.; Ognibene, F.P.; Cannon, R.O. Role of Circulating Nitrite and S-Nitrosohemoglobin in the Regulation of Regional Blood Flow in Humans. Proc. Natl. Acad. Sci. USA 2000, 97, 11482–11487. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.; Bhadrakom, S. Reactions of Strips of Rabbit Aorta to Epinephrine, Isopropylarterenol, Sodium Nitrite and Other Drugs. J. Pharmacol. Exp. Ther. 1953, 2, 129–143. [Google Scholar]
- Brücken, A.; Derwall, M.; Bleilevens, C.; Stoppe, C.; Götzenich, A.; Gaisa, N.T.; Weis, J.; Nolte, K.W.; Rossaint, R.; Ichinose, F.; et al. Brief Inhalation of Nitric Oxide Increases Resuscitation Success and Improves 7-Day-Survival after Cardiac Arrest in Rats: A Randomized Controlled Animal Study. Crit. Care 2015, 19, 408. [Google Scholar] [CrossRef]
- Wu, M.; Pritchard, K.A.; Kaminski, P.M.; Fayngersh, R.P.; Hintze, T.H.; Wolin, M.S. Involvement of Nitric Oxide and Nitrosothiols in Relaxation of Pulmonary Arteries to Peroxynitrite. Am. J. Physiol. 1994, 266, H2108–H2113. [Google Scholar] [CrossRef]
- Liu, S.; Beckman, J.S.; Ku, D.D. Peroxynitrite, a Product of Superoxide and Nitric Oxide, Produces Coronary Vasorelaxation in Dogs. J. Pharmacol. Exp. Ther. 1994, 268, 1114–1121. [Google Scholar]
- Wei, E.P.; Kontos, H.A.; Beckman, J.S.; Faraci, F.M. Antioxidants Inhibit ATP-Sensitive Potassium Channels in Cerebral Arterioles. Stroke 1998, 29, 817–823. [Google Scholar] [CrossRef][Green Version]
- Nossaman, B.D.; Kadowitz, P.J. Potential Benefits of Peroxynitrite. Open Pharmacol. J. 2008, 2, 31–53. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Daneva, Z.; Marziano, C.; Ottolini, M.; Chen, Y.L.; Baker, T.M.; Kuppusamy, M.; Zhang, A.; Ta, H.Q.; Reagan, C.E.; Mihalek, A.D.; et al. Caveolar Peroxynitrite Formation Impairs Endothelial TRPV4 Channels and Elevates Pulmonary Arterial Pressure in Pulmonary Hypertension. Proc. Natl. Acad. Sci. USA 2021, 118, e2023130118. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, M.; Hong, K.; Cope, E.L.; Daneva, Z.; Delalio, L.J.; Sokolowski, J.D.; Marziano, C.; Nguyen, N.Y.; Altschmied, J.; Haendeler, J.; et al. Local Peroxynitrite Impairs Endothelial Transient Receptor Potential Vanilloid 4 Channels and Elevates Blood Pressure in Obesity. Circulation 2020, 141, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Ronson, R.S.; Nakamura, M.; Vinten-Johansen, J. The Cardiovascular Effects and Implications of Peroxynitrite. Cardiovasc. Res. 1999, 44, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.L.; Irvine, J.C.; Tare, M.; Apostolopoulos, J.; Favaloro, J.L.; Triggle, C.R.; Kemp-Harper, B.K. A Role for Nitroxyl (HNO) as an Endothelium-Derived Relaxing and Hyperpolarizing Factor in Resistance Arteries. Br. J. Pharmacol. 2009, 157, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.L.; Lumsden, N.G.; Farry, J.; Jefferis, A.M.; Kemp-Harper, B.K.; Chin-Dusting, J.P.F. Nitroxyl: A Vasodilator of Human Vessels That Is Not Susceptible to Tolerance. Clin. Sci. 2015, 129, 179–187. [Google Scholar] [CrossRef]
- Paolocci, N.; Jackson, M.I.; Lopez, B.E.; Miranda, K.; Tocchetti, C.G.; Wink, D.A.; Hobbs, A.J.; Fukuto, J.M. The Pharmacology of Nitroxyl (HNO) and Its Therapeutic Potential: Not Just the Janus Face of NO. Pharmacol. Ther. 2007, 113, 442–458. [Google Scholar] [CrossRef]
- Favaloro, J.L.; Kemp-Harper, B.K. The Nitroxyl Anion (HNO) Is a Potent Dilator of Rat Coronary Vasculature. Cardiovasc. Res. 2007, 73, 587–596. [Google Scholar] [CrossRef]
- Kemp-Harper, B.K.; Horowitz, J.D.; Ritchie, R.H. Therapeutic Potential of Nitroxyl (HNO) Donors in the Management of Acute Decompensated Heart Failure. Drugs 2016, 76, 1337–1348. [Google Scholar] [CrossRef]
- Zhu, G.; Groneberg, D.; Sikka, G.; Hori, D.; Ranek, M.J.; Nakamura, T.; Takimoto, E.; Paolocci, N.; Berkowitz, D.E.; Friebe, A.; et al. Soluble Guanylate Cyclase Is Required for Systemic Vasodilation But Not Positive Inotropy Induced by Nitroxyl in the Mouse. Hypertension 2014, 65, 385–392. [Google Scholar] [CrossRef]
- Eberhardt, M.; Dux, M.; Namer, B.; Miljkovic, J.; Cordasic, N.; Will, C.; Kichko, T.I.; De La Roche, J.; Fischer, M.; Suárez, S.A.; et al. H2S and NO Cooperatively Regulate Vascular Tone by Activating a Neuroendocrine HNO–TRPA1–CGRP Signalling Pathway. Nat. Commun. 2014, 5, 4381. [Google Scholar] [CrossRef]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 90. [Google Scholar] [CrossRef]
- Gryglewski, R.J.; Palmer, R.M.J.; Moncada, S. Superoxide Anion Is Involved in the Breakdown of Endothelium-Derived Vascular Relaxing Factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef]
- Zhang, M.S.; Liang, J.H.; Yang, M.J.; Ren, Y.R.; Cheng, D.H.; Wu, Q.H.; He, Y.; Yin, J. Low Serum Superoxide Dismutase Is Associated With a High Risk of Cognitive Impairment After Mild Acute Ischemic Stroke. Front. Aging Neurosci. 2022, 14, 834114. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Marcos, M.A.; Blázquez-Medela, A.M.; Gamella-Pozuelo, L.; Recio-Rodriguez, J.I.; García-Ortiz, L.; Martínez-Salgado, C. Serum Superoxide Dismutase Is Associated with Vascular Structure and Function in Hypertensive and Diabetic Patients. Oxid. Med. Cell. Longev. 2016, 2016, 9124676. [Google Scholar] [CrossRef] [PubMed]
- Fesharaki-Zadeh, A. Oxidative Stress in Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 13000. [Google Scholar] [CrossRef] [PubMed]
- Younus, H. Therapeutic Potentials of Superoxide Dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- McGirt, M.J.; Parra, A.; Sheng, H.; Higuchi, Y.; Oury, T.D.; Laskowitz, D.T.; Pearlstein, R.D.; Warner, D.S. Attenuation of Cerebral Vasospasm after Subarachnoid Hemorrhage in Mice Overexpressing Extracellular Superoxide Dismutase. Stroke 2002, 33, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Hazell, A.S.; Faim, S.; Wertheimer, G.; Silva, V.R.; Marques, C.S. The Impact of Oxidative Stress in Thiamine Deficiency: A Multifactorial Targeting Issue. Neurochem. Int. 2013, 62, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Lavrnja, I. Thiamine and Benfotiamine: Focus on Their Therapeutic Potential. Heliyon 2023, 9, e21839. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Glutathione Peroxidases. In Comprehensive Toxicology, 2nd ed.; Volume 4: Biotransformation; Elsevier Science: Amsterdam, The Netherlands, 2010; pp. 229–242. [Google Scholar] [CrossRef]
- Angstwurm, M.W.A.; Engelmann, L.; Zimmermann, T.; Lehmann, C.; Spes, C.H.; Abel, P.; Strauß, R.; Meier-Hellmann, A.; Insel, R.; Radke, J.; et al. Selenium in Intensive Care (SIC): Results of a Prospective Randomized, Placebo-Controlled, Multiple-Center Study in Patients with Severe Systemic Inflammatory Response Syndrome, Sepsis, and Septic Shock. Crit. Care Med. 2007, 35, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Jaff, S.; Zeraattalab-Motlagh, S.; Amiri Khosroshahi, R.; Gubari, M.; Mohammadi, H.; Djafarian, K. The Effect of Selenium Therapy in Critically Ill Patients: An Umbrella Review of Systematic Reviews and Meta-Analysis of Randomized Controlled Trials. Eur. J. Med. Res. 2023, 28, 104. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Matuz-Mares, D.; Riveros-Rosas, H.; Vázquez-Meza, H.; Vilchis-Landeros, M.M. Glutathione Participation in the Prevention of Cardiovascular Diseases. Antioxidants 2021, 10, 1220. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F.M. Effects of N-Acetylcysteine, Oral Glutathione (GSH) and a Novel Sublingual Form of GSH on Oxidative Stress Markers: A Comparative Crossover Study. Redox Biol. 2015, 6, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Watanabe, K.; Ko, K.; Higashi, K.; Kogure, N.; Kitajima, M.; Takayama, H.; Takao, K.; Sugita, Y.; Sakamoto, A.; et al. Protective Effects of Brain Infarction by N-Acetylcysteine Derivatives. Stroke 2018, 49, 1727–1733. [Google Scholar] [CrossRef]
- Sabetghadam, M.; Mazdeh, M.; Abolfathi, P.; Mohammadi, Y.; Mehrpooya, M. Evidence for a Beneficial Effect of Oral N-Acetylcysteine on Functional Outcomes and Inflammatory Biomarkers in Patients with Acute Ischemic Stroke. Neuropsychiatr. Dis. Treat. 2020, 16, 1265–1278. [Google Scholar] [CrossRef]
- Olesen, H.Ø.; Pors, S.E.; Jensen, L.B.; Grønning, A.P.; Lemser, C.E.; Nguyen Heimbürger, M.T.H.; Mamsen, L.S.; Getreu, N.; Christensen, S.T.; Andersen, C.Y.; et al. N-Acetylcysteine Protects Ovarian Follicles from Ischemia-Reperfusion Injury in Xenotransplanted Human Ovarian Tissue. Hum. Reprod. 2021, 36, 429–443. [Google Scholar] [CrossRef]
- Cayuela, N.C.; Koike, M.K.; Jacysyn, J.d.F.; Rasslan, R.; Cerqueira, A.R.A.; Costa, S.K.P.; Diniz-Júnior, J.A.P.; Utiyama, E.M.; de Souza Montero, E.F. N-Acetylcysteine Reduced Ischemia and Reperfusion Damage Associated with Steatohepatitis in Mice. Int. J. Mol. Sci. 2020, 21, 4106. [Google Scholar] [CrossRef]
- Raghu, G.; Berk, M.; Campochiaro, P.A.; Jaeschke, H.; Marenzi, G.; Richeldi, L.; Wen, F.-Q.; Nicoletti, F.; Calverley, P.M.A. The Multifaceted Therapeutic Role of N-Acetylcysteine (NAC) in Disorders Characterized by Oxidative Stress. Curr. Neuropharmacol. 2021, 19, 1202–1224. [Google Scholar] [CrossRef]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of Vitamin C: Expanding the Focus from Antioxidant to Endogenous Neuromodulator. Pharmacol. Res. 2019, 146, 104321. [Google Scholar] [CrossRef]
- Harrison, F.E.; May, J.M. Vitamin C Function in the Brain: Vital Role of the Ascorbate Transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate Regulation and Its Neuroprotective Role in the Brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Grossmann, M.; Dobrev, D.; Himmel, H.M.; Ravens, U.; Kirch, W. Ascorbic Acid–Induced Modulation of Venous Tone in Humans. Hypertension 2001, 37, 949–954. [Google Scholar] [CrossRef] [PubMed][Green Version]
- May, J.M. How Does Ascorbic Acid Prevent Endothelial Dysfunction? Free Radic. Biol. Med. 2000, 28, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Barak, O.F.; Caljkusic, K.; Hoiland, R.L.; Ainslie, P.N.; Thom, S.R.; Yang, M.; Jovanov, P.; Dujic, Z. Differential Influence of Vitamin C on the Peripheral and Cerebral Circulation after Diving and Exposure to Hyperoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R759–R767. [Google Scholar] [CrossRef] [PubMed]
- Salvagno, M.; Taccone, F.S.; Bogossian, E.G. Tissue Hyperoxia Is Not a Risk Factor for Cerebral Vasospasm Following Subarachnoid Hemorrhage. J. Neurol. Sci. 2023, 445, 120535. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, M.; Kodama, N.; Toda, N. Suppression of the Cerebral Vasospastic Actions of Oxyhemoglobin by Ascorbic Acid. Neurosurgery 1991, 28, 33. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.; Jamil, R.T.; Attia, F.N. Vitamin C (Ascorbic Acid). In Encyclopedia of Toxicology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 962–963. [Google Scholar] [CrossRef]
- Engin, K.N. Alpha-Tocopherol: Looking beyond an Antioxidant. Mol. Vis. 2009, 15, 855–860. [Google Scholar]
- La Torre, M.E.; Villano, I.; Monda, M.; Messina, A.; Cibelli, G.; Valenzano, A.; Pisanelli, D.; Panaro, M.A.; Tartaglia, N.; Ambrosi, A.; et al. Role of Vitamin E and the Orexin System in Neuroprotection. Brain Sci. 2021, 11, 1098. [Google Scholar] [CrossRef]
- Motoyama, T.; Kawano, H.; Kugiyama, K.; Hirashima, O.; Ohgushi, M.; Tsunoda, R.; Moriyama, Y.; Miyao, Y.; Yoshimura, M.; Ogawa, H.; et al. Vitamin E Administration Improves Impairment of Endothelium-Dependent Vasodilation in Patients with Coronary Spastic Angina. J. Am. Coll. Cardiol. 1998, 32, 1672–1679. [Google Scholar] [CrossRef]
- Rasool, A.H.G.; Rahman, A.R.A.; Yuen, K.H.; Wong, A.R. Arterial Compliance and Vitamin E Blood Levels with a Self Emulsifying Preparation of Tocotrienol Rich Vitamin E. Arch. Pharm. Res. 2008, 31, 1212–1217. [Google Scholar] [CrossRef]
- Heitzer, T.; Ylä Herttuala, S.; Wild, E.; Luoma, J.; Drexler, H. Effect of Vitamin E on Endothelial Vasodilator Function in Patients with Hypercholesterolemia, Chronic Smoking or Both. J. Am. Coll. Cardiol. 1999, 33, 499–505. [Google Scholar] [CrossRef]
- Cheng, P.; Wang, L.; Ning, S.; Liu, Z.; Lin, H.; Chen, S.; Zhu, J. Vitamin E Intake and Risk of Stroke: A Meta-Analysis. Br. J. Nutr. 2018, 120, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Loh, H.C.; Lim, R.; Lee, K.W.; Ooi, C.Y.; Chuan, D.R.; Looi, I.; Kah Hay, Y.; Abdul Karim Khan, N. Effects of Vitamin E on Stroke: A Systematic Review with Meta-Analysis and Trial Sequential Analysis. Stroke Vasc. Neurol. 2021, 6, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Pfister, H.W.; Ködel, U.; Dirnagl, U.; Haberl, R.L.; Ruckdeschel, G.; Einhäupl, K.M. Effect of Catalase on Regional Cerebral Blood Flow and Brain Edema during the Early Phase of Experimental Pneumococcal Meningitis. J. Infect. Dis. 1992, 166, 1442–1445. [Google Scholar] [CrossRef]
- Iida, Y.; Katusic, Z.S.; Wei, E.P. Mechanisms of Cerebral Arterial Relaxations to Hydrogen Peroxide. Stroke 2000, 31, 2224–2230. [Google Scholar] [CrossRef] [PubMed]
- Bondonno, N.P.; Bondonno, C.P.; Blekkenhorst, L.C.; Considine, M.J.; Maghzal, G.; Stocker, R.; Woodman, R.J.; Ward, N.C.; Hodgson, J.M.; Croft, K.D. Flavonoid-Rich Apple Improves Endothelial Function in Individuals at Risk for Cardiovascular Disease: A Randomized Controlled Clinical Trial. Mol. Nutr. Food Res. 2018, 62, 1700674. [Google Scholar] [CrossRef]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P.E. The Neuroprotective Potential of Flavonoids: A Multiplicity of Effects. Genes Nutr. 2008, 3, 115–126. [Google Scholar] [CrossRef]
- Gül, Ş.; Aydoğmuş, E.; Bahadir, B.; Büyükuysal, Ç.; Güven, B. Neuroprotective Effects of Quercetin on Cerebral Vasospasm Following Experimental Subarachnoid Haemorrhage in Rats. Turk. J. Med. Sci. 2020, 50, 1106–1110. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, J.; Yang, F.; Li, S.; Ma, W.; Chang, X.; Yang, L. Neuroprotective Effects of Quercetin on Ischemic Stroke: A Literature Review. Front. Pharmacol. 2022, 13, 854249. [Google Scholar] [CrossRef]
- Lee, J.M.; Jeong, S.W.; Kim, M.Y.; Park, J.B.; Kim, M.S. The Effect of Vitamin D Supplementation in Patients with Acute Traumatic Brain Injury. World Neurosurg. 2019, 126, e1421–e1426. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Yuan, X.; Li, D.; Fan, F.; Guo, X.; Xu, Y.; Guan, S. Vitamin D Level Is Associated with Rupture of Intracranial Aneurysm in Patients with Subarachnoid Hemorrhage. Front. Neurol. 2022, 13, 890950. [Google Scholar] [CrossRef]
- Della Nera, G.; Sabatino, L.; Gaggini, M.; Gorini, F.; Vassalle, C. Vitamin D Determinants, Status, and Antioxidant/Anti-Inflammatory-Related Effects in Cardiovascular Risk and Disease: Not the Last Word in the Controversy. Antioxidants 2023, 12, 948. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, S.; Porri, D.; De Giuseppe, R.; Manuelli, M.; Alessio, F.; Cena, H. The Controversial Role of Vitamin D as an Antioxidant: Results from Randomised Controlled Trials. Nutr. Res. Rev. 2019, 32, 99–105. [Google Scholar] [CrossRef]
- Dao, D.Q.; Ngo, T.C.; Thong, N.M.; Nam, P.C. Is Vitamin A an Antioxidant or a Pro-Oxidant? J. Phys. Chem. B 2017, 121, 9348–9357. [Google Scholar] [CrossRef]
- Hummel, R.; Ulbrich, S.; Appel, D.; Li, S.; Hirnet, T.; Zander, S.; Bobkiewicz, W.; Gölz, C.; Schäfer, M.K.E. Administration of All-Trans Retinoic Acid after Experimental Traumatic Brain Injury Is Brain Protective. Br. J. Pharmacol. 2020, 177, 5208–5223. [Google Scholar] [CrossRef] [PubMed]
- Bonney, S.; Harrison-Uy, S.; Mishra, S.; MacPherson, A.M.; Choe, Y.; Li, D.; Jaminet, S.C.; Fruttiger, M.; Pleasure, S.J.; Siegenthaler, J.A. Diverse Functions of Retinoic Acid in Brain Vascular Development. J. Neurosci. 2016, 36, 7786–7801. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Johnson, R.J. Uric Acid: The Oxidant–Antioxidant Paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619. [Google Scholar] [CrossRef]
- Halvorson, B.; Frisbee, J.; Halvorson, B.; Frisbee, J. Cerebral Vascular Tone Regulation: Integration and Impact of Disease. In Basic and Clinical Understanding of Microcirculation; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Hsieh, H.J.; Liu, C.A.; Huang, B.; Tseng, A.H.; Wang, D.L. Shear-Induced Endothelial Mechanotransduction: The Interplay between Reactive Oxygen Species (ROS) and Nitric Oxide (NO) and the Pathophysiological Implications. J. Biomed. Sci. 2014, 21, 3. [Google Scholar] [CrossRef]
- Demchenko, I.T.; Boso, A.E.; O’Neill, T.J.; Bennett, P.B.; Piantadosi, C.A. Nitric Oxide and Cerebral Blood Flow Responses to Hyperbaric Oxygen. J. Appl. Physiol. 2000, 88, 1381–1389. [Google Scholar] [CrossRef]
- Zhilyaev, S.Y.; Moskvin, A.N.; Platonova, T.F.; Gutsaeva, D.R.; Churilina, I.V.; Demchenko, I.T. Hyperoxic Vasoconstriction in the Brain Is Mediated by Inactivation of Nitric Oxide by Superoxide Anions. Neurosci. Behav. Physiol. 2003, 33, 783–787. [Google Scholar] [CrossRef]
- Jelinek, M.; Jurajda, M.; Duris, K. Oxidative Stress in the Brain: Basic Concepts and Treatment Strategies in Stroke. Antioxidants 2021, 10, 1886. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Patel, A.B.; Kioutchoukova, I.P.; Diaz, M.J.; Valenti, D.; Nguyen, A.; Patel, A.B.; Kioutchoukova, I.P.; Diaz, M.J.; Lucke-Wold, B. Mechanisms of Mitochondrial Oxidative Stress in Brain Injury: From Pathophysiology to Therapeutics. Oxygen 2023, 3, 163–178. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative Stress in Neurodegenerative Diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Villalba, N.; Sonkusare, S.K.; Longden, T.A.; Tran, T.L.; Sackheim, A.M.; Nelson, M.T.; Wellman, G.C.; Freeman, K. Traumatic Brain Injury Disrupts Cerebrovascular Tone Through Endothelial Inducible Nitric Oxide Synthase Expression and Nitric Oxide Gain of Function. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2014, 3, e001474. [Google Scholar] [CrossRef] [PubMed]
- Szarka, N.; Pabbidi, M.R.; Amrein, K.; Czeiter, E.; Berta, G.; Pohoczky, K.; Helyes, Z.; Ungvari, Z.; Koller, A.; Buki, A.; et al. Traumatic Brain Injury Impairs Myogenic Constriction of Cerebral Arteries: Role of Mitochondria-Derived H2O2 and TRPV4-Dependent Activation of BKca Channels. J. Neurotrauma 2018, 35, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Spranger, M.; Krempien, S.; Schwab, S.; Donneberg, S.; Hacke, W. Superoxide Dismutase Activity in Serum of Patients With Acute Cerebral Ischemic Injury. Stroke 1997, 28, 2425–2428. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yin, L.; Song, Y.; Zhang, M.; Lu, Y.; Wang, S. The Use of Hydrocortisone, Ascorbic Acid and Thiamine in Patients with Sepsis and Septic Shock—A Systematic Review. J. Pharm. Pract. 2023, 36, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, F.; Masse, M.-H.; Menard, J.; Sprague, S.; Pinto, R.; Heyland, D.K.; Cook, D.J.; Battista, M.-C.; Day, A.G.; Guyatt, G.H.; et al. Intravenous Vitamin C in Adults with Sepsis in the Intensive Care Unit. N. Engl. J. Med. 2022, 386, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Bowton, D.L.; Bertels, N.H.; Prough, D.S.; Stump, D.A. Cerebral Blood Flow Is Reduced in Patients with Sepsis Syndrome. Crit. Care Med. 1989, 17, 399–403. [Google Scholar] [CrossRef]
- Margaritelis, N.V.; Paschalis, V.; Theodorou, A.A.; Vassiliou, V.; Kyparos, A.; Nikolaidis, M.G. Rapid Decreases of Key Antioxidant Molecules in Critically Ill Patients: A Personalized Approach. Clin. Nutr. 2020, 39, 1146–1154. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model/Technique | Investigations on Cerebral Vasculature |
|---|---|
| Animal models (e.g., rats, rabbits, etc.) [25,26,27] | ROS/RNS effects on cerebral blood flow, microperfusion and autoregulation |
| In vitro studies [28,29,30] | Molecular mechanisms modulating the endothelial and smooth muscle cell response |
| Use of antioxidants [31,32,33] | Therapeutic neuroprotective therapies and modulation of vascular tone in oxidative stress conditions |
| Diagnostic tools | Non-invasive measurement of the effects of ROS/RNS on cerebral blood flow and metabolism |
| Pharmacological interventions | Investigation of the causal relationship among ROS, RNS, antioxidants, and vascular tone |
| Mechanisms | ROS/RNS Involved | Effect on Cerebral Vascular Tone |
|---|---|---|
| Interaction with ion channels | O2−, H2O2, HNO, ONOO− | Modulation of K+ channels, leading to vasodilation or vasoconstriction [13,36,105,116] |
| Interaction with nitric oxide | O2− | Inactivation of NO, leading to vasoconstriction [44,45] |
| Other endothelial and myogenic mechanisms | O2−, H2O2, NO, ONOO− | Vasodilation/vasoconstriction [28,46,60] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvagno, M.; Sterchele, E.D.; Zaccarelli, M.; Mrakic-Sposta, S.; Welsby, I.J.; Balestra, C.; Taccone, F.S. Oxidative Stress and Cerebral Vascular Tone: The Role of Reactive Oxygen and Nitrogen Species. Int. J. Mol. Sci. 2024, 25, 3007. https://doi.org/10.3390/ijms25053007
Salvagno M, Sterchele ED, Zaccarelli M, Mrakic-Sposta S, Welsby IJ, Balestra C, Taccone FS. Oxidative Stress and Cerebral Vascular Tone: The Role of Reactive Oxygen and Nitrogen Species. International Journal of Molecular Sciences. 2024; 25(5):3007. https://doi.org/10.3390/ijms25053007
Chicago/Turabian StyleSalvagno, Michele, Elda Diletta Sterchele, Mario Zaccarelli, Simona Mrakic-Sposta, Ian James Welsby, Costantino Balestra, and Fabio Silvio Taccone. 2024. "Oxidative Stress and Cerebral Vascular Tone: The Role of Reactive Oxygen and Nitrogen Species" International Journal of Molecular Sciences 25, no. 5: 3007. https://doi.org/10.3390/ijms25053007
APA StyleSalvagno, M., Sterchele, E. D., Zaccarelli, M., Mrakic-Sposta, S., Welsby, I. J., Balestra, C., & Taccone, F. S. (2024). Oxidative Stress and Cerebral Vascular Tone: The Role of Reactive Oxygen and Nitrogen Species. International Journal of Molecular Sciences, 25(5), 3007. https://doi.org/10.3390/ijms25053007