
GATOR1 Mutations Impair PI3 Kinase-Dependent Growth Factor Signaling Regulation of mTORC1

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Epilepsy-Linked NPRL2 Mutations Differentially Effect mTORC1 Signal Transduction
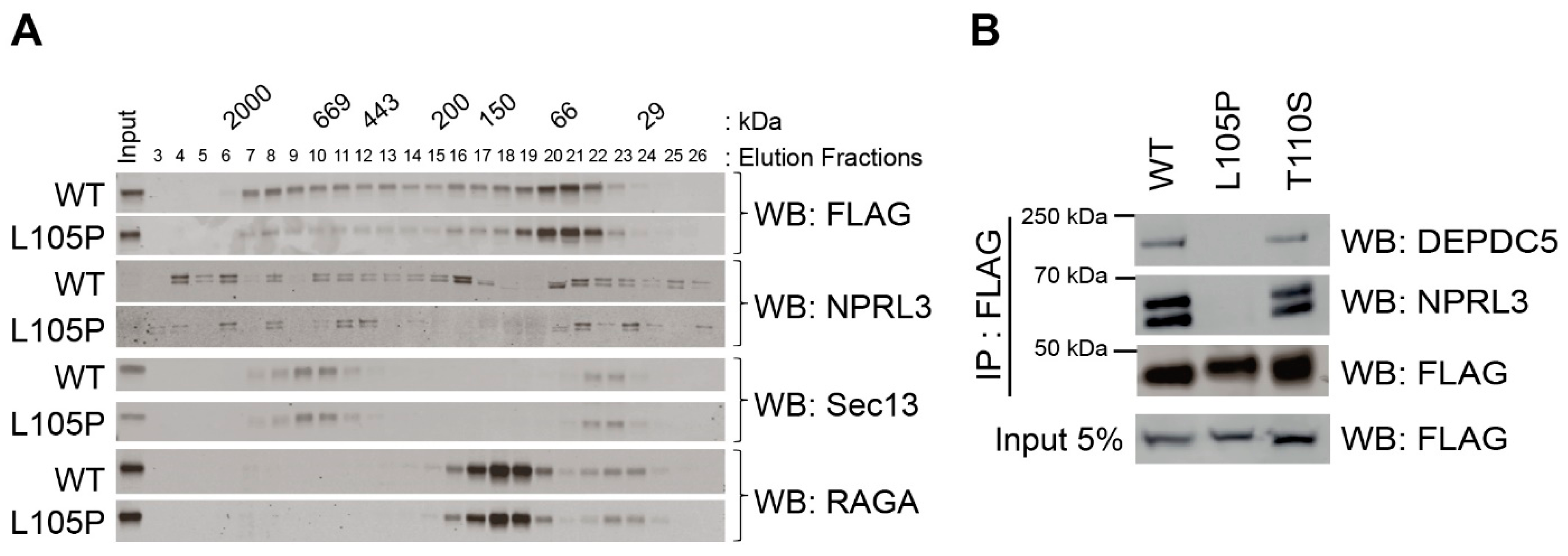
2.2. NPRL2-L105P Impairs GATOR1 Complex Formation
2.3. GATOR1 Mediates PI3K-Dependent Regulation of mTORC1
2.4. Loss of NPRL2 Uncouples PI3K-Dependent Regulation of mTORC1 Functions
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Site-Directed Mutagenesis
4.3. Lentiviral Production
4.4. Generation of Stable Cell Lines
4.5. Western Blotting
4.6. Fast Protein Liquid Chromatography
4.7. Co-Immunoprecipitation
4.8. Rheb-GTP-Binding Assays
4.9. Mass Spectrometry Protein Identification
4.10. AHA Labeling of Nascent Translation
4.11. TFEB Localization
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baldassari, S.; Picard, F.; Verbeek, N.E.; van Kempen, M.; Brilstra, E.H.; Lesca, G.; Conti, V.; Guerrini, R.; Bisulli, F.; Licchetta, L.; et al. The landscape of epilepsy-related GATOR1 variants. Genet. Med. 2019, 21, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Ricos, M.G.; Hodgson, B.L.; Pippucci, T.; Saidin, A.; Ong, Y.S.; Heron, S.E.; Licchetta, L.; Bisulli, F.; Bayly, M.A.; Hughes, J.; et al. Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann. Neurol. 2016, 79, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Burger, B.J.; Rose, S.; Bennuri, S.C.; Gill, P.S.; Tippett, M.L.; Delhey, L.; Melnyk, S.; Frye, R.E. Autistic Siblings with Novel Mutations in Two Different Genes: Insight for Genetic Workups of Autistic Siblings and Connection to Mitochondrial Dysfunction. Front. Pediatr. 2017, 5, 219. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.A.; Borlot, F.; Cossette, P.; Minassian, B.A.; Andrade, D.M. Two definite cases of sudden unexpected death in epilepsy in a family with a DEPDC5 mutation. Neurol. Genet. 2015, 1, e28. [Google Scholar] [CrossRef] [PubMed]
- Ribierre, T.; Deleuze, C.; Bacq, A.; Baldassari, S.; Marsan, E.; Chipaux, M.; Muraca, G.; Roussel, D.; Navarro, V.; Leguern, E.; et al. Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Investig. 2018, 128, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Valenstein, M.L.; Gu, X.; Sabatini, D.M. Arg-78 of Nprl2 catalyzes GATOR1-stimulated GTP hydrolysis by the Rag GTPases. J. Biol. Chem. 2019, 294, 2970–2975. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Davis, R.W. A genome-wide screen for regulators of TORC1 in response to amino acid starvation reveals a conserved Npr2/3 complex. PLoS Genet. 2009, 5, e1000515. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Bourdeau Julien, I.; Sephton, C.F.; Dutchak, P.A. Metabolic Networks Influencing Skeletal Muscle Fiber Composition. Front. Cell Dev. Biol. 2018, 6, 125. [Google Scholar] [CrossRef]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR-Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9, 655731. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Sampson, J.R.; Pal, D.K.; Bateman, J.M. The role of mTOR signalling in neurogenesis, insights from tuberous sclerosis complex. Semin. Cell Dev. Biol. 2016, 52, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Isotani, S.; Hara, K.; Tokunaga, C.; Inoue, H.; Avruch, J.; Yonezawa, K. Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase alpha in vitro. J. Biol. Chem. 1999, 274, 34493–34498. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Dutchak, P.A.; Laxman, S.; Estill, S.J.; Wang, C.; Wang, Y.; Wang, Y.; Bulut, G.B.; Gao, J.; Huang, L.J.; Tu, B.P. Regulation of Hematopoiesis and Methionine Homeostasis by mTORC1 Inhibitor NPRL2. Cell Rep. 2015, 12, 371–379. [Google Scholar] [CrossRef]
- Sun, Y.; Wan, L.; Yan, H.; Li, Z.; Yang, G. Phenotypic and Genotypic Characterization of NPRL2-Related Epilepsy: Two Case Reports and Literature Review. Front. Neurol. 2021, 12, 780799. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Huang, R.K.; Brignole, E.J.; Condon, K.J.; Valenstein, M.L.; Chantranupong, L.; Bomaliyamu, A.; Choe, A.; Hong, C.; Yu, Z.; et al. Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature 2018, 556, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.E.; Nieto Guil, A.F.; Robertson, L.J.; Piltz, S.G.; Hughes, J.N.; Thomas, P.Q. Functional screening of GATOR1 complex variants reveals a role for mTORC1 deregulation in FCD and focal epilepsy. Neurobiol. Dis. 2020, 134, 104640. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef]
- Yao, Y.; Hong, S.; Ikeda, T.; Mori, H.; MacDougald, O.A.; Nada, S.; Okada, M.; Inoki, K. Amino Acids Enhance Polyubiquitination of Rheb and Its Binding to mTORC1 by Blocking Lysosomal ATXN3 Deubiquitinase Activity. Mol. Cell 2020, 80, 437–451.e6. [Google Scholar] [CrossRef]
- Gollwitzer, P.; Grutzmacher, N.; Wilhelm, S.; Kummel, D.; Demetriades, C. A Rag GTPase dimer code defines the regulation of mTORC1 by amino acids. Nat. Cell Biol. 2022, 24, 1394–1406. [Google Scholar] [CrossRef]
- Wang, G.; Chen, L.; Lei, X.; Qin, S.; Geng, H.; Zheng, Y.; Xia, C.; Yao, J.; Meng, T.; Deng, L. Role of FLCN Phosphorylation in Insulin-Mediated mTORC1 Activation and Tumorigenesis. Adv. Sci. 2023, 10, e2206826. [Google Scholar] [CrossRef]
- Hui, J.B.; Silva, J.C.H.; Pelaez, M.C.; Sevigny, M.; Venkatasubramani, J.P.; Plumereau, Q.; Chahine, M.; Proulx, C.D.; Sephton, C.F.; Dutchak, P.A. NPRL2 Inhibition of mTORC1 Controls Sodium Channel Expression and Brain Amino Acid Homeostasis. eNeuro 2022, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dutchak, P.A.; Estill-Terpack, S.J.; Plec, A.A.; Zhao, X.; Yang, C.; Chen, J.; Ko, B.; Deberardinis, R.J.; Yu, Y.; Tu, B.P. Loss of a Negative Regulator of mTORC1 Induces Aerobic Glycolysis and Altered Fiber Composition in Skeletal Muscle. Cell Rep. 2018, 23, 1907–1914. [Google Scholar] [CrossRef]
- Ding, J.; Ji, J.; Rabow, Z.; Shen, T.; Folz, J.; Brydges, C.R.; Fan, S.; Lu, X.; Mehta, S.; Showalter, M.R.; et al. A metabolome atlas of the aging mouse brain. Nat. Commun. 2021, 12, 6021. [Google Scholar] [CrossRef]
- Korenke, G.C.; Eggert, M.; Thiele, H.; Nurnberg, P.; Sander, T.; Steinlein, O.K. Nocturnal frontal lobe epilepsy caused by a mutation in the GATOR1 complex gene NPRL3. Epilepsia 2016, 57, e60–e63. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Makrythanasis, P.; Navarro, V.; Ishida, S.; de Bellescize, J.; Ville, D.; Weckhuysen, S.; Fosselle, E.; Suls, A.; De Jonghe, P.; et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology 2014, 82, 2101–2106. [Google Scholar] [CrossRef]
- Bartolini, E.; Ferrari, A.R.; Fiori, S.; Della Vecchia, S. Glycaemic Imbalances in Seizures and Epilepsy of Paediatric Age: A Literature Review. J. Clin. Med. 2023, 12, 2580. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.C.; Bartlett, J.J.; Mercer, A.; Slade, L.; Surette, M.; Ballabio, A.; Flibotte, S.; Hussein, B.; Rodrigues, B.; Kienesberger, P.C.; et al. Loss of function of transcription factor EB remodels lipid metabolism and cell death pathways in the cardiomyocyte. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165832. [Google Scholar] [CrossRef] [PubMed]
- Yuskaitis, C.J.; Rossitto, L.A.; Gurnani, S.; Bainbridge, E.; Poduri, A.; Sahin, M. Chronic mTORC1 inhibition rescues behavioral and biochemical deficits resulting from neuronal Depdc5 loss in mice. Hum. Mol. Genet. 2019, 28, 2952–2964. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Zhao, D.; Sawada, Y.; Hiraoka, Y.; Mashimo, T.; Tanaka, K. Dorsal telencephalon-specific Nprl2- and Nprl3-knockout mice: Novel mouse models for GATORopathy. Hum. Mol. Genet. 2022, 31, 1519–1530. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muller, M.; Bélanger, J.; Hadj-Aissa, I.; Zhang, C.; Sephton, C.F.; Dutchak, P.A. GATOR1 Mutations Impair PI3 Kinase-Dependent Growth Factor Signaling Regulation of mTORC1. Int. J. Mol. Sci. 2024, 25, 2068. https://doi.org/10.3390/ijms25042068
Muller M, Bélanger J, Hadj-Aissa I, Zhang C, Sephton CF, Dutchak PA. GATOR1 Mutations Impair PI3 Kinase-Dependent Growth Factor Signaling Regulation of mTORC1. International Journal of Molecular Sciences. 2024; 25(4):2068. https://doi.org/10.3390/ijms25042068
Chicago/Turabian StyleMuller, Maéline, Jasmine Bélanger, Imane Hadj-Aissa, Conghao Zhang, Chantelle F. Sephton, and Paul A. Dutchak. 2024. "GATOR1 Mutations Impair PI3 Kinase-Dependent Growth Factor Signaling Regulation of mTORC1" International Journal of Molecular Sciences 25, no. 4: 2068. https://doi.org/10.3390/ijms25042068
APA StyleMuller, M., Bélanger, J., Hadj-Aissa, I., Zhang, C., Sephton, C. F., & Dutchak, P. A. (2024). GATOR1 Mutations Impair PI3 Kinase-Dependent Growth Factor Signaling Regulation of mTORC1. International Journal of Molecular Sciences, 25(4), 2068. https://doi.org/10.3390/ijms25042068