

Genetic Landscape of Factor VII Deficiency: Insights from a Comprehensive Analysis of Pathogenic Variants and Their Impact on Coagulation Activity


 ,
,  ,
,
Abstract
1. Introduction
2. Results
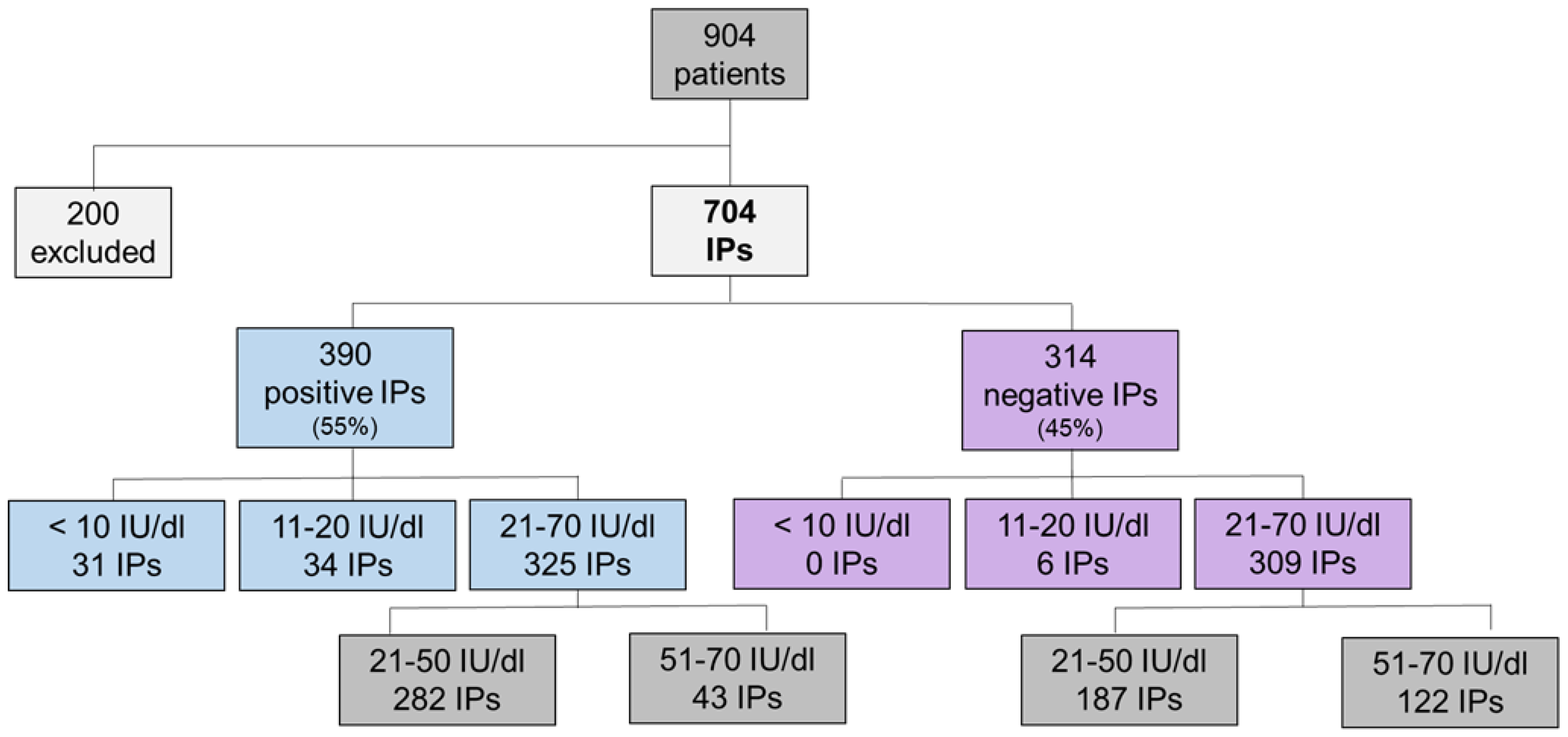
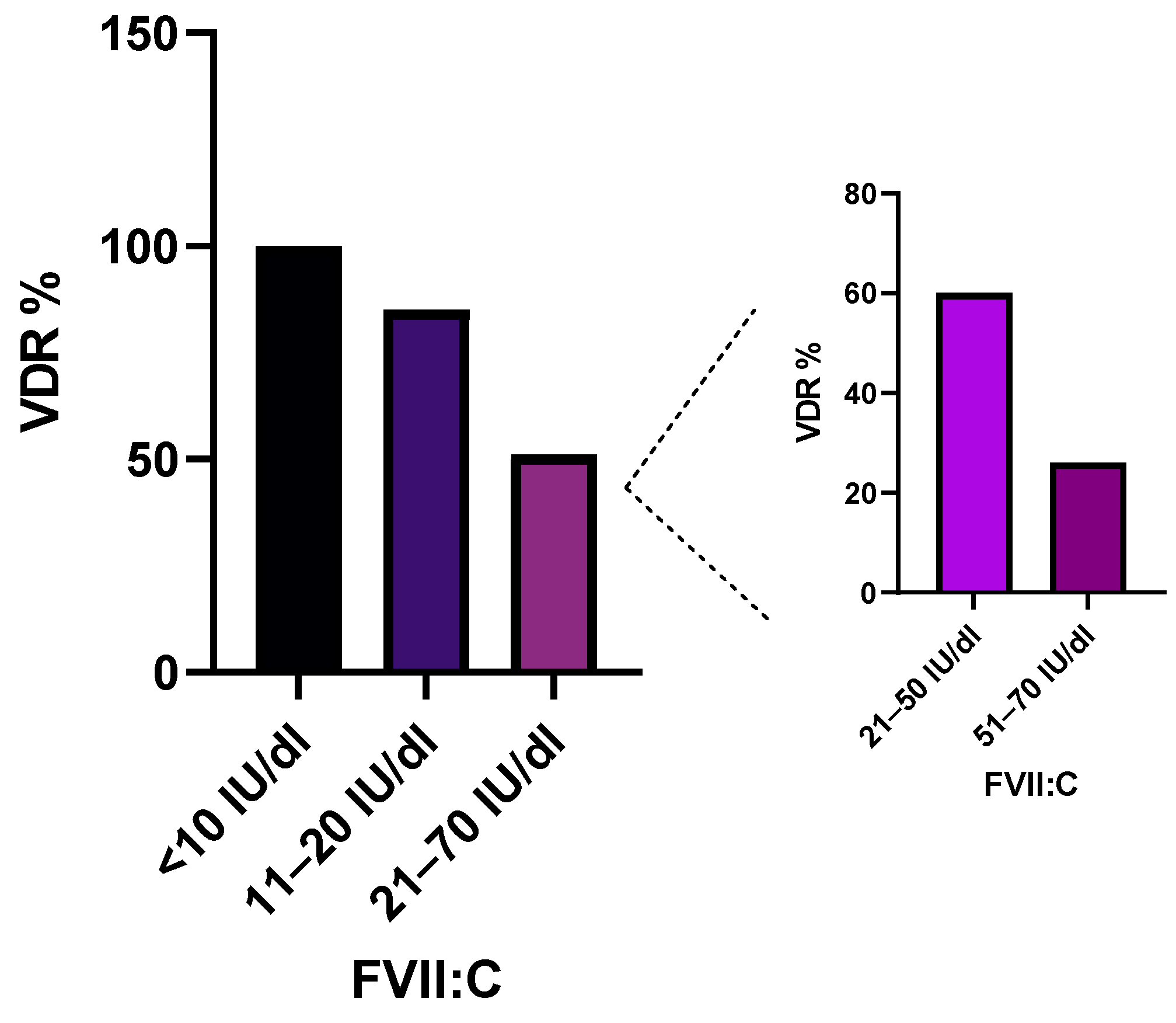
2.1. Association of Variant Detection Rate with FVII:C Levels
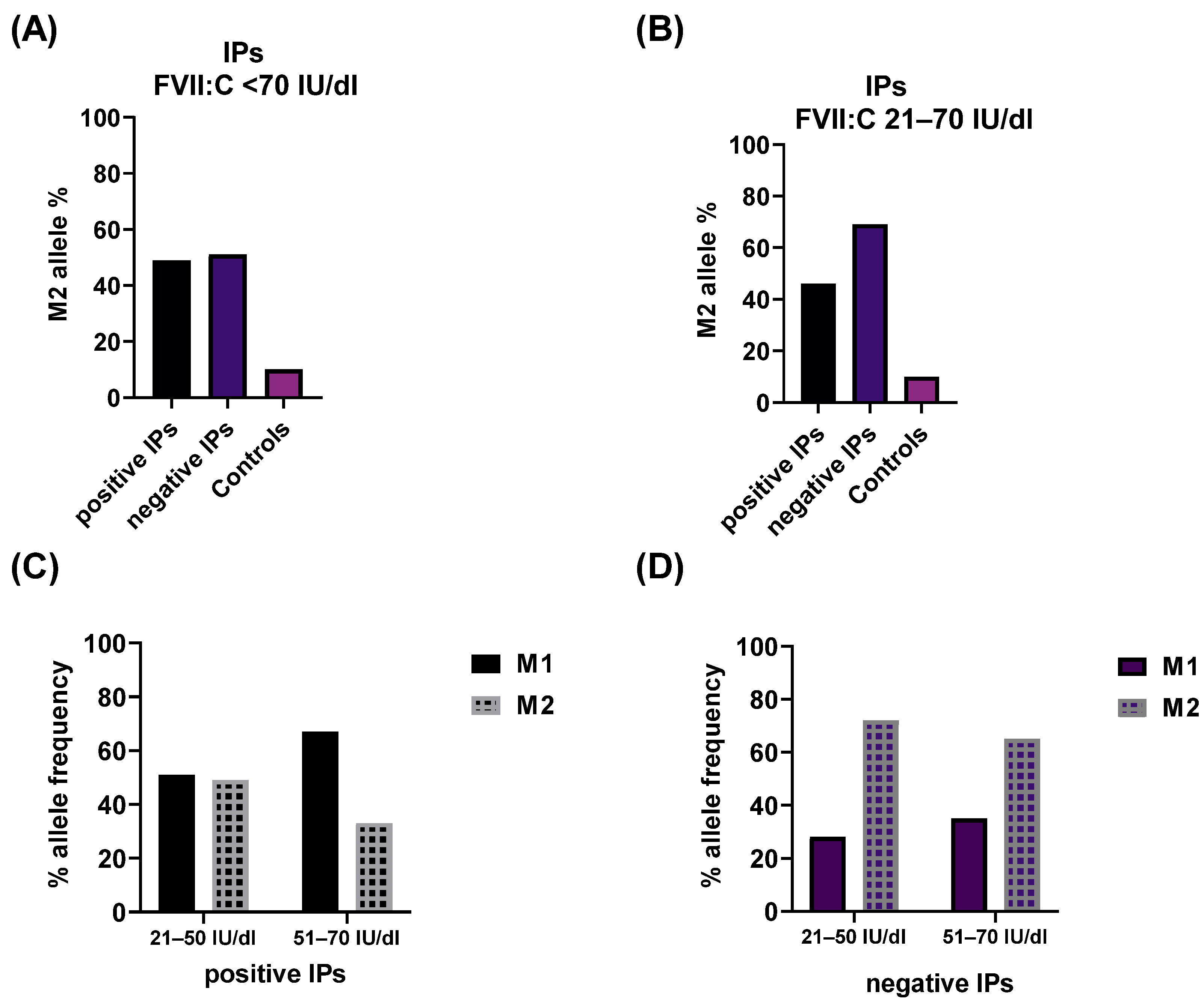
2.2. Impact of the rs6046 Variant on FVII Activity
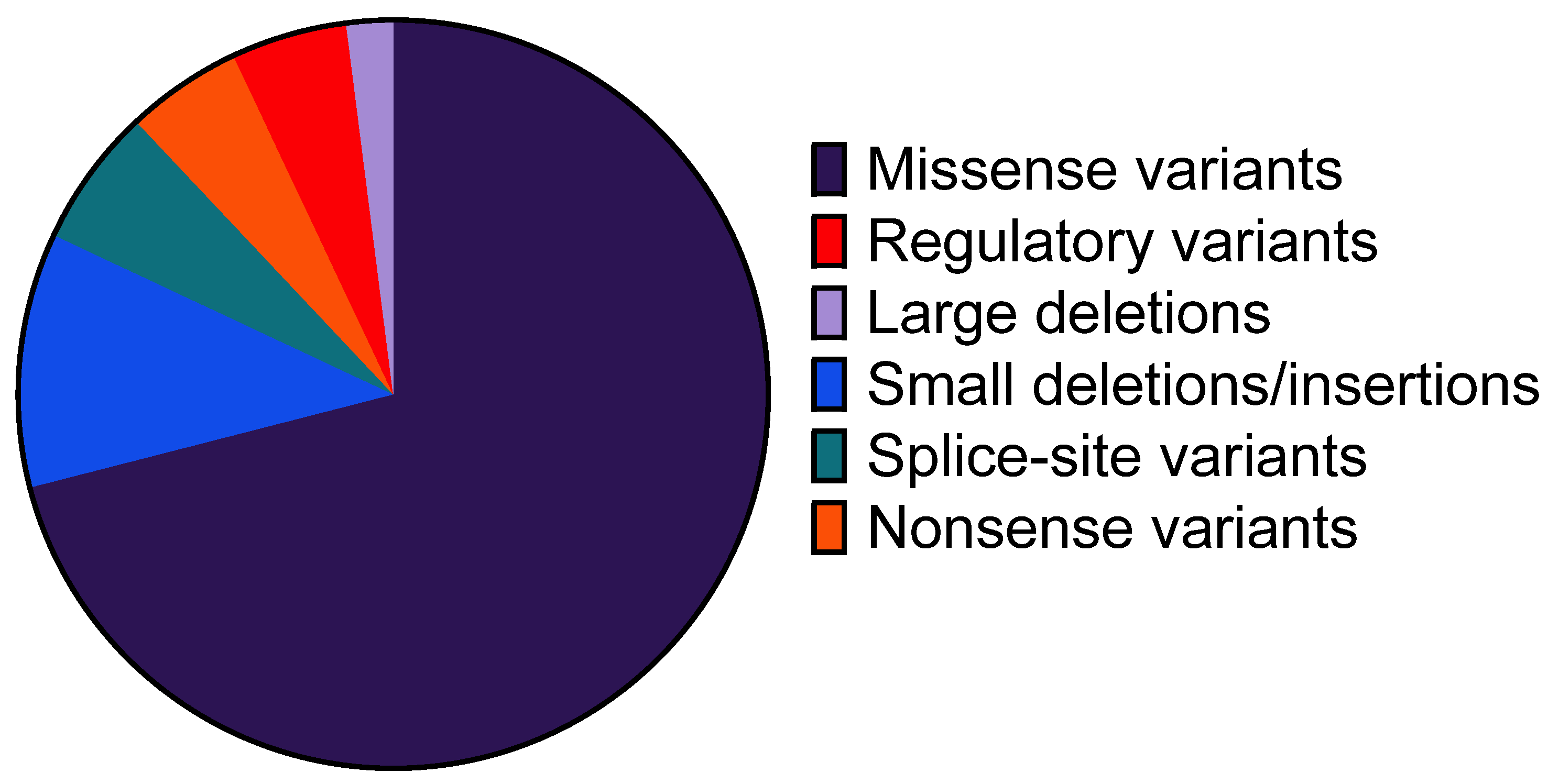
2.3. Profile of Identified Genetic Variants
2.4. Genetic Variants Detected in Multiple IPs
2.5. Genetic Variants Detected in a Single IP
3. Discussion
4. Materials and Methods
4.1. Patient and Control Cohorts
4.2. Molecular Genetics Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mariani, G.; Bernardi, F. Factor VII Deficiency. Semin. Thromb. Hemost. 2009, 35, 400–406. [Google Scholar] [CrossRef]
- Perry, D.J. Factor VII Deficiency. Brit J. Haematol. 2002, 118, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, M.N.D.; Dolce, A.; Mariani, G. Bleeding symptoms at disease presentation and prediction of ensuing bleeding in inherited FVII deficiency. Thromb. Haemost. 2013, 109, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Mariani, G.; Herrmann, F.H.; Dolce, A.; Batorova, A.; Etro, D.; Peyvandi, F.; Wulff, K.; Schved, J.F.; Auerswald, G.; Ingerslev, J.; et al. Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. Thromb. Haemost. 2005, 93, 481–487. [Google Scholar] [CrossRef]
- Pavlova, A.; Preisler, B.; Driesen, J.; de Moerloose, P.; Zieger, B.; Hutker, S.; Dengler, K.; Harbrecht, U.; Oldenburg, J. Congenital combined deficiency of coagulation factors VII and X—Different genetic mechanisms. Haemophilia 2015, 21, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Preisler, B.; Pezeshkpoor, B.; Banchev, A.; Fischer, R.; Zieger, B.; Scholz, U.; Rühl, H.; Kemkes-Matthes, B.; Schmitt, U.; Redlich, A.; et al. Familial Multiple Coagulation Factor Deficiencies (FMCFDs) in a Large Cohort of Patients-A Single-Center Experience in Genetic Diagnosis. J. Clin. Med. 2021, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Giansily-Blaizot, M.; Rallapalli, P.M.; Perkins, S.J.; Kemball-Cook, G.; Hampshire, D.J.; Gomez, K.; Ludlam, C.A.; McVey, J.H. The EAHAD blood coagulation factor VII variant database. Hum. Mutat. 2020, 41, 1209–1219. [Google Scholar] [CrossRef]
- Herrmann, F.H.; Wulff, K.; Auerswald, G.; Schulman, S.; Astermark, J.; Batorova, A.; Kreuz, W.; Pollmann, H.; Ruiz-Saez, A.; de Bosch, N.; et al. Factor VII deficiency: Clinical manifestation of 717 subjects from Europe and Latin America with mutations in the factor 7 gene. Haemophilia 2009, 15, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Millar, D.S.; Kemball-Cook, G.; McVey, J.H.; Tuddenham, E.G.; Mumford, A.D.; Attock, G.B.; Reverter, J.C.; Lanir, N.; Parapia, L.A.; Reynaud, J.; et al. Molecular analysis of the genotype-phenotype relationship in factor VII deficiency. Hum. Genet. 2000, 107, 327–342. [Google Scholar] [CrossRef]
- Green, F.; Kelleher, C.; Wilkes, H.; Temple, A.; Meade, T.; Humphries, S. A common genetic polymorphism associated with lower coagulation factor VII levels in healthy individuals. Arterioscler. Thromb. 1991, 11, 540–546. [Google Scholar] [CrossRef]
- Bernardi, F.; Marchetti, G.; Pinotti, M.; Arcieri, P.; Baroncini, C.; Papacchini, M.; Zepponi, E.; Ursicino, N.; Chiarotti, F.; Mariani, G. Factor VII Gene Polymorphisms Contribute About One Third of the Factor VII Level Variation in Plasma. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 72–76. [Google Scholar] [CrossRef]
- Hunault, M.; Arbini, A.A.; Lopaciuk, S.; Carew, J.A.; Bauer, K.A. The Arg353Gln polymorphism reduces the level of coagulation factor VII. In vivo and in vitro studies. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2825–2829. [Google Scholar] [CrossRef]
- Ferraresi, P.; Balestra, D.; Guittard, C.; Buthiau, D.; Pan-Petesh, B.; Maestri, I.; Farah, R.; Pinotti, M.; Giansily-Blaizot, M. Next-generation sequencing and recombinant expression characterized aberrant splicing mechanisms and provided correction strategies in factor VII deficiency. Haematologica 2020, 105, 829–837. [Google Scholar] [CrossRef]
- McVey, J.H.; Boswell, E.; Mumford, A.D.; Kemball-Cook, G.; Tuddenham, E.G. Factor VII deficiency and the FVII mutation database. Hum. Mutat. 2001, 17, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Sevenet, P.-O.; Kaczor, D.A.; Depasse, F. Factor VII Deficiency: From Basics to Clinical Laboratory Diagnosis and Patient Management. Clin. Appl. Thromb. Hemost. 2017, 23, 703–710. [Google Scholar] [CrossRef]
- Fromovich-Amit, Y.; Zivelin, A.; Rosenberg, N.; Tamary, H.; Landau, M.; Seligsohn, U. Characterization of mutations causing factor VII deficiency in 61 unrelated Israeli patients. J. Thromb. Haemost. 2004, 2, 1774–1781. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, G.; Riccardi, F.; Rivolta, G.F.; Martorana, D.; Di Perna, C.; Percesepe, A.; Tagliaferri, A. F7 gene variants modulate protein levels in a large cohort of patients with factor VII deficiency. Results from a genotype-phenotype study. Thromb. Haemost. 2017, 117, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Rabelo, F.Y.; Jardim, L.L.; Landau, M.B.; Gadelha, T.; Corrêa, M.F.B.; Pereira, I.F.M.; Rezende, S.M. The molecular basis of low activity levels of coagulation factor VII: A Brazilian cohort. Haemophilia 2015, 21, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Elmahmoudi, H.; Ben-lakhal, F.; Elborji, W.; Jlizi, A.; Zahra, K.; Sassi, R.; Zorgan, M.; Meddeb, B.; Elgaaied Ben Ammar, A.; Gouider, E. Identification of genetic defects underlying FVII deficiency in 10 patients belonging to eight unrelated families of the North provinces from Tunisia. Diagn. Pathol. 2012, 7, 92. [Google Scholar] [CrossRef]
- Peyvandi, F.; Duga, S.; Akhavan, S.; Mannucci, P.M. Rare coagulation deficiencies. Haemophilia 2002, 8, 308–321. [Google Scholar] [CrossRef]
- Peyvandi, F.; Mannucci, P.M.; Asti, D.; Abdoullahi, M.; Di Rocco, N.; Sharifian, R. Clinical manifestations in 28 Italian and Iranian patients with severe factor VII deficiency. Haemophilia 1997, 3, 242–246. [Google Scholar] [CrossRef]
- Peyvandi, F.; Menegatti, M.; Palla, R. Rare Bleeding Disorders: Worldwide Efforts for Classification, Diagnosis, and Management. Semin. Thromb. Hemost. 2013, 39, 579–584. [Google Scholar] [CrossRef]
- Caspers, M.; Pavlova, A.; Driesen, J.; Harbrecht, U.; Klamroth, R.; Kadar, J.; Fischer, R.; Kemkes-Matthes, B.; Oldenburg, J. Deficiencies of antithrombin, protein C and protein S—Practical experience in genetic analysis of a large patient cohort. Thromb. Haemost. 2012, 108, 247–257. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.B.; Hay, C.R.M.; Shanks, D.; Mitchell, M.J.; McVey, J.H. The importance of tissue factor source in the management of Factor VII deficiency. Thromb. Haemost. 2007, 97, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Halimeh, S.; Koch, L.; Kenet, G.; Kuta, P.; Rahmfeld, T.; Stoll, M.; Nowak-Göttl, U. Genotype-Phenotype Relationship among 785 Unrelated White Women with Inherited Congenital Factor VII Deficiency: A Three-Center Database Study. J. Clin. Med. 2023, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Goodeve, A.C.; Reitsma, P.H.; McVey, J.H. Nomenclature of genetic variants in hemostasis. J. Thromb. Haemost. 2011, 9, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nr. | Exon/Intron | HGVS (Nucleotide Position) | HGVS (Protein Position) | Type of Variant | Classification | FVII:C (IU/dl) |
|---|---|---|---|---|---|---|
| 1 | Promotor | c.-4C>A | - | regulatory | VUS | 31 |
| 2 | 1 | c.44T>C | p.Leu15Pro ++ | missense | likely pathogenic | 37 |
| 3 | 1 | c.48_64dup17ins | p.Gly17Alafs | dup/ins | likely pathogenic | 38 |
| 4 | 1 | c.65G>A | p.Gly22Asp | missense | VUS | 43 |
| 5 | 3 | c.143A>T | p.Gln48Leu + | missense | likely pathogenic | 20 |
| 6 | 3 | c.218T>C # | p.Leu73Pro | missense | VUS | 1 |
| 7 | 3 | c.220dupG | p.Glu74Glyfs | duplication | likely pathogenic | 35 |
| 8 | 3 | c.281_291del10bp | p.Ala94Glyfs | deletion | likely pathogenic | 40 |
| 9 | 4 | c.316+1G>A | - | splice-site | likely pathogenic | 43 |
| 10 | 5 | c.343T>G | p.Cys115Gly + | missense | likely pathogenic | 30 |
| 11 | 5 | c.408C>G | p.Phe136Leu | missense | VUS | 33 |
| 12 | 5 | c.412G>C | p.Gly138Arg | missense | VUS | 35 |
| 13 | 6 | c.430C>G | p.His144Asp | missense | VUS | 43 |
| 14 | 6 | c.512C>T § | p.Ser171Phe ++ | missense | likely pathogenic | 1 |
| 15 | 6 | c.514T>C | p.Cys172Arg | missense | VUS | 32 |
| 16 | 6 | c.526G>A | p.Glu176Lys | missense | VUS | 48 |
| 17 | 6 | c.538_539delCT | p.Leu180Alafs | deletion | likely pathogenic | 54 |
| 18 | 6 | c.557C>T | p.Ser186Phe | missense | VUS | 29 |
| 19 | 6 | c.565C>T | p.Pro189Ser ++++ | missense | likely pathogenic | 50 |
| 20 | 6 | c.548A>T | p.Asp183Val | missense | VUS | 28 |
| 21 | 7 | c.587G>C | p.Gly196Ala | missense | VUS | 33 |
| 22 | 7 | c.665G>A | p.Gly222Glu | missense | VUS | 30 |
| 23 | 7 | c.667G>T | p.Glu223Ter | nonsense | likely pathogenic | 57 |
| 24 | 7 | c.676T>G | p.Trp226Gly | missense | VUS | 36 |
| 25 | 7 | c.681+1G>T | - | splice-site | likely pathogenic | 52 |
| 26 | 8 | c.691_693delTTG # | p.Leu231del | deletion | likely pathogenic | 8 |
| 27 | 8 | c.718G>T | p.Gly240Trp + | missense | likely pathogenic | 30 |
| 28 | 8 | c.728T>C | p.Ile243Thr | missense | VUS | 29 |
| 29 | 8 | c.739T>C | p.Trp247Arg | missense | VUS | 26 |
| 30 | 8 | c.806-3C>G | - | splice-site | likely pathogenic | 54 |
| 31 | 9 | c.808G>A | p.Glu270Lys | missense | VUS | 48 |
| 32 | 9 | c.823G>A | p.Glu275Lys | missense | VUS | 41 |
| 33 | 9 | c.843G>T # | p.Gln281His | missense | VUS | 1 |
| 34 | 9 | c.903C>G | p.His301Gln +++ | missense | likely pathogenic | 59 |
| 35 | 9 | c.904G>A | p.Asp302Asn | missense | VUS | 30 |
| 36 | 9 | c.911C>T | p.Ala304Val | missense | VUS | 30 |
| 37 | 9 | c.930G>T | p.Gln310His ++ | missense | likely pathogenic | 43 |
| 38 | 9 | c.944C>T | p.Thr315Ile +++ | missense | likely pathogenic | 19 |
| 39 | 9 | c.955G>A | p.Val319Met | missense | VUS | 50 |
| 40 | 9 | c.977G>A | p.Arg326Gln | missense | VUS | 37 |
| 41 | 9 | c.1160T>C | p.Met387hr | missense | VUS | 42 |
| 42 | 9 | c.1168G>A | p.Ala390Thr | missense | VUS | 43 |
| 43 | 9 | c.1078C>T | p.Leu360Phe | missense | VUS | 64 |
| 44 | 9 | c.1089_1090delCC | p. Arg364Alafs | deletion | likely pathogenic | 19 |
| 45 | 9 | c.1235A>G | p.Tyr412Cys ++ | missense | likely pathogenic | 43 |
| 46 | 9 | c.1250A>G | p.Tyr417Cys | missense | VUS | 45 |
| 47 | 9 | c.1274G>C | p.Gly425Ala | missense | VUS | 40 |
| 48 | 9 | c.1262T>C | p.Ile421Thr | missense | VUS | 45 |
| 49 | 9 | c.1306G>A | p.Val436Met | missense | VUS | 47 |
| 50 | 9 | c.1313C>A # | p.Thr438Asn | missense | VUS | 8 |
| 51 | 9 | c.1316G>C # | p.Arg439Thr | missense | VUS | 2 |
| 52 | 9 | c.1329C>G | p.Tyr443Ter | nonsense | likely pathogenic | 14 |
| 53 | 9 | c.1354C>T | p.Arg452Cys | missense | VUS | 37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preisler, B.; Pezeshkpoor, B.; Merzenich, A.; Ohlenforst, S.; Rühl, H.; Ivaškevičius, V.; Scholz, U.; Bönigk, H.; Eberl, W.; Zieger, B.; et al. Genetic Landscape of Factor VII Deficiency: Insights from a Comprehensive Analysis of Pathogenic Variants and Their Impact on Coagulation Activity. Int. J. Mol. Sci. 2024, 25, 2384. https://doi.org/10.3390/ijms25042384
Preisler B, Pezeshkpoor B, Merzenich A, Ohlenforst S, Rühl H, Ivaškevičius V, Scholz U, Bönigk H, Eberl W, Zieger B, et al. Genetic Landscape of Factor VII Deficiency: Insights from a Comprehensive Analysis of Pathogenic Variants and Their Impact on Coagulation Activity. International Journal of Molecular Sciences. 2024; 25(4):2384. https://doi.org/10.3390/ijms25042384
Chicago/Turabian StylePreisler, Barbara, Behnaz Pezeshkpoor, Anja Merzenich, Sandra Ohlenforst, Heiko Rühl, Vytautas Ivaškevičius, Ute Scholz, Hagen Bönigk, Wolfgang Eberl, Barbara Zieger, and et al. 2024. "Genetic Landscape of Factor VII Deficiency: Insights from a Comprehensive Analysis of Pathogenic Variants and Their Impact on Coagulation Activity" International Journal of Molecular Sciences 25, no. 4: 2384. https://doi.org/10.3390/ijms25042384
APA StylePreisler, B., Pezeshkpoor, B., Merzenich, A., Ohlenforst, S., Rühl, H., Ivaškevičius, V., Scholz, U., Bönigk, H., Eberl, W., Zieger, B., Pavlova, A., & Oldenburg, J. (2024). Genetic Landscape of Factor VII Deficiency: Insights from a Comprehensive Analysis of Pathogenic Variants and Their Impact on Coagulation Activity. International Journal of Molecular Sciences, 25(4), 2384. https://doi.org/10.3390/ijms25042384