
Synthesis and Biological Evaluation of Novel Amino and Amido Substituted Pentacyclic Benzimidazole Derivatives as Antiproliferative Agents

 , , and
, , and
Abstract
1. Introduction
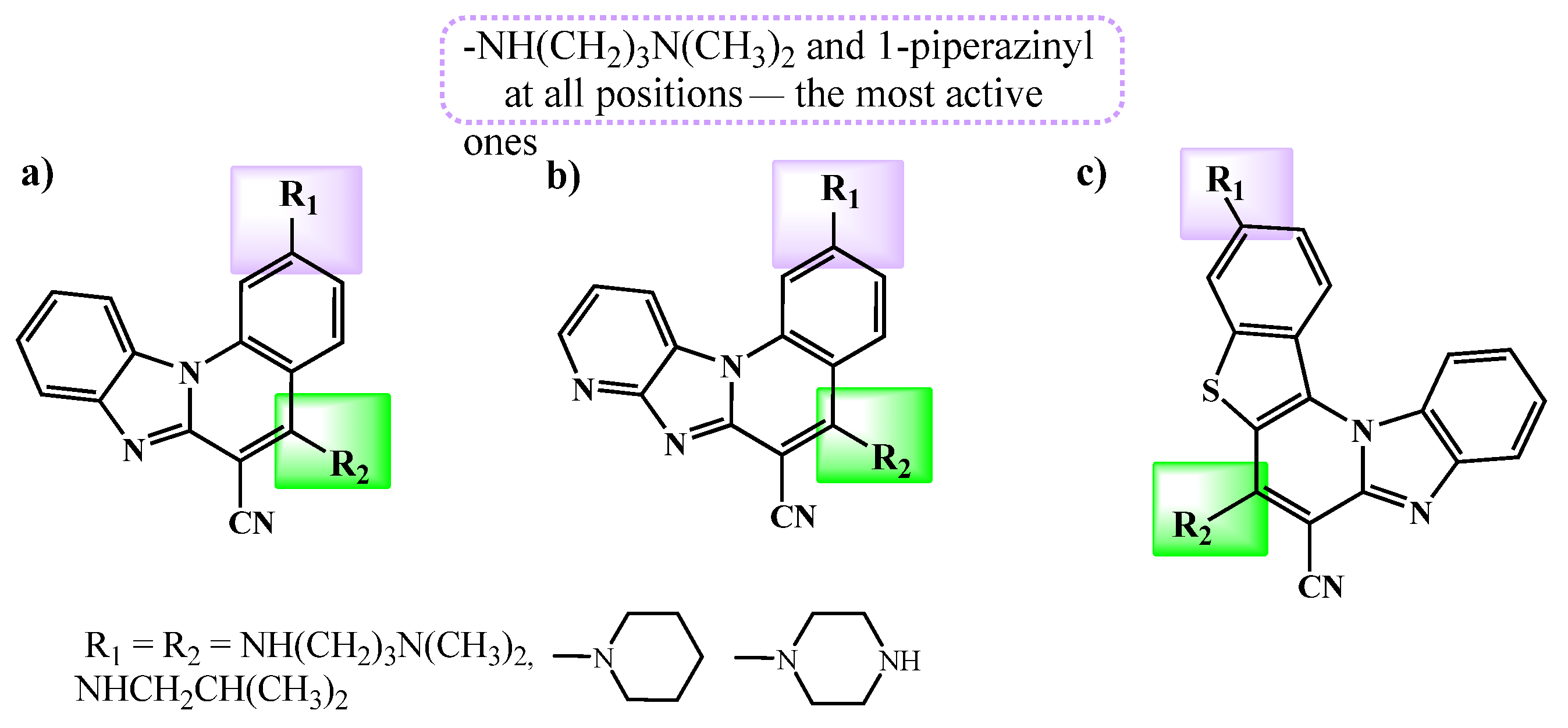
2. Results and Discussion
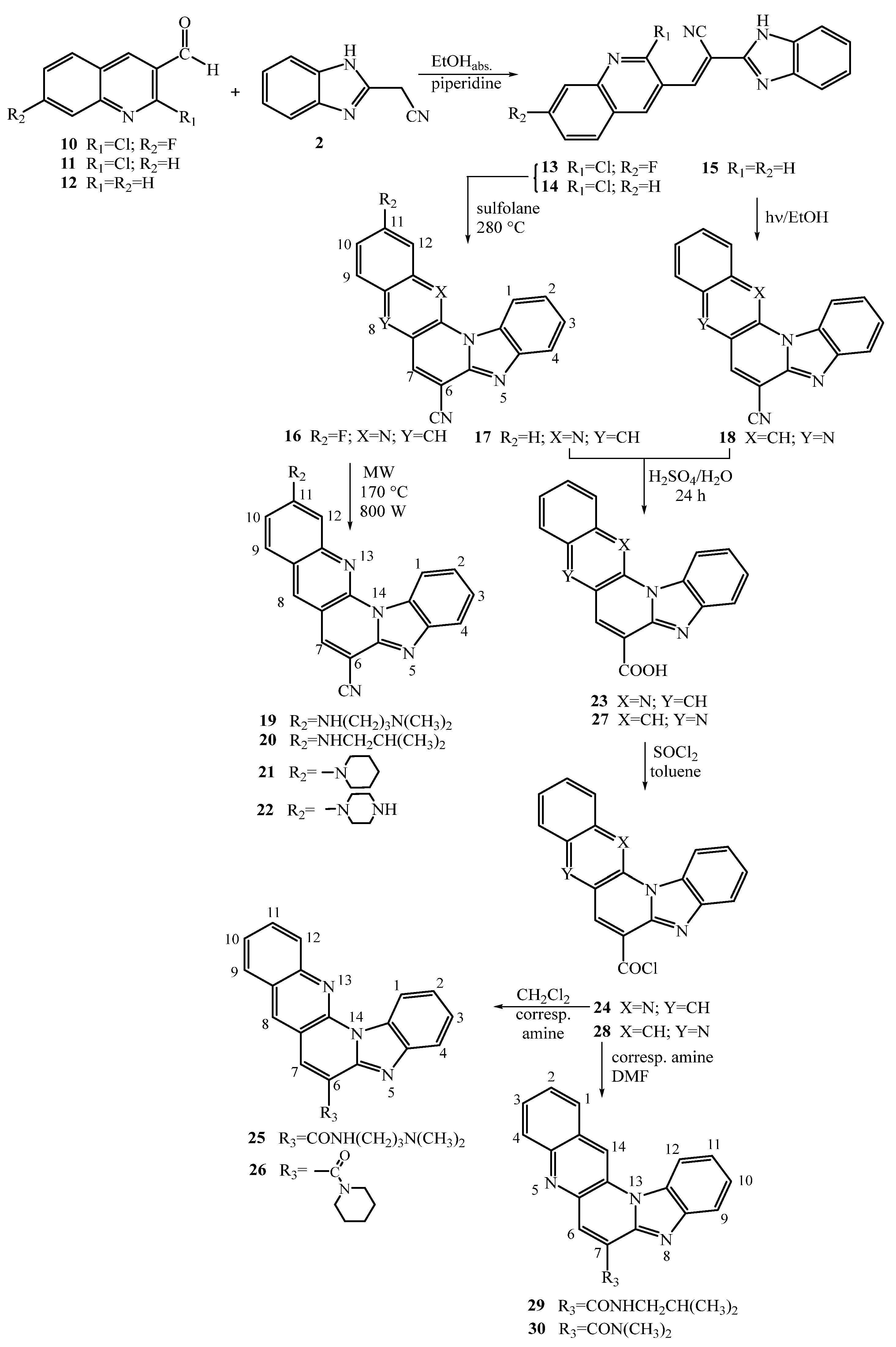
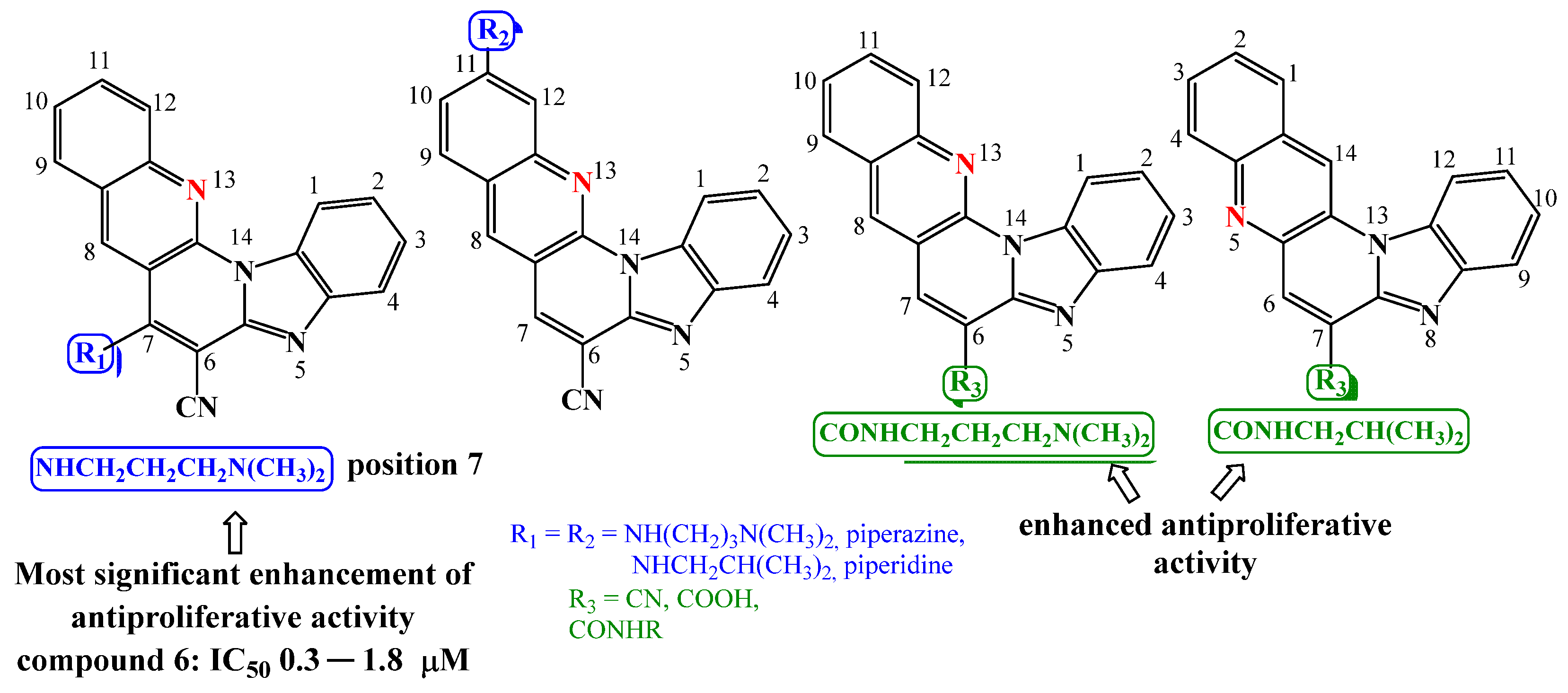
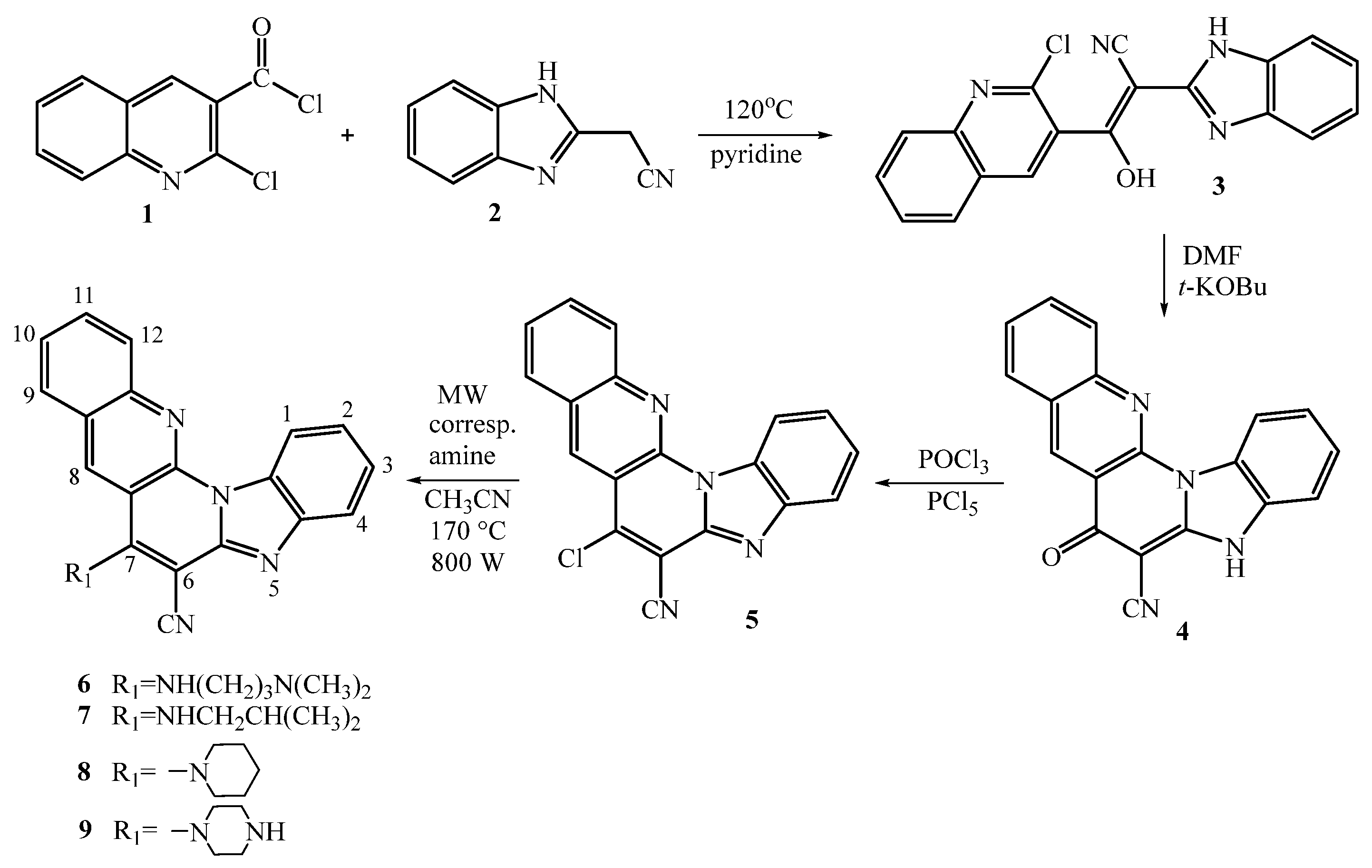
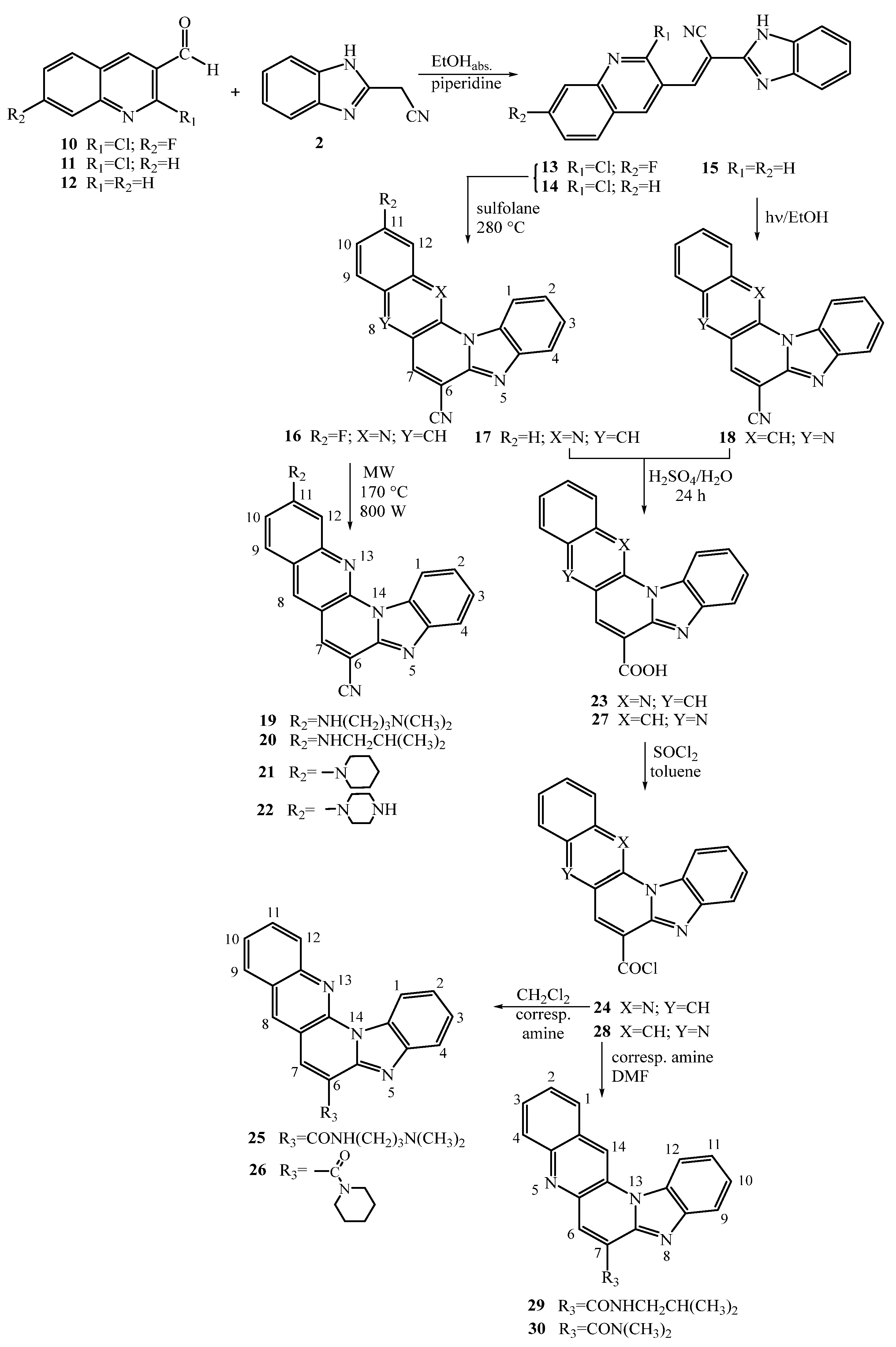
2.1. Chemistry
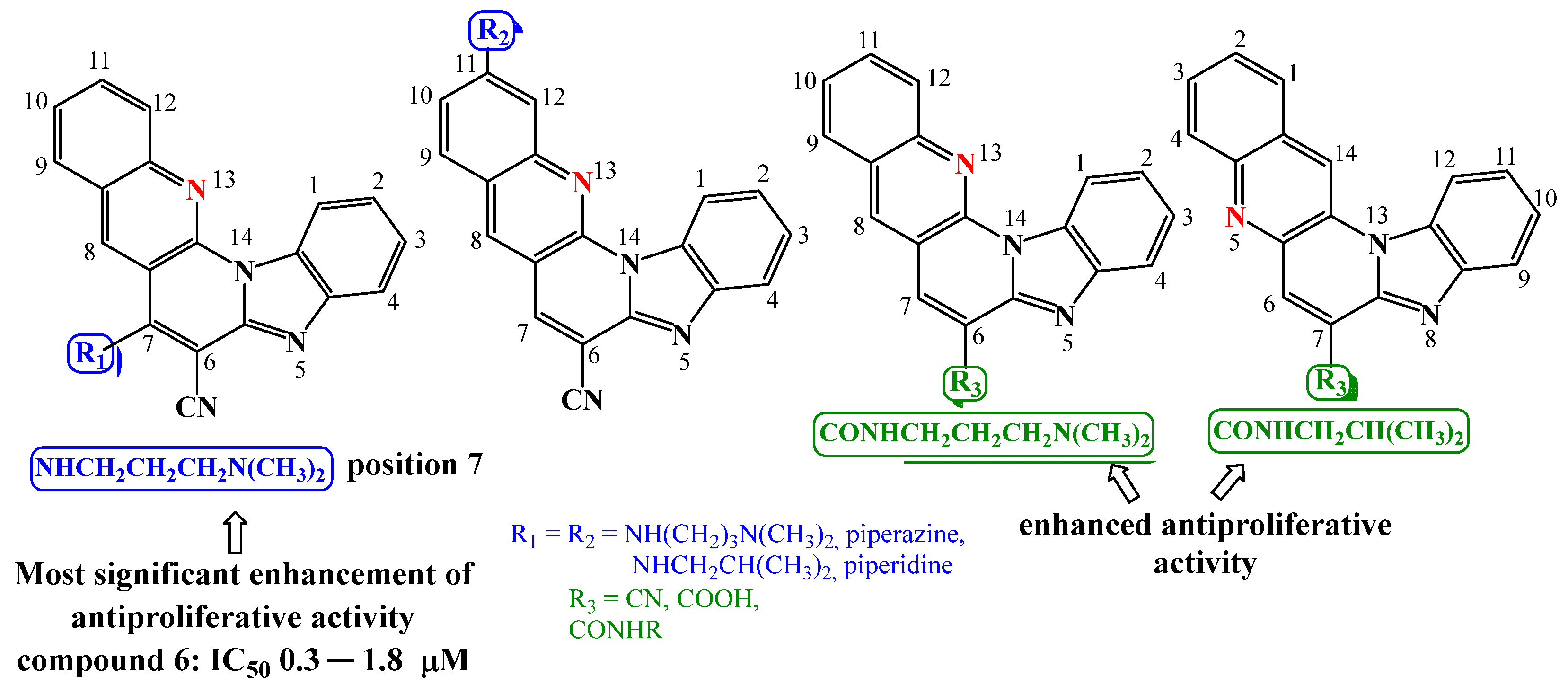
2.2. Biological Activity
2.2.1. Antiproliferative Activity In Vitro
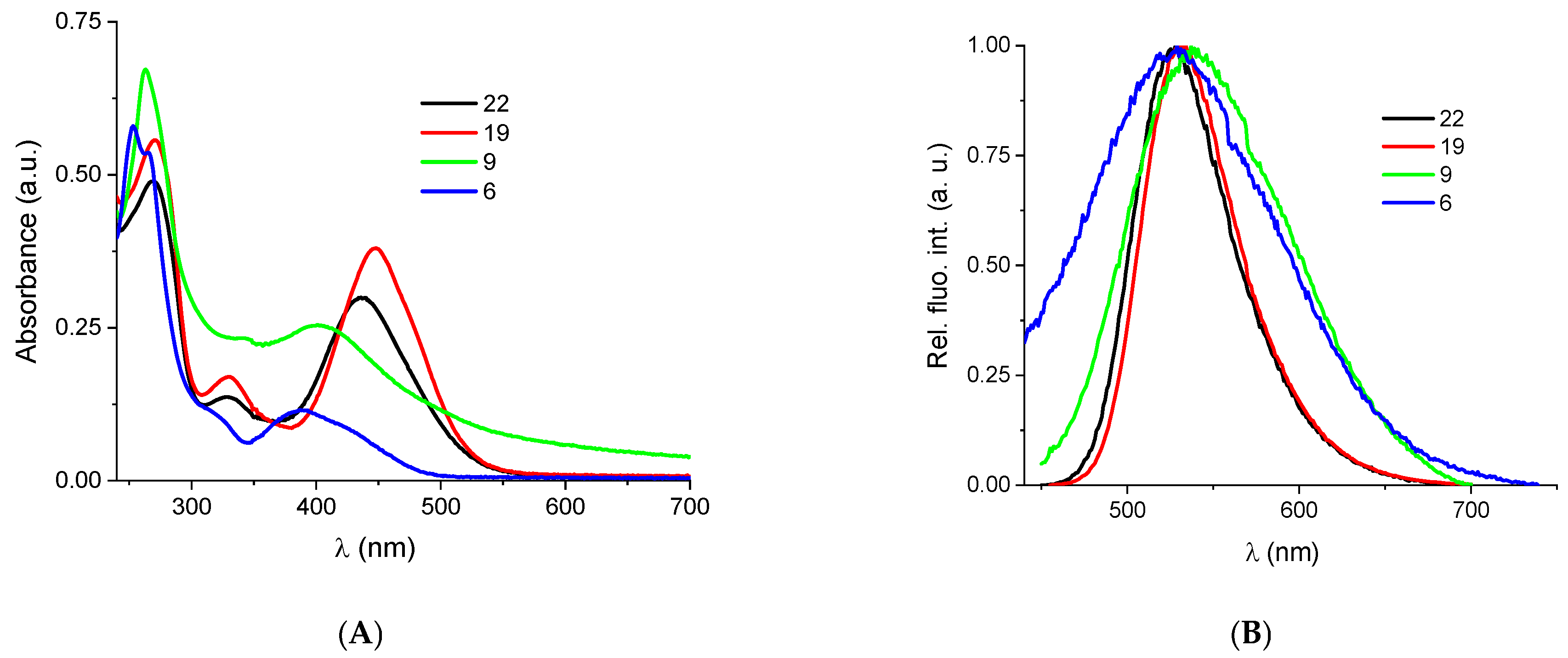
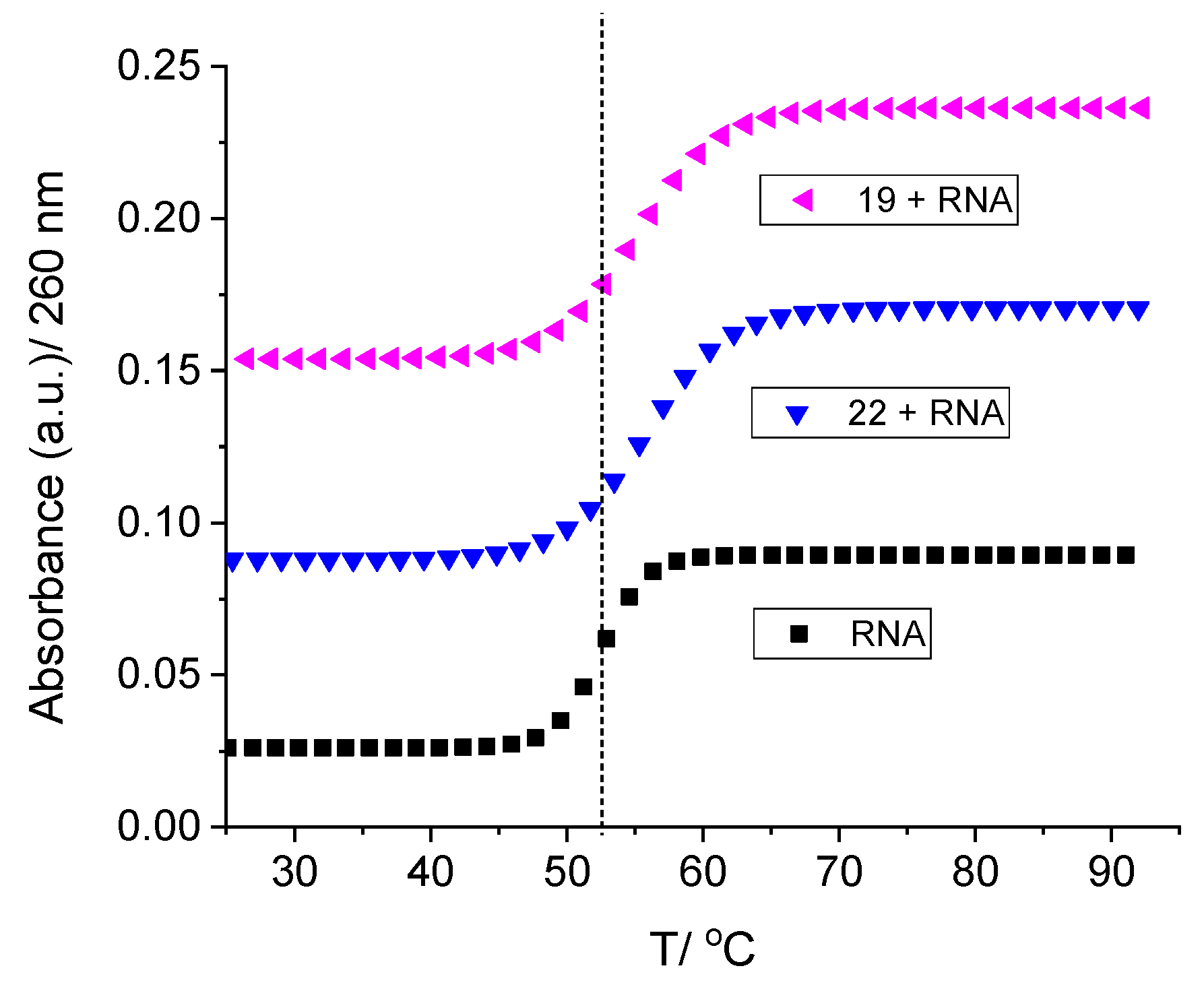
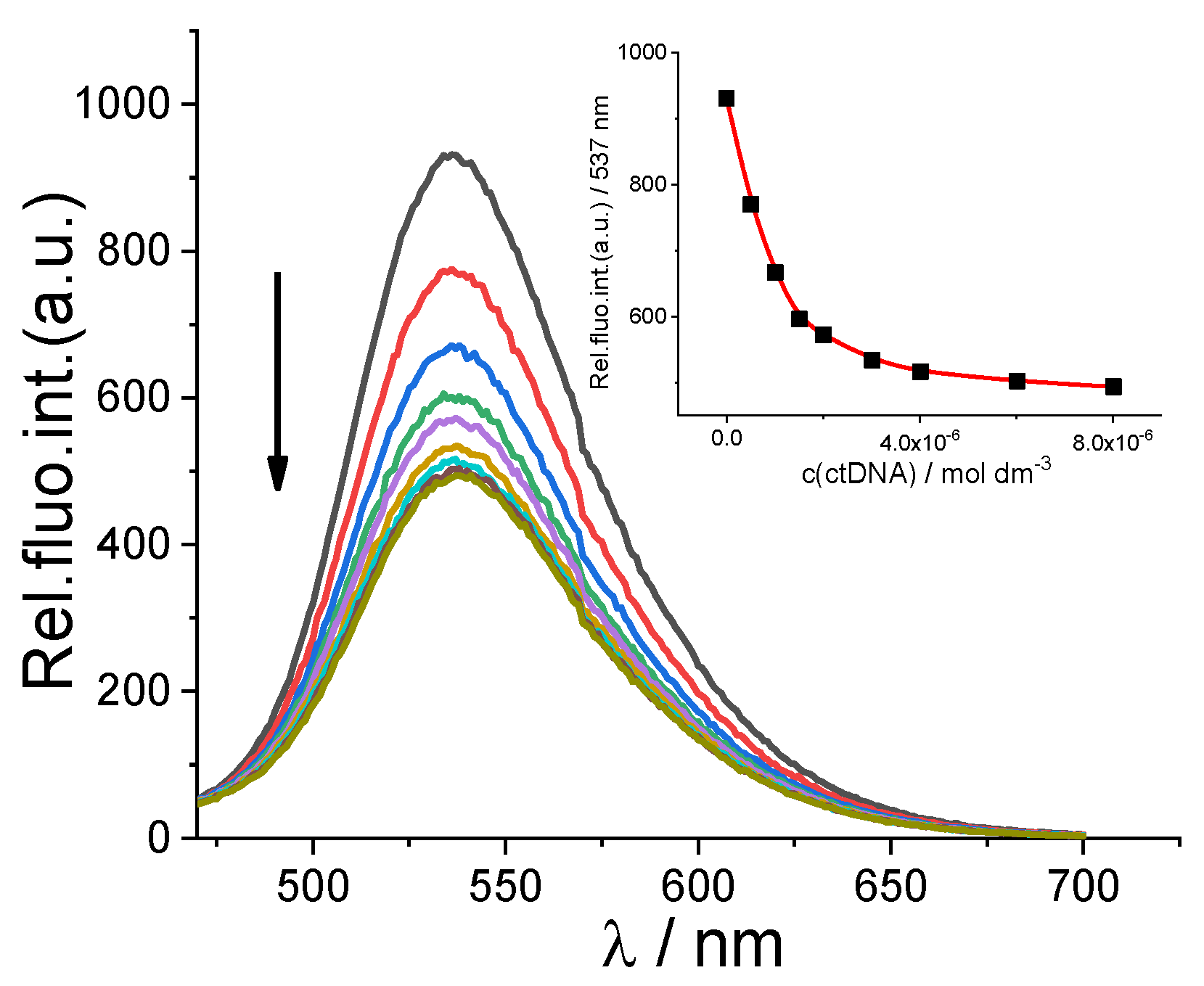
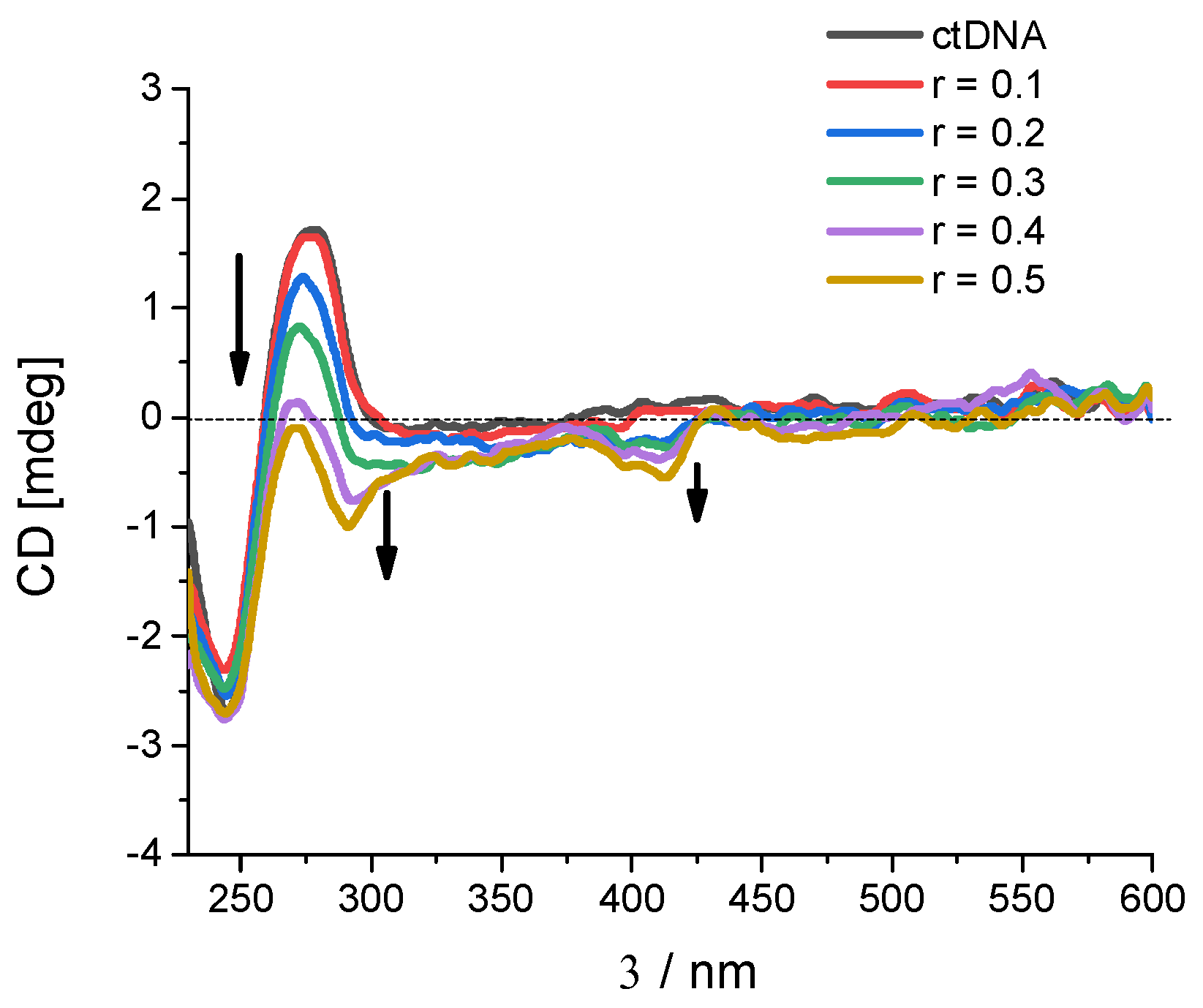
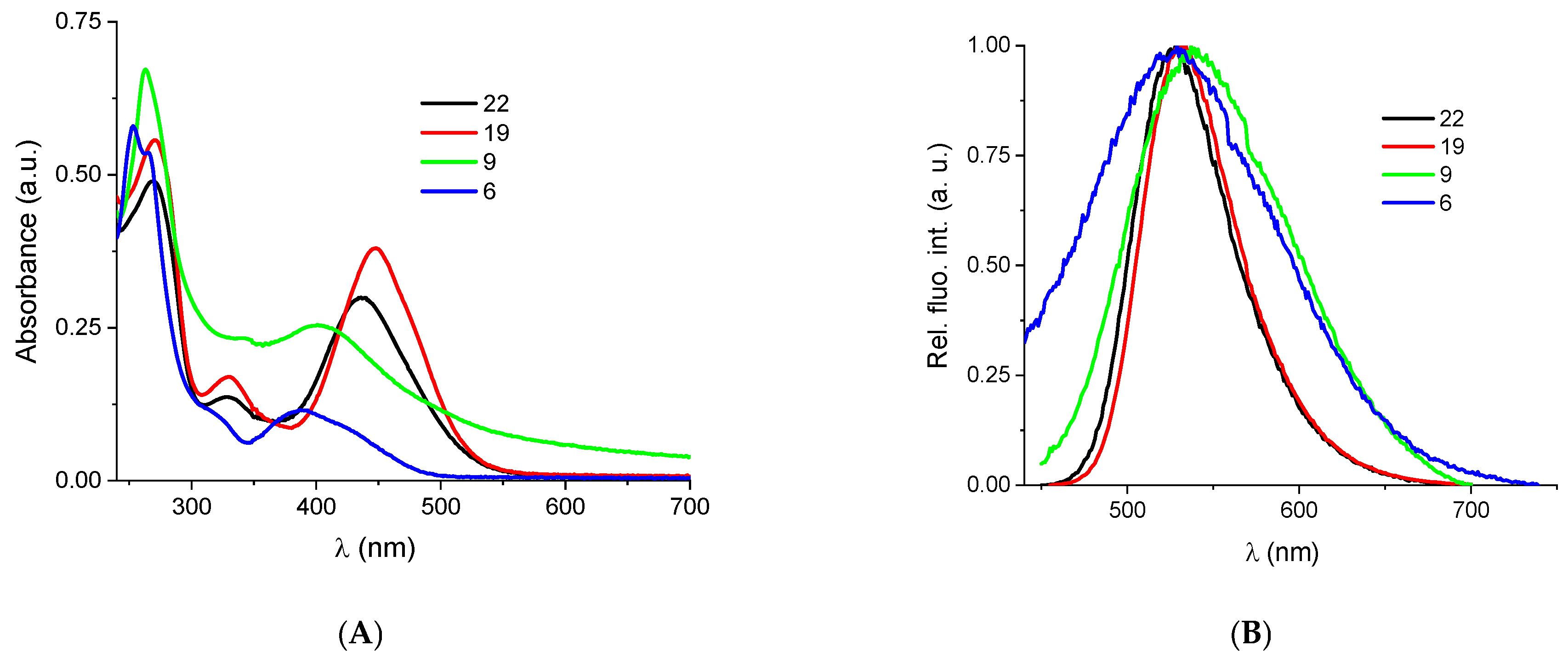
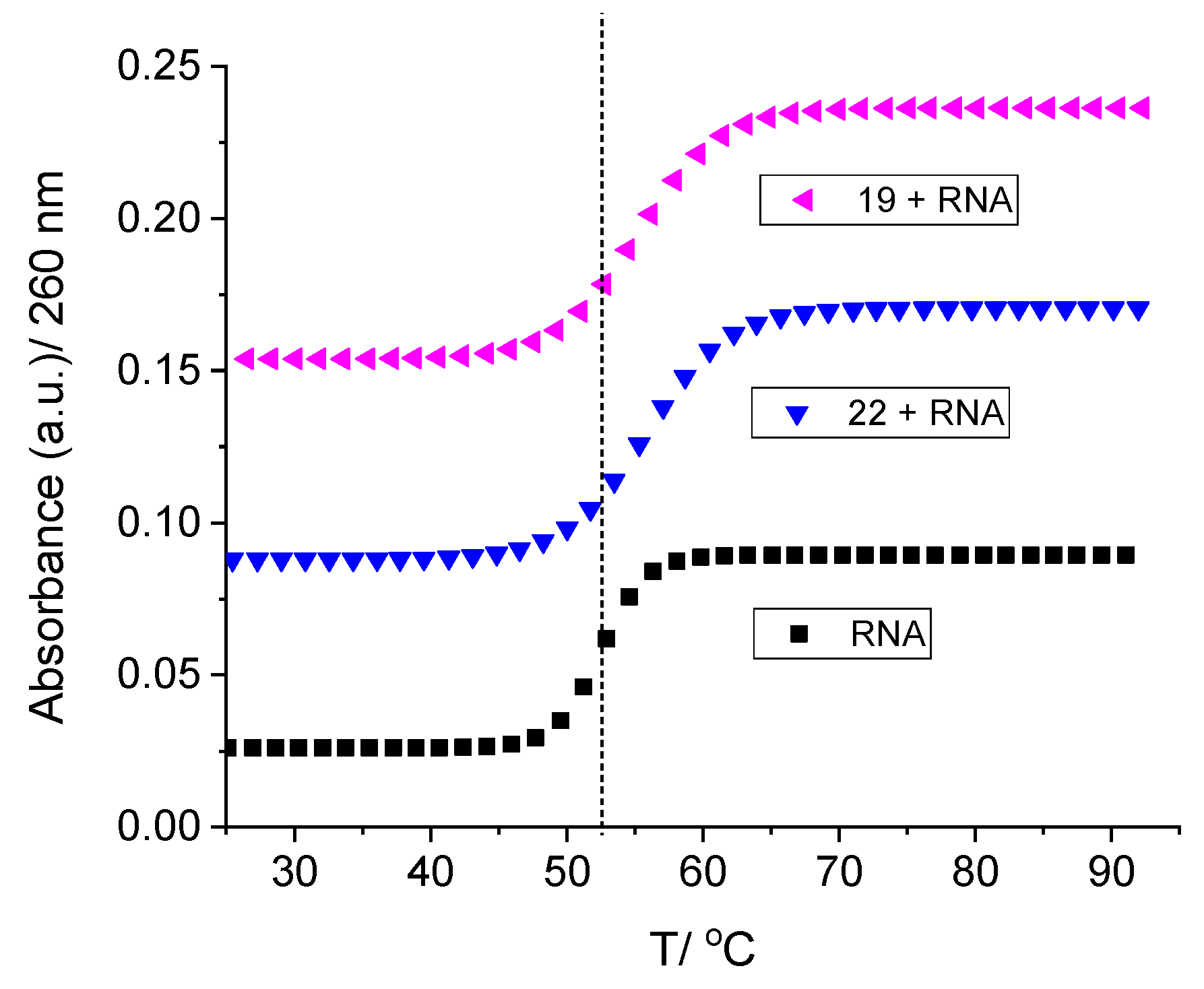
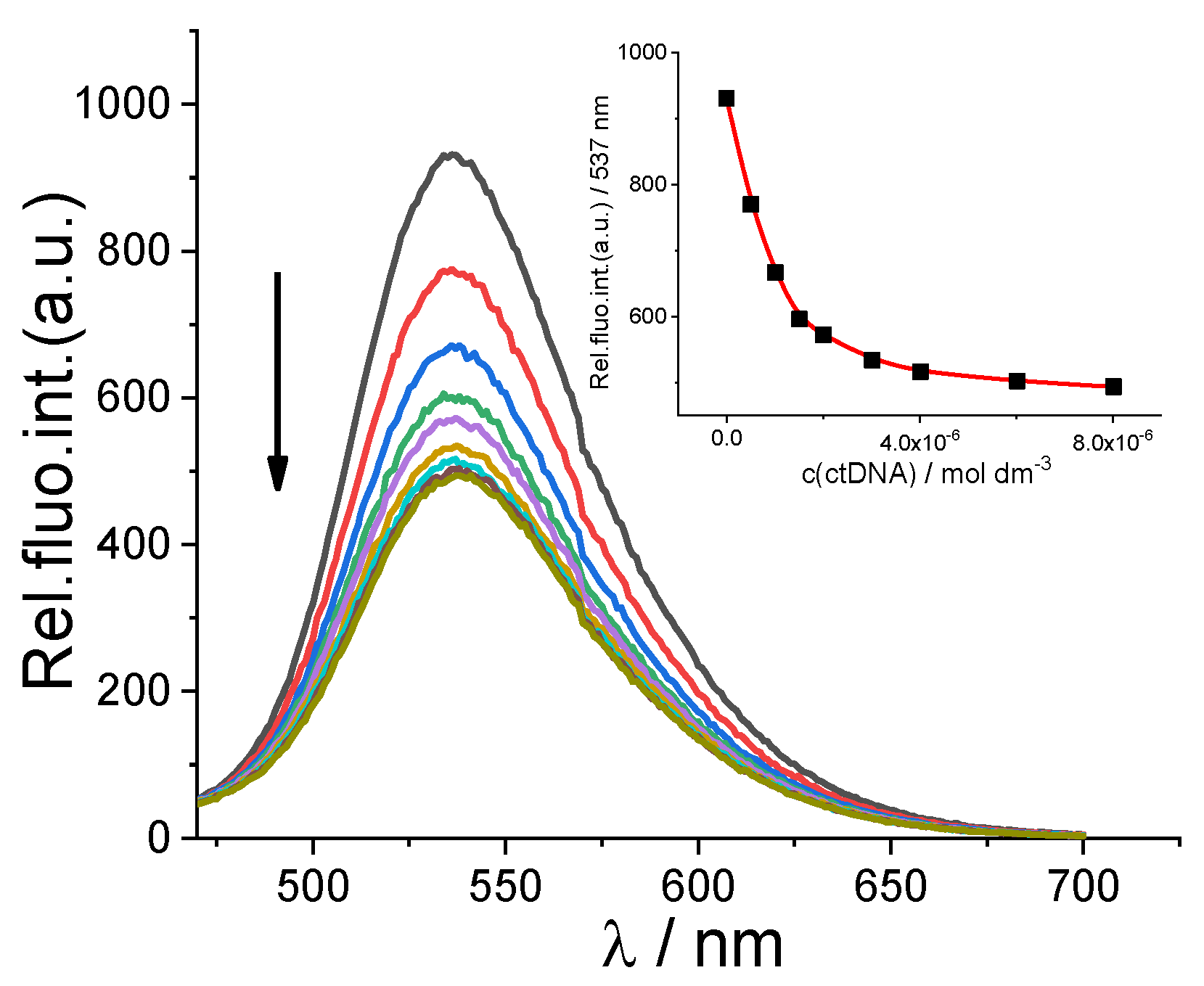
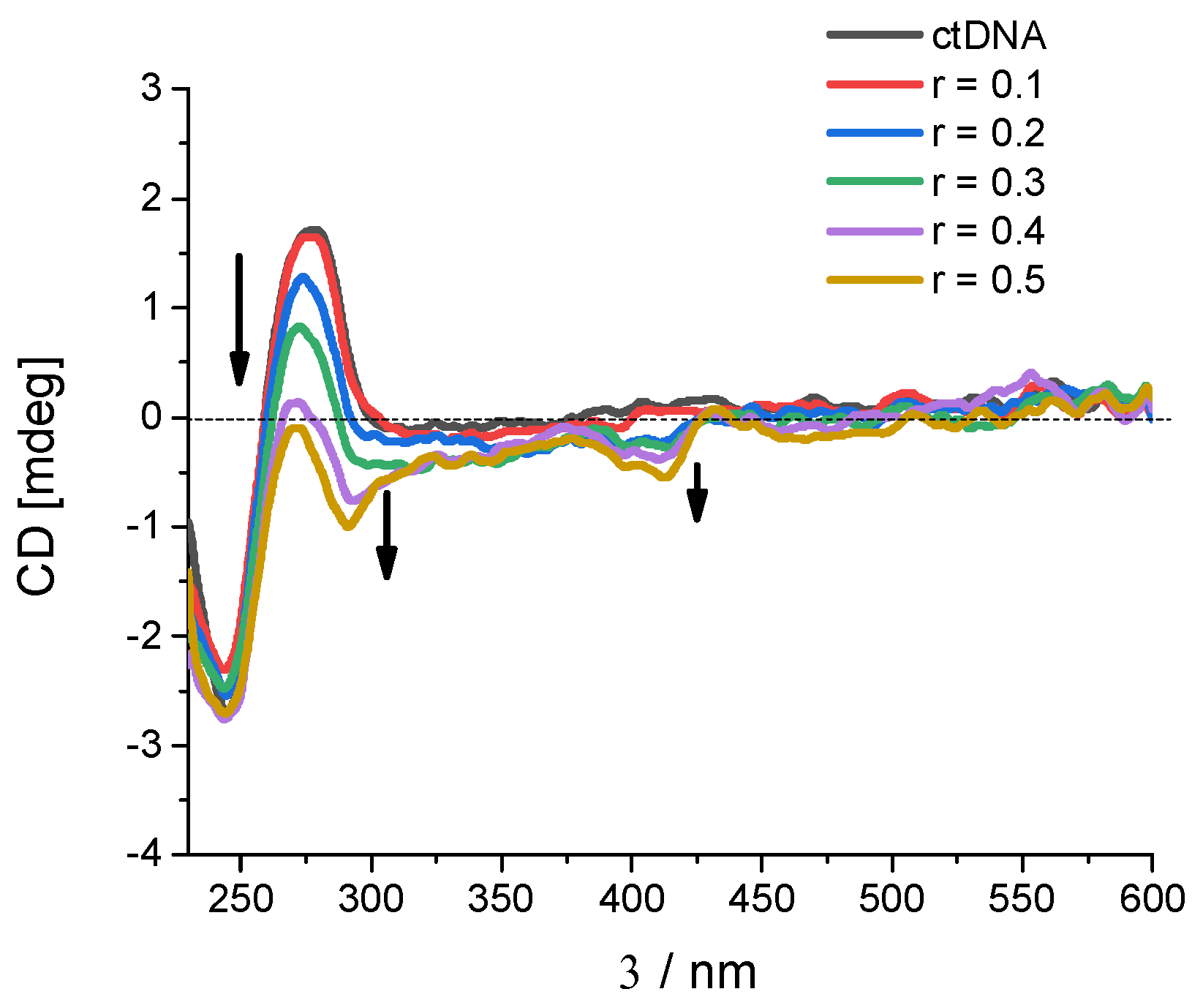
2.2.2. Binding with Nucleic Acids
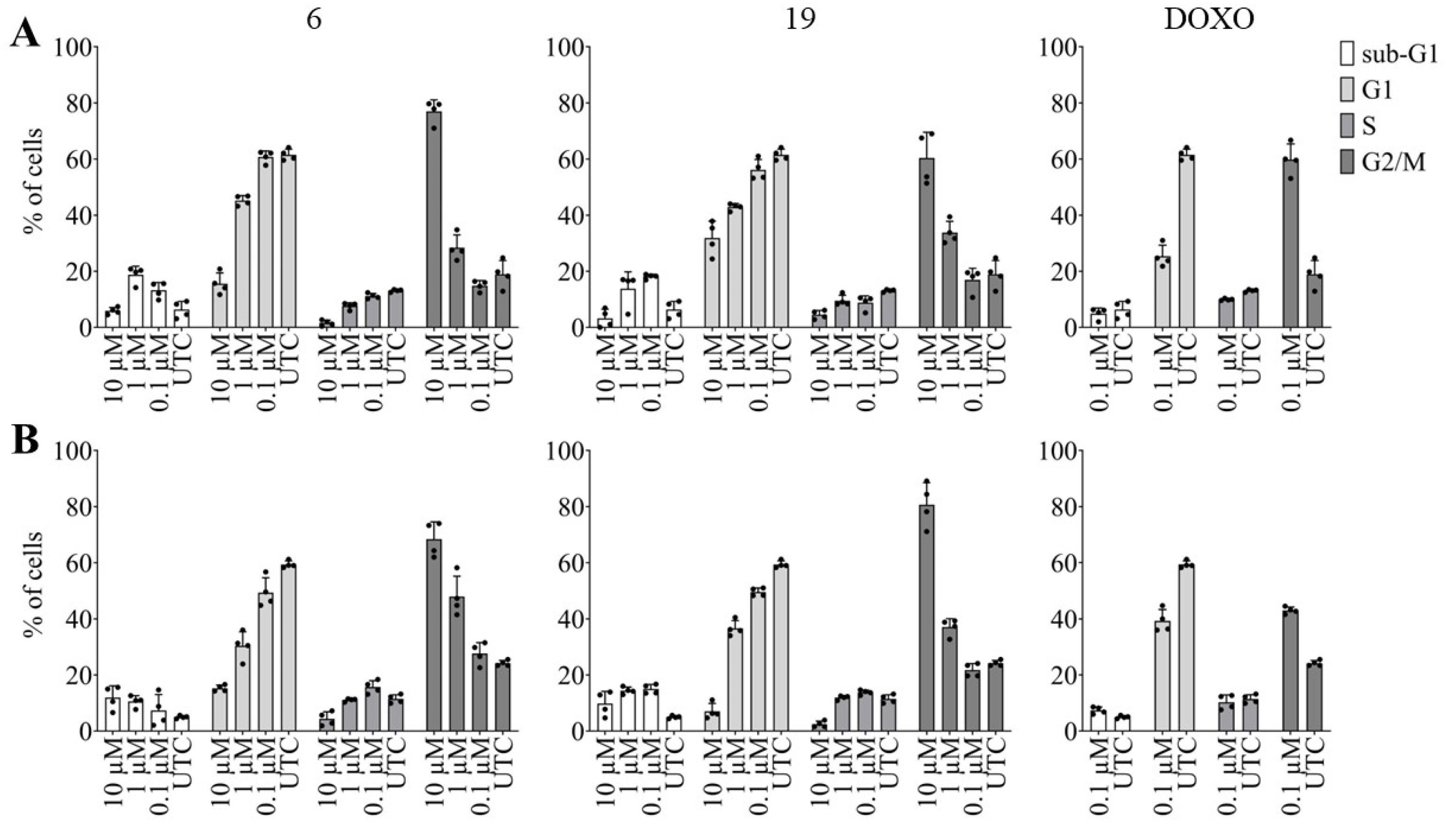
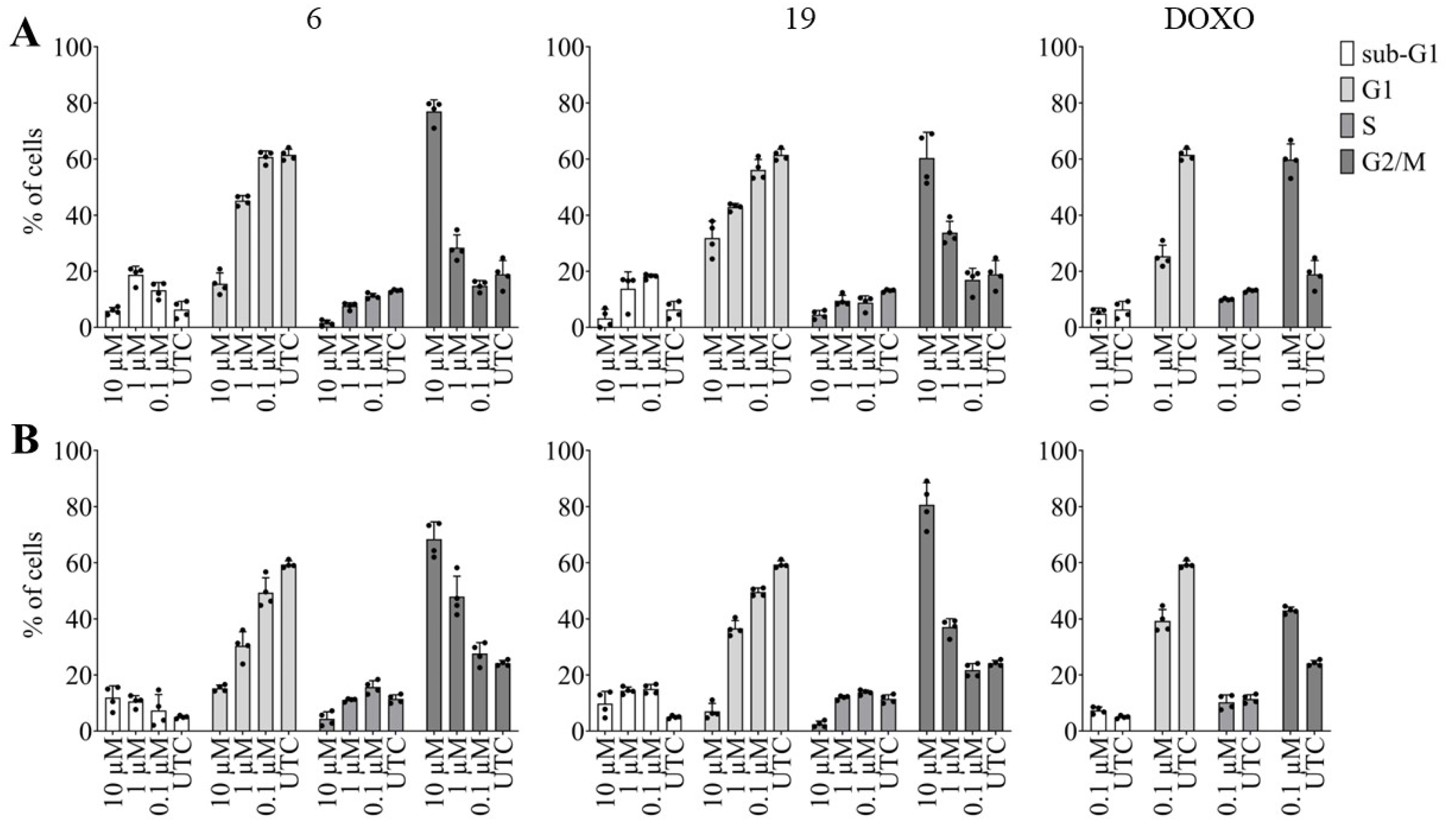
2.2.3. Effects of Pentacyclic Benzimidazole Derivatives on Cancer Cell Cycle Regulation
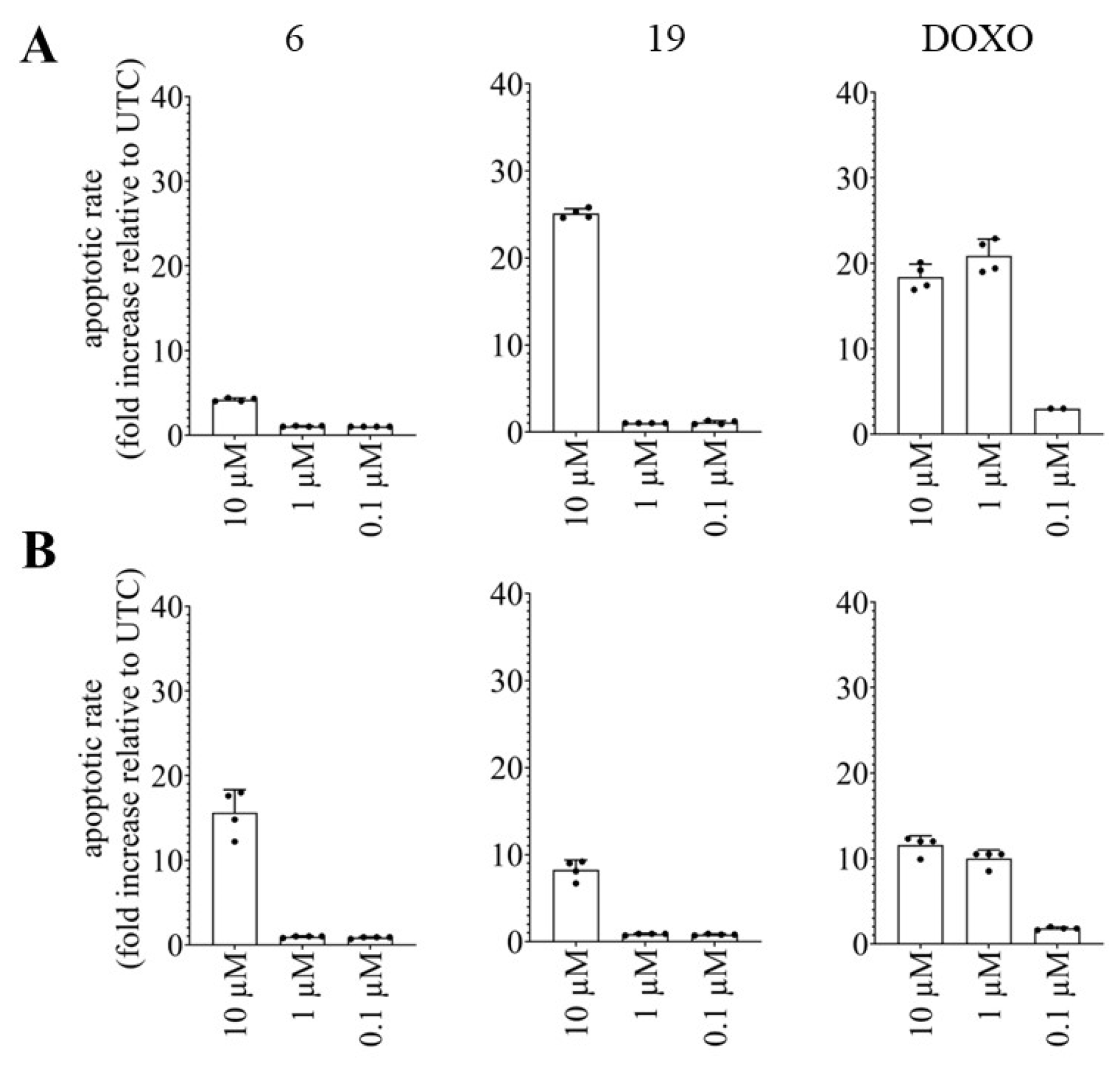
2.2.4. Effects of Pentacyclic Benzimidazole Derivatives on Caspase-3 and -7 Activity
3. Materials and Methods
3.1. Chemistry—General Methods
3.2. Synthesis of 7-Amino Substituted Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]-naphthyridine-6-carbonitriles
3.2.1. (E)-2-benzimidazolyl-1-(2-chloroquinolin-3-yl)-2-isocyanoethen-1-ol 3
3.2.2. 7-Oxo-5,7-dihydrobenzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo-nitrile 4
3.2.3. 7-Chlorobenzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonitrile 5
3.2.4. General Method for Preparation of Compounds 6–9
7-((3-N,N-(dimethylamino)propyl)amino)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]-naphthyridi- ne-6-carbonitrile 6
7-(N-isobutylamino)benzo[g]benzo[4,5]imidazo [1,2-a][1,8]naphthyridine-6-carbo- nitrile 7
7-(Piperidin-1-yl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo- Nitrile 8
7-(Piperazin-1-yl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo- Nitrile 9
3.3. Synthesis of 11-Amino Substituted Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]-naphthyridine-6-carbonitriles
3.3.1. (E)-2-(1H-benzo[d]imidazol-2-yl)-3-(2-chloro-7-fluoroquinolin-3-yl)acrylonitrile 13
3.3.2. 11-Fluorobenzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonitrile 16
3.3.3. General Method for Preparation of Compounds 19–22
11-((3-N,N-(dimethylamino)propyl)amino)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]-naphthyridine-6-carbonitrile 19
11-(N-isobutylamino)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonitrile 20
11-(Piperidin-1-yl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo-nitrile 21
11-(Piperazin-1-yl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo-nitrile 22
3.4. Synthesis of Amido Substituted benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridines and benzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridines
3.4.1. General Method for the Synthesis of Compounds 14 and 15
(E)-2-(1H-benzo[d]imidazol-2-yl)-3-(2-chloroquinolin-3-yl)acrylonitrile 14
(E)-2-(1H-benzo[d]imidazol-2-yl)-3-(quinolin-3-yl)acrylonitrile 15
3.4.2. Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonitrile 17
3.4.3. Benzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carbonitrile 18
3.4.4. General Method for the Synthesis of Compounds 23 and 27
Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carboxylic acid 23
Benzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carboxylic acid 27
3.4.5. General Method for the Synthesis of Compounds 24 and 28
Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonyl Chloride 24
Benzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carbonyl Chloride 28
3.4.6. General Method for the Synthesis of Compounds 25 and 26
N-(3-N,N-(dimethylamino)propyl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]-naphthyridine-6-carboxamide 25
Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridin-6-yl(piperidin-1-yl)metha-none 26
3.4.7. General Method for the Synthesis of Compounds 29 and 30
N-isobutylbenzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carboxamide 29
N,N-dimethylbenzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carbox-amide 30
3.5. Antiproliferative Activity
3.5.1. Cell Culture and Reference Compounds
3.5.2. Proliferation Assays
3.5.3. High Content Imaging Analysis of Cell Cycle Distribution
3.5.4. Caspase-3/7 Activity
3.6. Binding with Nucleic Acids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, A.; Singh, A.K.; Singh, H.; Vijayan, V.; Kumar, D.; Naik, J.; Thareja, S.; Yadav, J.P.; Pathak, P.; Grishina, M.; et al. Nitrogen Containing Heterocycles as Anticancer Agents: A Medicinal Chemistry Perspective. Pharmaceuticals 2023, 16, 299. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: An overview. RSC Adv. 2020, 10, 44247. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef]
- Lang, D.K.; Kaur, R.; Arora, R.; Saini, B.; Arora, S. Nitrogen-Containing Heterocycles as Anticancer Agents: An Overview. Anticancer Agents Med. Chem. 2020, 20, 2150–2168. [Google Scholar] [CrossRef]
- Nehra, B.; Mathew, B.; Chawla, P.A. A Medicinal Chemist’s Perspective Towards Structure Activity Relationship of Heterocycle Based Anticancer Agents. Curr. Top. Med. Chem. 2022, 22, 493–528. [Google Scholar] [CrossRef] [PubMed]
- Vušak, D.; Perin, N.; Martin-Kleiner, I.; Kralj, M.; Karminski-Zamola, G.; Hranjec, M.; Bertoša, B. Synthesis and antiproliferative activity of amino-substituted benzimidazo[1,2-a]quinolines as mesylate salts designed by 3D-QSAR analysis. Mol. Divers. 2017, 21, 621. [Google Scholar] [CrossRef]
- Meščić Macan, A.; Perin, N.; Jakopec, S.; Mioč, M.; Radić Stojković, M.; Kralj, M.; Hranjec, M.; Raić-Malić, S. Synthesis, antiproliferative activity and DNA/RNA-binding properties of mono- and bis-(1,2,3-triazolyl)-appended benzimidazo[1,2-a]quinoline derivatives. Eur. J. Med. Chem. 2020, 185, 111845. [Google Scholar] [CrossRef]
- Perin, N.; Babić, D.; Kassal, P.; Čikoš, A.; Hranjec, M.; Vianello, R. Spectroscopic and Computational Study of the Protonation Equilibria of Amino-Substituted benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazoles as Novel pH-Sensing Materials. Chemosensors 2022, 10, 21. [Google Scholar] [CrossRef]
- Akhtar, W.; Faraz Khan, M.; Verma, G.; Shaquiquzzaman, M.; Rizvi, M.A.; Mehdi, S.H.; Akhter, M.; Mumtaz Alam, M. Therapeutic evolution of benzimidazole derivatives in the last quinquennial period. Eur. J. Med. Chem. 2017, 126, 705–753. [Google Scholar] [CrossRef]
- Venugopal, S.; Sharma, V.; Mehra, A.; Singh, I.; Singh, G. DNA intercalators as anticancer agents. Chem. Biol. Drug Des. 2022, 100, 580–598. [Google Scholar] [CrossRef]
- Sharma, V.; Gupta, M.; Kumar, P.; Sharma, A. A Comprehensive Review on Fused Heterocyclic as DNA Intercalators: Promising Anticancer Agents. Curr. Pharm. Des. 2021, 27, 15–42. [Google Scholar] [CrossRef] [PubMed]
- Simon, Z.; Voiculetz, N.; Motoc, I. Specific Interaction and Biological Recognition Processes; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Xu, J.; Wang, G.A.; Gao, L.; Wu, L.; Lei, Q.; Deng, H.; Li, F. Enabling programmable dynamic DNA chemistry using small-molecule DNA binders. Nat. Commun. 2023, 14, 4248. [Google Scholar] [CrossRef] [PubMed]
- Sischka, A.; Toensing, K.; Eckel, R.; Wilking, S.D.; Sewald, N.; Ros, R.; Anselmetti, D. Molecular mechanisms and kinetics between DNA and DNA binding ligands. Biophys. J. 2005, 88, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Mekheimer, R.A.; Al-Sheikh, M.A.; Medrasi, H.Y.; Sadek, K.U. Advancements in the synthesis of fused tetracyclic quinoline derivatives. RSC Adv. 2020, 10, 19867–19935. [Google Scholar] [CrossRef] [PubMed]
- Ajani, O.O.; Iyaye, K.T.; Ademosun, O.T. Recent advances in chemistry and therapeutic potential of functionalized quinoline motifs—A review. RSC Adv. 2022, 12, 18594–18614. [Google Scholar] [CrossRef] [PubMed]
- Shiro, T.; Fukaya, T.; Tobe, M. The chemistry and biological activity of heterocycle-fused quinoline derivatives: A review. Eur. J. Med. Chem. 2015, 97, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Perin, N.; Martin-Kleiner, I.; Nhili, R.; Laine, W.; David-Cordonnier, M.-H.; Vugrek, O.; Karminski-Zamola, G.; Kralj, M.; Hranjec, M. Biological activity and DNA binding studies of 2-substituted benzimidazo[1,2-a]quinolines bearing different amino side chains. Med. Chem. Comm. 2013, 4, 1537–1550. [Google Scholar] [CrossRef]
- Perin, N.; Nhili, R.; Ester, K.; Laine, W.; Karminski-Zamola, G.; Kralj, M.; David-Cordonnier, M.-H.; Hranjec, M. Synthesis, antiproliferative activity and DNA binding properties of novel 5-aminobenzimidazo[1,2-a]quinoline-6-carbonitriles. Eur. J. Med. Chem. 2014, 80, 218–227. [Google Scholar] [CrossRef]
- Perin, N.; Nhili, R.; Cindrić, M.; Bertoša, B.; Vušak, D.; Martin-Kleiner, I.; Laine, W.; Karminski-Zamola, G.; Kralj, M.; David-Cordonnier, M.-H.; et al. Amino substituted benzimidazo[1,2-a]quinolines: Antiproliferative potency, 3D QSAR study and DNA binding properties. Eur. J. Med. Chem. 2016, 122, 530–545. [Google Scholar] [CrossRef]
- Lončar, B.; Perin, N.; Mioč, M.; Boček, I.; Grgić, L.; Kralj, M.; Tomić, S.; Radić Stojković, M.; Hranjec, M. Novel amino substituted tetracyclic imidazo[4,5-b]pyridine derivatives: Design, synthesis, antiproliferative activity and DNA/RNA binding study. Eur. J. Med. Chem. 2021, 217, 113342. [Google Scholar] [CrossRef]
- Hranjec, M.; Lučić, B.; Ratkaj, I.; Kraljević Pavelić, S.; Piantanida, I.; Pavelić, K.; Karminski-Zamola, G. Novel Imidazo[4,5-b]Pyridine and Triaza-Benzo[c]Fluorene Derivatives: Synthesis, Antiproliferative Activity and DNA Binding Studies. Eur. J. Med. Chem. 2011, 46, 2748–2758. [Google Scholar] [CrossRef] [PubMed]
- Perin, N.; Bobanović, K.; Zlatar, I.; Jelić, D.; Kelava, V.; Koštrun, S.; Gabelica Marković, V.; Brajša, K.; Hranjec, M. Antiproliferative activity of amino substituted benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazoles explored by 2D and 3D cell culture system. Eur. J. Med. Chem. 2017, 125, 722–735. [Google Scholar] [CrossRef]
- Hranjec, M.; Piantanida, I.; Kralj, M.; Šuman, L.; Pavelić, K.; Karminski-Zamola, G. Novel amidino-substituted thienyl- and furyl-vinyl-benzimidazole derivatives and their photochemical conversion into corresponding diaza-cyclopenta[c]fluorenes. Synthesis, interactions with DNA and RNA and antitumor evaluation. Part 4. J. Med. Chem. 2008, 51, 4899–4910. [Google Scholar] [CrossRef] [PubMed]
- Brajša, K.; Vujasinović, I.; Jelić, D.; Trzun, M.; Zlatar, I.; Karminski-Zamola, G.; Hranjec, M. Antitumor activity of amidino-substituted benzimidazole and benzimidazo[1,2-a]quinoline derivatives tested in 2D and 3D cell culture systems. J. Enzym. Inhib. Med. Chem. 2016, 31, 1139–1145. [Google Scholar] [CrossRef]
- Perin, N.; Alić, J.; Liekens, S.; Van Aerschot, A.; Vervaeke, P.; Gadakh, B.; Hranjec, M. Different positions of amide side chains on the benzimidazo[1,2-a]quinoline skeleton strongly influenced biological activity. New J. Chem. 2018, 42, 7096–7104. [Google Scholar] [CrossRef]
- Cindrić, M.; Jambon, S.; Harej, A.; Depauw, S.; David-Cordonnier, M.-H.; Kraljević Pavelić, S.; Karminski-Zamola, G.; Hranjec, M. Novel amidino substituted benzimidazole and benzothiazole benzo[b]thieno-2-carboxamides exert strong antiproliferative and DNA binding properties. Eur. J. Med. Chem. 2017, 136, 468–479. [Google Scholar] [CrossRef]
- Saenger, W. Principles of Nucleic Acid Structure; Springer: New York, NY, USA, 1984. [Google Scholar]
- Cantor, C.R. Techniques for the Study of Biological Structure and Function; W. H. Freeman: San Francisco, CA, USA, 1980. [Google Scholar]
- Neidle, S. Oxford Handbook of Nucleic Acid Structure; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Mergny, J.L.; Lacroix, L. Analysis of thermal melting curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef] [PubMed]
- Scatchard, G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- McGhee, J.D.; von Hippel, P.H. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Berova, N.; Nakanishi, K.; Woody, R.W. Circular Dichroism Principles and Applications, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar]
- Smidlehner, T.; Piantanida, I.; Pescitelli, G. Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids—Tutorial. Beilstein J. Org. Chem. 2017, 14, 84–105. [Google Scholar] [CrossRef]
- Chaires, J.B.; Dattagupta, N.; Crothers, D.M. Studies on Interaction of Anthracycline Antibiotics and Deoxyribonucleic-Acid—Equilibrium Binding-Studies on Interaction of Daunomycin with Deoxyribonucleic-Acid. Biochemistry 1982, 17, 3933–3940. [Google Scholar] [CrossRef] [PubMed]
- Chalikian, T.V.; Volker, J.; Plum, G.E.; Breslauer, K.J. A more unified picture for the thermodynamics of nucleic acid duplex melting: A characterization by calorimetric and volumetric techniques. Proc. Natl. Acad. Sci. USA 1999, 14, 7853–7858. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | IC50/μM * | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell Line | |||||||||
| Capan-1 | HCT-116 | Hap-1 | NCI-H460 | DND-41 | HL-60 | K-562 | MM.1S | Z-138 | |
| 5 | 5.8 ± 2.5 | 16.5 ± 0.7 | 23.1 ± 0.1 | 29.5 ± 2.5 | 4.9 ± 0.2 | 5.2 ± 1.3 | 64.1 ± 3.4 | 12.9 ± 0.8 | 2.4 ± 0.3 |
| 6 | 0.3 ± 0.1 | 1.8 ± 0.6 | 1.2 ± 0.2 | 1.0 ± 0.8 | 0.5 ± 0.0 | 0.5 ± 0.1 | 0.3 ± 0.0 | 0.6 ± 0.0 | 0.4 ± 0.1 |
| 7 | ≥44.3 | >100 | 34.2 ± 2.9 | 41.4 ± 4.0 | ≥86.4 | 28.5 ± 4.5 | >100 | >100 | 34.4 ± 2.3 |
| 8 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 9 | 2.3 ± 0.1 | 5.6 ± 2.1 | 5.5 ± 2.1 | 5.2 ± 2.8 | 2.0 ± 0.3 | 3.5 ± 2.9 | 3.0 ± 1.9 | 5.6 ± 3.7 | 1.5 ± 0.1 |
| 16 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 17 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 18 | 27.2 ± 2.1 | 41.9 ± 5.7 | 22.2 ± 0.5 | 32.5 ± 8.1 | 62.3 ± 3.3 | 12.5 ± 2.3 | 16.3 ± 2.5 | >100 | 36.0 ± 7.0 |
| 19 | 1.0 ± 0.5 | 5.9 ± 1.9 | 6.5 ± 1.3 | 1.1 ± 0.3 | 1.3 ± 0.9 | 1.7 ± 0.2 | 0.4 ± 0.0 | 2.4 ± 0.5 | 0.6 ± 0.3 |
| 20 | >100 | ≥85.1 | >100 | 30.5 ± 2.5 | >100 | >100 | >100 | >100 | >100 |
| 21 | 78.8 ± 8.4 | >100 | >100 | 47.6 ± 2.5 | >100 | >100 | >100 | >100 | >100 |
| 22 | 1.2 ± 0.2 | 6.2 ± 0.9 | 5.9 ± 2.1 | 5.4 ± 2.2 | 2.1 ± 0.1 | 2.0 ± 0.2 | 8.4 ± 0.7 | 2.9 ± 0.1 | 1.4 ± 0.2 |
| 25 | 5.8 ± 2.4 | 5.5 ± 2.1 | 28.5 ± 5.2 | 7.8 ± 1.0 | 4.4 ± 2.6 | 4.2 ± 1.7 | 5.3 ± 4.2 | 11.9 ± 0.9 | 2.1 ± 0.4 |
| 26 | 47.4 ± 8.1 | >100 | 49.8 ± 4.8 | ≥70.4 | >100 | >100 | >100 | >100 | 84.9 ± 6.6 |
| 29 | ≥41.7 | ≥49.8 | 15.9 ± 2.5 | 23.4 ± 2.5 | >100 | ≥61.2 | >100 | >100 | 49.1 ± 2.5 |
| 30 | 36.2 ± 1.3 | 44.4 ± 0.4 | 25.5 ± 4.9 | 26.6 ± 0.8 | ≥86.1 | 49.5 ± 6.4 | >100 | ≥92.1 | 44.4 ± 8.6 |
| Etoposide | 0.2 ± 0.1 | 3.1 ± 0.5 | 0.2 ± 0.0 | 5.6 ± 0.9 | 0.4 ± 0.2 | 1.6 ± 1.0 | 9.0 ± 1.1 | 0.8 ± 0.5 | 0.9 ± 0.5 |
| ctDNA | rArU | |||
|---|---|---|---|---|
| logKa | ΔTm/°C | logKa | ΔTm/°C | |
| 22 | 6.8 | 2.3 | 7.9 | 4.9 |
| 19 | 6.8 | 2.3 | 8.6 | 1.1 |
| 9 | - b | 1.6 | - e | - e |
| 6 | - b | 1.8 | - e | - e |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perin, N.; Gulin, M.; Kos, M.; Persoons, L.; Daelemans, D.; Fabijanić, I.; Stojković, M.R.; Hranjec, M. Synthesis and Biological Evaluation of Novel Amino and Amido Substituted Pentacyclic Benzimidazole Derivatives as Antiproliferative Agents. Int. J. Mol. Sci. 2024, 25, 2288. https://doi.org/10.3390/ijms25042288
Perin N, Gulin M, Kos M, Persoons L, Daelemans D, Fabijanić I, Stojković MR, Hranjec M. Synthesis and Biological Evaluation of Novel Amino and Amido Substituted Pentacyclic Benzimidazole Derivatives as Antiproliferative Agents. International Journal of Molecular Sciences. 2024; 25(4):2288. https://doi.org/10.3390/ijms25042288
Chicago/Turabian StylePerin, Nataša, Marjana Gulin, Marija Kos, Leentje Persoons, Dirk Daelemans, Ivana Fabijanić, Marijana Radić Stojković, and Marijana Hranjec. 2024. "Synthesis and Biological Evaluation of Novel Amino and Amido Substituted Pentacyclic Benzimidazole Derivatives as Antiproliferative Agents" International Journal of Molecular Sciences 25, no. 4: 2288. https://doi.org/10.3390/ijms25042288
APA StylePerin, N., Gulin, M., Kos, M., Persoons, L., Daelemans, D., Fabijanić, I., Stojković, M. R., & Hranjec, M. (2024). Synthesis and Biological Evaluation of Novel Amino and Amido Substituted Pentacyclic Benzimidazole Derivatives as Antiproliferative Agents. International Journal of Molecular Sciences, 25(4), 2288. https://doi.org/10.3390/ijms25042288