

Double Heterozygosity for Rare Deleterious Variants in the BRCA1 and BRCA2 Genes in a Hungarian Patient with Breast Cancer

 , , and
, , and 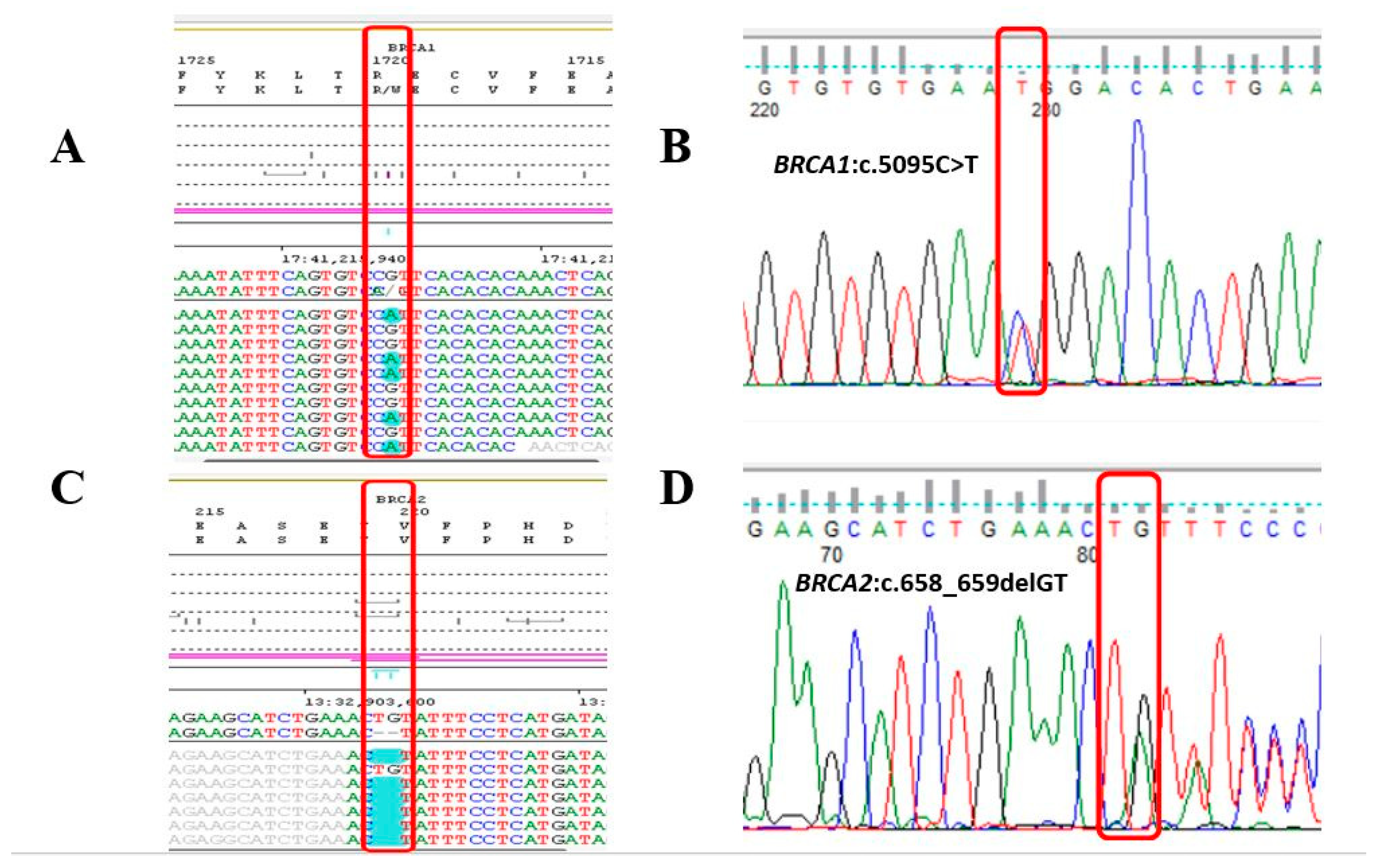{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Case History
2.2. Molecular Genetic Testing of BRCA1 and BRCA2 Genes
2.3. Cascade Screening
3. Discussion
4. Methods
Molecular Genetic Methods and Data Analysis
- Molecular genetic tests were performed on genomic DNA samples isolated from peripheral blood leukocytes using QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Coding regions and exon/intron boundaries of BRCA1 and BRCA2 genes were analyzed using Devyser BRCA (Devyser, Hägersten, Sweden) Next Generation DNA library preparation kit. CNV analysis was also performed based on coverage data. Bidirectional DNA sequencing was performed using Illumina MiSeq sequencer (San Diego, CA, USA).
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Paul, A.; Paul, S. The Breast Cancer Susceptibility Genes (BRCA) in Breast and Ovarian Cancers. Front. Biosci. 2014, 19, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R. Hereditary Breast and Ovarian Cancer (HBOC): Review of Its Molecular Characteristics, Screening, Treatment, and Prognosis. Breast Cancer 2021, 28, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Le, H.P.; Heyer, W.-D.; Liu, J. Guardians of the Genome: BRCA2 and Its Partners. Genes 2021, 12, 1229. [Google Scholar] [CrossRef] [PubMed]
- Krammer, J.; Pinker-Domenig, K.; Robson, M.E.; Gonen, M.; Bernard-Davila, B.; Morris, E.A.; Mangino, D.A.; Jochelson, M.S. Breast Cancer Detection and Tumor Characteristics in BRCA1 and BRCA2 Mutation Carriers. Breast Cancer Res. Treat. 2017, 163, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Caliendo, G.; D’Elia, G.; Resse, M.; Casamassimi, A.; Minucci, P.B.; Dello Ioio, C.; Cioffi, M.; Molinari, A.M. Five Italian Families with Two Mutations in BRCA Genes. Genes 2020, 11, 1451. [Google Scholar] [CrossRef]
- Leegte, B.; van der Hout, A.H.; Deffenbaugh, A.M.; Bakker, M.K.; Mulder, I.M.; ten Berge, A.; Leenders, E.P.; Wesseling, J.; de Hullu, J.; Hoogerbrugge, N.; et al. Phenotypic Expression of Double Heterozygosity for BRCA1 and BRCA2 Germline Mutations. J. Med. Genet. 2005, 42, e20. [Google Scholar] [CrossRef]
- Lavie, O.; Narod, S.; Lejbkowicz, F.; Dishon, S.; Goldberg, Y.; Gemer, O.; Rennert, G. Double Heterozygosity in the BRCA1 and BRCA2 Genes in the Jewish Population. Ann. Oncol. 2011, 22, 964–966. [Google Scholar] [CrossRef]
- Ramus, S.J.; Friedman, L.S.; Gayther, S.A.; Ponder, B.A.J.; Bobrow, L.G.; van der Looji, M.; Papp, J.; Olah, E. A Breast/Ovarian Cancer Patient with Germline Mutations in Both BRCA1 and BRCA2. Nat. Genet. 1997, 15, 14–15. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Friebel, T.M.; Mitra, N.; Wan, F.; Chen, S.; Andrulis, I.L.; Apostolou, P.; Arnold, N.; Arun, B.K.; Barrowdale, D.; et al. Inheritance of Deleterious Mutations at Both BRCA1 and BRCA2 in an International Sample of 32,295 Women. Breast Cancer Res. 2016, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Alhuqail, A.-J.; Alzahrani, A.; Almubarak, H.; Al-Qadheeb, S.; Alghofaili, L.; Almoghrabi, N.; Alhussaini, H.; Park, B.H.; Colak, D.; Karakas, B. High Prevalence of Deleterious BRCA1 and BRCA2 Germline Mutations in Arab Breast and Ovarian Cancer Patients. Breast Cancer Res. Treat. 2018, 168, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Laraqui, A.; Uhrhammer, N.; Lahlou-Amine, I.; EL Rhaffouli, H.; El Baghdadi, J.; Dehayni, M.; Moussaoui, R.D.; Ichou, M.; Sbitti, Y.; Al Bouzidi, A.; et al. Mutation Screening of the BRCA1 Gene in Early Onset and Familial Breast/Ovarian Cancer in Moroccan Population. Int. J. Med. Sci. 2012, 10, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Kluska, A.; Balabas, A.; Paziewska, A.; Kulecka, M.; Nowakowska, D.; Mikula, M.; Ostrowski, J. New Recurrent BRCA1/2 Mutations in Polish Patients with Familial Breast/Ovarian Cancer Detected by next Generation Sequencing. BMC Med. Genom. 2015, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Tian, L.; Kähkönen, M.; Schwartzentruber, J.; Kircher, M.; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium; Majewski, J.; Dyment, D.A.; Innes, A.M.; et al. Biallelic Mutations in BRCA1 Cause a New Fanconi Anemia Subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef]
- Vallon-Christersson, J.; Cayanan, C.; Haraldsson, K.; Loman, N.; Bergthorsson, J.T.; Brøndum-Nielsen, K.; Gerdes, A.-M.; Møller, P.; Kristoffersson, U.; Olsson, H.; et al. Functional analysis of BRCA1 C-terminal missense mutations identified in breast and ovarian cancer families. Hum. Mol. Genet. 2001, 10, 353–360. [Google Scholar] [CrossRef]
- Coquelle, N.; Green, R.; Glover, J.N.M. Impact of BRCA1 BRCT Domain Missense Substitutions on Phosphopeptide Recognition. Biochemistry 2011, 50, 4579–4589. [Google Scholar] [CrossRef]
- Jakubowska, A.; Scott, R.; Menkiszak, J.; Gronwald, J.; Byrski, T.; Huzarski, T.; Górski, B.; Cybulski, C.; Debniak, T.; Kowalska, E.; et al. A High Frequency of BRCA2 Gene Mutations in Polish Families with Ovarian and Stomach Cancer. Eur. J. Hum. Genet. 2003, 11, 955–958. [Google Scholar] [CrossRef]
- Laitman, Y.; Friebel, T.M.; Yannoukakos, D.; Fostira, F.; Konstantopoulou, I.; Figlioli, G.; Bonanni, B.; Manoukian, S.; Zuradelli, M.; Tondini, C.; et al. The Spectrum of BRCA1 and BRCA2 Pathogenic Sequence Variants in Middle Eastern, North African, and South European Countries. Hum. Mutat. 2019, 40, e1–e23. [Google Scholar] [CrossRef]
- Miguel, I.; Rodrigues, F.; Fragoso, S.; Freixo, J.; Clara, A.; Luís, A.; Bento, S.; Fernandes, M.; Bacelar, F.; Câmara, S.; et al. Hereditary Breast Cancer and Ancestry in the Madeira Archipelago: An Exploratory Study. Ecancermedicalscience 2021, 15, 1261. [Google Scholar] [CrossRef]
- Miele, E.; Mastronuzzi, A.; Po, A.; Carai, A.; Alfano, V.; Serra, A.; Colafati, G.S.; Strocchio, L.; Antonelli, M.; Buttarelli, F.R.; et al. Characterization of Medulloblastoma in Fanconi Anemia: A Novel Mutation in the BRCA2 Gene and SHH Molecular Subgroup. Biomark. Res. 2015, 3, 13. [Google Scholar] [CrossRef]
- Heidemann, S.; Fischer, C.; Engel, C.; Fischer, B.; Harder, L.; Schlegelberger, B.; Niederacher, D.; Goecke, T.O.; Doelken, S.C.; Dikow, N.; et al. Double Heterozygosity for Mutations in BRCA1 and BRCA2 in German Breast Cancer Patients: Implications on Test Strategies and Clinical Management. Breast Cancer Res. Treat. 2012, 134, 1229–1239. [Google Scholar] [CrossRef]
- Brose, M.S.; Rebbeck, T.R.; Calzone, K.A.; Stopfer, J.E.; Nathanson, K.L.; Weber, B.L. Cancer Risk Estimates for BRCA1 Mutation Carriers Identified in a Risk Evaluation Program. J. Natl. Cancer Inst. 2002, 94, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Le Page, C.; Rahimi, K.; Rodrigues, M.; Heinzelmann-Schwarz, V.; Recio, N.; Tommasi, S.; Bataillon, G.; Portelance, L.; Golmard, L.; Meunier, L.; et al. Clinicopathological Features of Women with Epithelial Ovarian Cancer and Double Heterozygosity for BRCA1 and BRCA2: A Systematic Review and Case Report Analysis. Gynecol. Oncol. 2020, 156, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Parmigiani, G. Meta-Analysis of BRCA1 and BRCA2 Penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Golmard, L.; Delnatte, C.; Laugé, A.; Moncoutier, V.; Lefol, C.; Abidallah, K.; Tenreiro, H.; Copigny, F.; Giraudeau, M.; Guy, C.; et al. Breast and Ovarian Cancer Predisposition Due to de Novo BRCA1 and BRCA2 Mutations. Oncogene 2016, 35, 1324–1327. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, I.; Provenzano, M.; Sorino, L.; Rodrigues, M.; Palka, G.; Stuppia, L. A New Case of “de Novo” BRCA1 Mutation in a Patient with Early-onset Breast Cancer. Clin. Case Rep. 2017, 5, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Alhopuro, P.; Vainionpää, R.; Anttonen, A.-K.; Aittomäki, K.; Nevanlinna, H.; Pöyhönen, M. Constitutional Mosaicism for a BRCA2 Mutation as a Cause of Early-Onset Breast Cancer. Fam. Cancer 2020, 19, 307–310. [Google Scholar] [CrossRef]
- Lieberman, S.; Lahad, A.; Tomer, A.; Koka, S.; BenUziyahu, M.; Raz, A.; Levy-Lahad, E. Familial Communication and Cascade Testing among Relatives of BRCA Population Screening Participants. Genet. Med. 2018, 20, 1446–1454. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madar, L.; Majoros, V.; Szűcs, Z.; Nagy, O.; Babicz, T.; Butz, H.; Patócs, A.; Balogh, I.; Koczok, K. Double Heterozygosity for Rare Deleterious Variants in the BRCA1 and BRCA2 Genes in a Hungarian Patient with Breast Cancer. Int. J. Mol. Sci. 2023, 24, 15334. https://doi.org/10.3390/ijms242015334
Madar L, Majoros V, Szűcs Z, Nagy O, Babicz T, Butz H, Patócs A, Balogh I, Koczok K. Double Heterozygosity for Rare Deleterious Variants in the BRCA1 and BRCA2 Genes in a Hungarian Patient with Breast Cancer. International Journal of Molecular Sciences. 2023; 24(20):15334. https://doi.org/10.3390/ijms242015334
Chicago/Turabian StyleMadar, László, Viktória Majoros, Zsuzsanna Szűcs, Orsolya Nagy, Tamás Babicz, Henriett Butz, Attila Patócs, István Balogh, and Katalin Koczok. 2023. "Double Heterozygosity for Rare Deleterious Variants in the BRCA1 and BRCA2 Genes in a Hungarian Patient with Breast Cancer" International Journal of Molecular Sciences 24, no. 20: 15334. https://doi.org/10.3390/ijms242015334
APA StyleMadar, L., Majoros, V., Szűcs, Z., Nagy, O., Babicz, T., Butz, H., Patócs, A., Balogh, I., & Koczok, K. (2023). Double Heterozygosity for Rare Deleterious Variants in the BRCA1 and BRCA2 Genes in a Hungarian Patient with Breast Cancer. International Journal of Molecular Sciences, 24(20), 15334. https://doi.org/10.3390/ijms242015334