
Parkinson’s Disease: Cells Succumbing to Lifelong Dopamine-Related Oxidative Stress and Other Bioenergetic Challenges



Abstract
1. Introduction
2. Progression of PD Pathology
2.1. Normal Age-Related Loss of SNc DA Neurons
2.2. The Braak Model of PD Staging
2.3. A Contributing Role of Lewy Bodies
2.4. Cellular and Regional Differences within the SNc
2.5. The Axonal Arbor Degenerates First
2.6. Degeneration of Other Catecholamine (CA) Neurons in the Midbrain

2.7. The Nucleus Accumbens (NAc)
2.8. Summary
3. Comparison of SNc DA Neurons between Humans and Other Species
3.1. An Evolutionary Ancient System
3.2. In Non-Human Mammals, SNc DA Neurons Preferentially Die upon Aging like in Humans, but Natural PD May Hardly Exist
3.3. Only in Humans Neuromelanin (NM) Has Abundantly Been Found
3.4. Artificial Animal PD Model Systems
3.5. Summary
4. The Bioenergetic Demands of SNc DA Neurons and a Role for Calcium in Their Vulnerability
4.1. Shared Features among Neurons Susceptible to PD
4.2. SNc DA Neurons Have a Massive Axonal Arbor in the Striatum Involved in “Volume Transmission” and Tonic Releasing of DA
4.3. Pacemaking Activity
5. Mitochondrial Dysfunction
5.1. Mitochondria in the SNc DA Neuron Cell Bodies; Energy Demands in the SNc DA Neurons Are Not Especially High
5.2. Mitochondria in the SNc DA Neuron Axonal Arbor in the Striatum
5.3. Calcium and Mitochondria
5.4. Familial PD Types Mediated by Mitochondrial Dysfunction
5.5. Animal PD Models Based on Mitochondrial Dysfunction
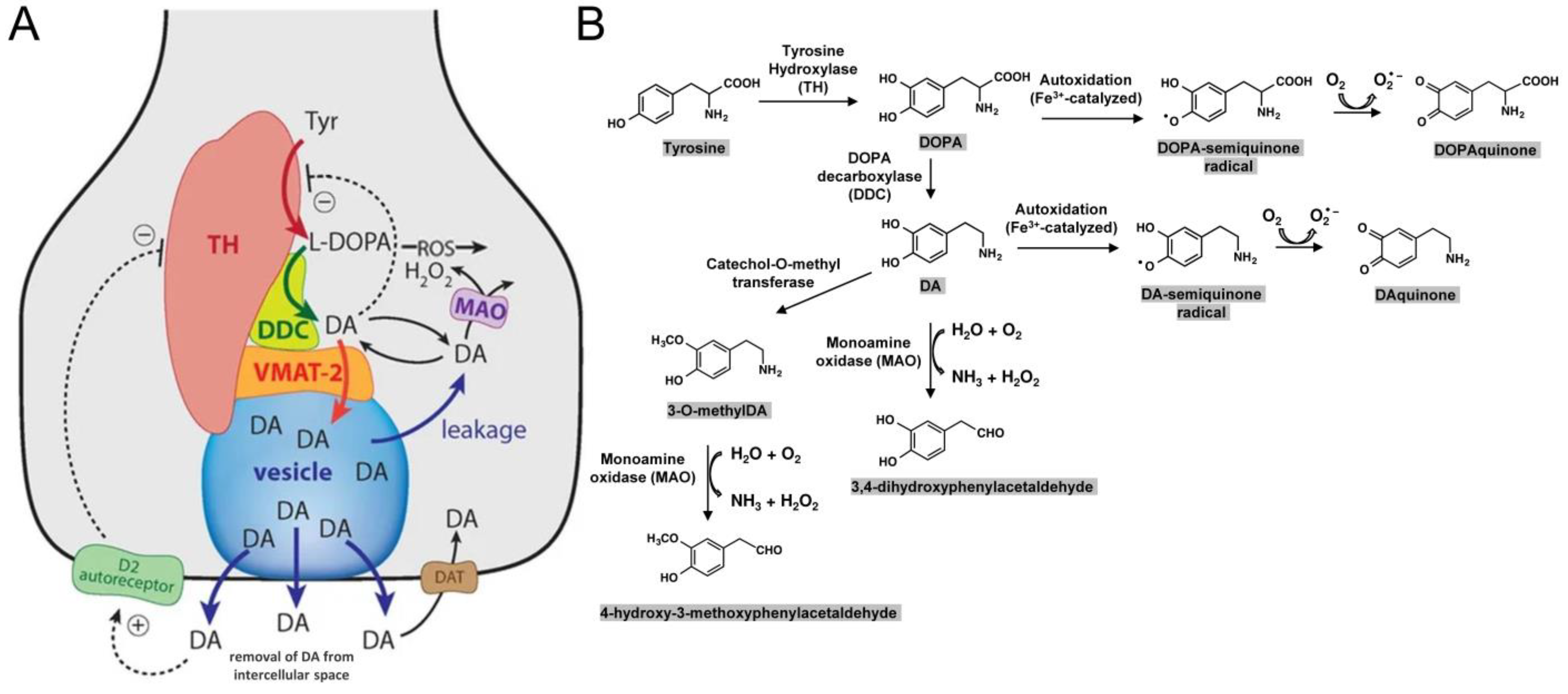
6. Intracellular Toxicity Directly Related to DA or Its Derivates
6.1. Vulnerability of Catecholaminergic Neurons in PD
6.2. Dopamine (DA)
6.3. The Pigment Neuromelanin (NM)
6.4. Summary
7. Risk Factors of PD
7.1. PD Risk Factors, General
7.2. Exposure to Toxins
7.3. Reduction in the Level of Reduced Glutathione Causes Oxidative Stress
7.4. Increased Iron Levels Cause Oxidative Stress
7.5. Aging Increases Various Risk Factors
7.6. Is Non-Secretion of DA a Risk Factor, and a Reason Why Regular Smoking May Be Protective against PD?
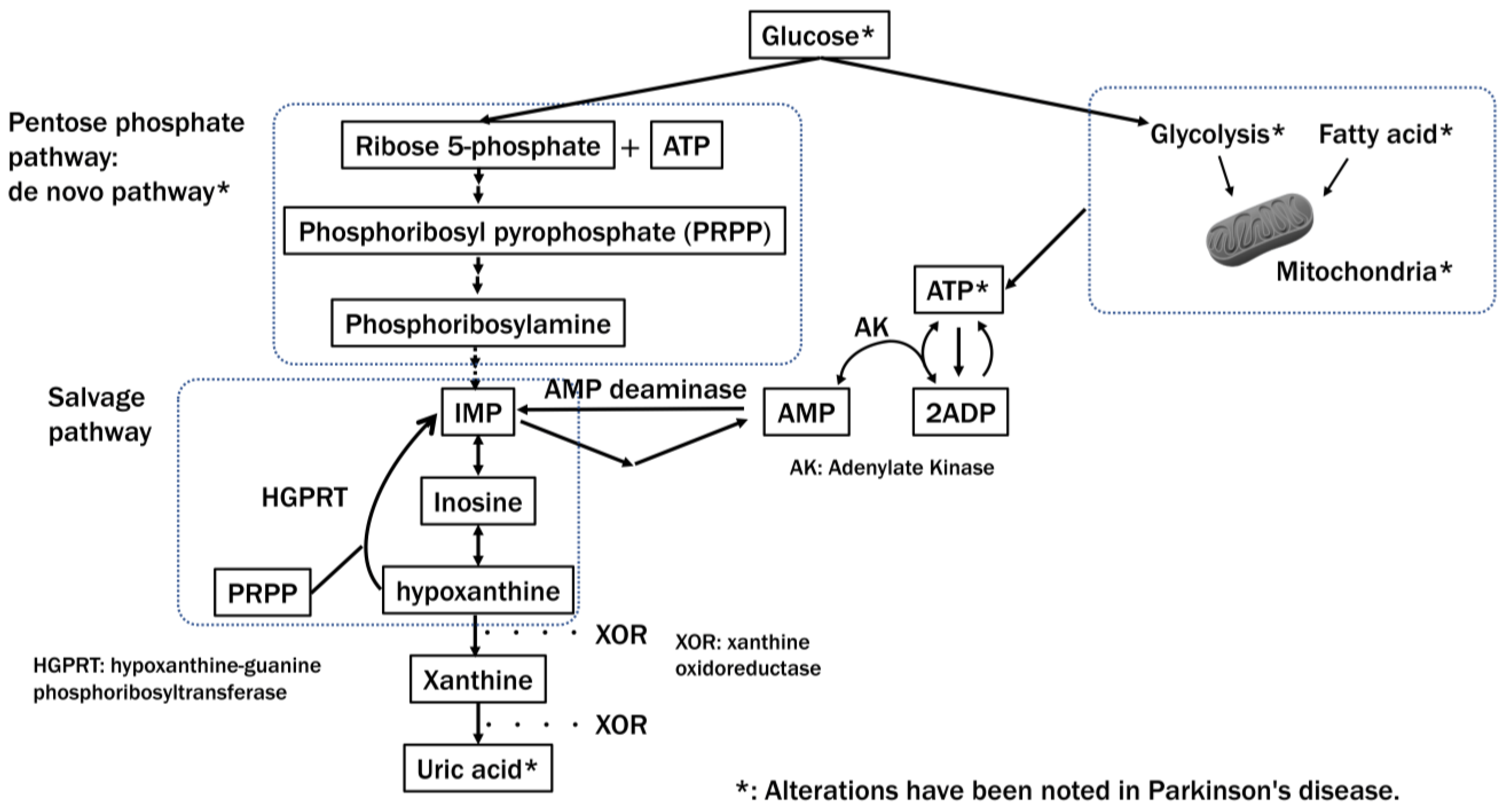
8. Energy Status and PD
8.1. Metabolic Alterations in PD
8.2. Disruption of the ATP-Producing System in PD
9. Potential Therapies and Questions That Need Addressing
10. Limitations of This Study
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| SNc | Substantia nigra pars compacta |
| DA | Dopamine |
| NE | Norepinephrine |
| LC | Locus coeruleus |
| NM | Neuromelanin |
| LB | Lewy bodies |
| TH | Tyrosine hydroxylase |
| CA | Catecholamine |
| VTA | Ventral tegmental area |
| Nac | Nucleus accumbens |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| OXPHOS | Oxidative phosphorylation |
| ROS | Reactive oxygen species |
| FDG | Fluorodeoxyglucose |
| ATP | Adenosine triphosphate |
References
- Berg, D.; Postuma, R.B.; Bloem, B.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.M.; Hardy, J.; Lang, A.E.; et al. Time to Redefine PD? Introductory Statement of the MDS Task Force on the Definition of Parkinson’s Disease. Mov. Disord. 2014, 29, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Hornykiewicz, O. A Brief History of Levodopa. J. Neurol. 2010, 257, S249–S252. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Sun, Z.; Peng, D.; Gianchandani, S.; Le, W.; Boltze, J.; Li, S. Cell-therapy for Parkinson’s disease: A systematic review and meta-analysis. J. Transl. Med. 2023, 21, 601. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef]
- Brichta, L.; Greengard, P. Molecular Determinants of Selective Dopaminergic Vulnerability in Parkinson’s Disease: An Update. Front. Neuroanat. 2014, 8, 152. [Google Scholar] [CrossRef]
- Riederer, P.; Nagatsu, T.; Youdim, M.B.H.; Wulf, M.; Dijkstra, J.M.; Sian-Huelsmann, J. Lewy Bodies, Iron, Inflammation and Neuromelanin: Pathological Aspects Underlying Parkinson’s Disease. J. Neural Transm. 2023, 130, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.K.; Pakkenberg, B. Histological Changes of the Dopaminergic Nigrostriatal System in Aging. Cell Tissue Res. 2004, 318, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain Dopamine and the Syndromes of Parkinson and Huntington. Clinical, Morphological and Neurochemical Correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Wuketich, S. Time Course of Nigrostriatal Degeneration in Parkinson’s Disease. A Detailed Study of Influential Factors in Human Brain Amine Analysis. J. Neural Transm. 1976, 38, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; O’Malley, K. Axon Degeneration in Parkinson’s Disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Schalkamp, A.-K.; Peall, K.J.; Harrison, N.A.; Sandor, C. Wearable Movement-Tracking Data Identify Parkinson’s Disease Years before Clinical Diagnosis. Nat. Med. 2023, 29, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Foffani, G.; Dehay, B.; Bezard, E.; Obeso, J.A. Motor and Non-Motor Circuit Disturbances in Early Parkinson Disease: Which Happens First? Nat. Rev. Neurosci. 2022, 23, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Watts, J.C. Calling α-Synuclein a Prion Is Scientifically Justifiable. Acta Neuropathol. 2019, 138, 505–508. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Jicha, G.A.; Liu, H.; Schmitt, F.A. Lewy Body Pathology in Normal Elderly Subjects. J. Neuropathol. Exp. Neurol. 2009, 68, 816–822. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s Disease: Possible Routes by Which Vulnerable Neuronal Types May Be Subject to Neuroinvasion by an Unknown Pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s Disease: A Dual-Hit Hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Borghammer, P.; Just, M.K.; Horsager, J.; Skjærbæk, C.; Raunio, A.; Kok, E.H.; Savola, S.; Murayama, S.; Saito, Y.; Myllykangas, L.; et al. A Postmortem Study Suggests a Revision of the Dual-Hit Hypothesis of Parkinson’s Disease. NPJ Park. Dis 2022, 8, 166. [Google Scholar] [CrossRef]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M.; et al. Brain-First versus Body-First Parkinson’s Disease: A Multimodal Imaging Case-Control Study. Brain 2020, 143, 3077–3088. [Google Scholar] [CrossRef]
- Parkkinen, L.; Pirttilä, T.; Alafuzoff, I. Applicability of Current Staging/categorization of Alpha-Synuclein Pathology and Their Clinical Relevance. Acta Neuropathol. 2008, 115, 399–407. [Google Scholar] [CrossRef]
- Koeglsperger, T.; Rumpf, S.-L.; Schließer, P.; Struebing, F.L.; Brendel, M.; Levin, J.; Trenkwalder, C.; Höglinger, G.U.; Herms, J. Neuropathology of Incidental Lewy Body & Prodromal Parkinson’s Disease. Mol. Neurodegener. 2023, 18, 32. [Google Scholar]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.A.; Schmidt, S.I.; Blaabjerg, M.; Meyer, M. Interaction between Parkin and α-Synuclein in PARK2-Mediated Parkinson’s Disease. Cells 2021, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Kingsbury, A.E.; Bandopadhyay, R.; Silveira-Moriyama, L.; Ayling, H.; Kallis, C.; Sterlacci, W.; Maeir, H.; Poewe, W.; Lees, A.J. Brain Stem Pathology in Parkinson’s Disease: An Evaluation of the Braak Staging Model. Mov. Disord. 2010, 25, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Yuan, L. Genetic Variants and Animal Models in SNCA and Parkinson Disease. Ageing Res. Rev. 2014, 15, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized Dopaminergic Neurons Are Differentially Susceptible to Degeneration in Parkinson’s Disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Graybiel, A.M.; Agid, Y. Selective Vulnerability of Pigmented Dopaminergic Neurons in Parkinson’s Disease. Acta Neurol. Scand. Suppl. 1989, 126, 19–22. [Google Scholar] [CrossRef]
- Nagatsu, T.; Nakashima, A.; Watanabe, H.; Ito, S.; Wakamatsu, K.; Zucca, F.A.; Zecca, L.; Youdim, M.; Wulf, M.; Riederer, P.; et al. The Role of Tyrosine Hydroxylase as a Key Player in Neuromelanin Synthesis and the Association of Neuromelanin with Parkinson’s Disease. J. Neural Transm. 2023, 130, 611–625. [Google Scholar] [CrossRef]
- Sulzer, D.; Surmeier, D.J. Neuronal Vulnerability, Pathogenesis, and Parkinson’s Disease. Mov. Disord. 2013, 28, 715–724. [Google Scholar] [CrossRef]
- Zecca, L.; Fariello, R.; Riederer, P.; Sulzer, D.; Gatti, A.; Tampellini, D. The Absolute Concentration of Nigral Neuromelanin, Assayed by a New Sensitive Method, Increases throughout the Life and Is Dramatically Decreased in Parkinson’s Disease. FEBS Lett. 2002, 510, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The Substantia Nigra of the Human Brain. II. Patterns of Loss of Dopamine-Containing Neurons in Parkinson’s Disease. Brain 1999, 122 Pt 8, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical Progression in Parkinson Disease and the Neurobiology of Axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [PubMed]
- Hornykiewicz, O. Biochemical Aspects of Parkinson’s Disease. Neurology 1998, 51, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Uneven Pattern of Dopamine Loss in the Striatum of Patients with Idiopathic Parkinson’s Disease. Pathophysiologic and Clinical Implications. N. Engl. J. Med. 1988, 318, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Alberico, S.L.; Cassell, M.D.; Narayanan, N.S. The Vulnerable Ventral Tegmental Area in Parkinson’s Disease. Basal Ganglia 2015, 5, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.; Yates, P.O.; Hawkes, J. The Pathology of the Human Locus Ceruleus. Clin. Neuropathol. 1983, 2, 1–7. [Google Scholar] [PubMed]
- Paredes-Rodriguez, E.; Vegas-Suarez, S.; Morera-Herreras, T.; De Deurwaerdere, P.; Miguelez, C. The Noradrenergic System in Parkinson’s Disease. Front. Pharmacol. 2020, 11, 435. [Google Scholar] [CrossRef]
- Gesi, M.; Soldani, P.; Giorgi, F.S.; Santinami, A.; Bonaccorsi, I.; Fornai, F. The Role of the Locus Coeruleus in the Development of Parkinson’s Disease. Neurosci. Biobehav. Rev. 2000, 24, 655–668. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single Nigrostriatal Dopaminergic Neurons Form Widely Spread and Highly Dense Axonal Arborizations in the Neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Watabe-Uchida, M.; Zhu, L.; Ogawa, S.K.; Vamanrao, A.; Uchida, N. Whole-Brain Mapping of Direct Inputs to Midbrain Dopamine Neurons. Neuron 2012, 74, 858–873. [Google Scholar] [CrossRef]
- Morales, M.; Margolis, E.B. Ventral Tegmental Area: Cellular Heterogeneity, Connectivity and Behaviour. Nat. Rev. Neurosci. 2017, 18, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Sakakibara, S.; Watanabe, E.; Yoshimoto, K.; Koibuchi, N. Vulnerability of Monoaminergic Neurons in the Brainstem of the Zitter Rat in Oxidative Stress. Prog. Brain Res. 2002, 136, 293–302. [Google Scholar]
- Mavridis, I.; Boviatsis, E.; Anagnostopoulou, S. The Human Nucleus Accumbens Suffers Parkinsonism-Related Shrinkage: A Novel Finding. Surg. Radiol. Anat. 2011, 33, 595–599. [Google Scholar] [CrossRef]
- Hanganu, A.; Bedetti, C.; Degroot, C.; Mejia-Constain, B.; Lafontaine, A.-L.; Soland, V.; Chouinard, S.; Bruneau, M.-A.; Mellah, S.; Belleville, S.; et al. Mild Cognitive Impairment Is Linked with Faster Rate of Cortical Thinning in Patients with Parkinson’s Disease Longitudinally. Brain 2014, 137, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.A. Dopamine, Learning and Motivation. Nat. Rev. Neurosci. 2004, 5, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Puig, M.V.; Rose, J.; Schmidt, R.; Freund, N. Dopamine Modulation of Learning and Memory in the Prefrontal Cortex: Insights from Studies in Primates, Rodents, and Birds. Front. Neural Circuits 2014, 8, 93. [Google Scholar] [CrossRef]
- Tolstenkov, O.; Mikhaleva, Y.; Glover, J.C. A Miniaturized Nigrostriatal-like Circuit Regulating Locomotor Performance in a Protochordate. Curr. Biol. 2023, 33, 3872–3883.e6. [Google Scholar] [CrossRef]
- Ardiel, E.L.; Rankin, C.H. An Elegant Mind: Learning and Memory in Caenorhabditis Elegans. Learn. Mem. 2010, 17, 191–201. [Google Scholar] [CrossRef]
- Pérez-Fernández, J.; Barandela, M.; Jiménez-López, C. The Dopaminergic Control of Movement-Evolutionary Considerations. Int. J. Mol. Sci. 2021, 22, 11284. [Google Scholar] [CrossRef]
- Yin, J.-A.; Liu, X.-J.; Yuan, J.; Jiang, J.; Cai, S.-Q. Longevity Manipulations Differentially Affect Serotonin/dopamine Level and Behavioral Deterioration in Aging Caenorhabditis Elegans. J. Neurosci. 2014, 34, 3947–3958. [Google Scholar] [CrossRef]
- Atkinson, L.E.; Liu, Y.; McKay, F.; Vandewyer, E.; Viau, C.; Irvine, A.; Rosa, B.A.; Li, Z.; Liang, Q.; Marks, N.J.; et al. Ascaris Suum Informs Extrasynaptic Volume Transmission in Nematodes. ACS Chem. Neurosci. 2021, 12, 3176–3188. [Google Scholar] [CrossRef]
- Fuxe, K.; Borroto-Escuela, D.O.; Romero-Fernandez, W.; Zhang, W.B.; Agnati, L.F. Volume transmission and its different forms in the central nervous system. Chin. J. Integr. Med. 2013, 19, 323–329. [Google Scholar] [CrossRef]
- Shin, M.; Wang, Y.; Borgus, J.R.; Venton, B.J. Electrochemistry at the Synapse. Annu. Rev. Anal. Chem. 2019, 12, 297–321. [Google Scholar] [CrossRef]
- Emborg, M.E.; Ma, S.Y.; Mufson, E.J.; Levey, A.I.; Taylor, M.D.; Brown, W.D.; Holden, J.E.; Kordower, J.H. Age-Related Declines in Nigral Neuronal Function Correlate with Motor Impairments in Rhesus Monkeys. J. Comp. Neurol. 1998, 401, 253–265. [Google Scholar] [CrossRef]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Hattori, N. Aging-Related Motor Function and Dopaminergic Neuronal Loss in C57BL/6 Mice. Mol. Brain 2020, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Su, L.-Y.; Yang, L.; Li, M.; Liu, Q.; Li, Z.; Hu, Y.; Li, H.; Wu, S.; Wang, W.; et al. A Cynomolgus Monkey with Naturally Occurring Parkinson’s Disease. Natl. Sci. Rev. 2021, 8, nwaa292. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yao, Y.-G.; Hu, X.-T. Biological Implications and Limitations of a Cynomolgus Monkey with Naturally Occurring Parkinson’s Disease. Zool Res 2021, 42, 138–140. [Google Scholar] [CrossRef]
- Adler, A. Melanin Pigment in the Brain of the Gorilla. J. Comp. Neurol. 1942, 76, 501–507. [Google Scholar] [CrossRef]
- Marsden, C.D. Pigmentation in the Nucleus Substantiae Nigrae of Mammals. J. Anat. 1961, 95, 256–261. [Google Scholar]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin Biosynthesis Is Driven by Excess Cytosolic Catecholamines Not Accumulated by Synaptic Vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain Tyrosinase Overexpression Implicates Age-Dependent Neuromelanin Production in Parkinson’s Disease Pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef]
- Raghanti, M.A.; Edler, M.K.; Stephenson, A.R.; Wilson, L.J.; Hopkins, W.D.; Ely, J.J.; Erwin, J.M.; Jacobs, B.; Hof, P.R.; Sherwood, C.C. Human-Specific Increase of Dopaminergic Innervation in a Striatal Region Associated with Speech and Language: A Comparative Analysis of the Primate Basal Ganglia. J. Comp. Neurol. 2016, 524, 2117–2129. [Google Scholar] [CrossRef]
- Raghanti, M.A.; Edler, M.K.; Stephenson, A.R.; Munger, E.L.; Jacobs, B.; Hof, P.R.; Sherwood, C.C.; Holloway, R.L.; Lovejoy, C.O. A Neurochemical Hypothesis for the Origin of Hominids. Proc. Natl. Acad. Sci. USA 2018, 115, E1108–E1116. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.M.M.; Zhu, Y.; Raghanti, M.A.; Kitchen, R.R.; Onorati, M.; Tebbenkamp, A.T.N.; Stutz, B.; Meyer, K.A.; Li, M.; Kawasawa, Y.I.; et al. Molecular and Cellular Reorganization of Neural Circuits in the Human Lineage. Science 2017, 358, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, A. Dopamine May Have given Humans Our Social Edge over Other Apes. Science 2018. Available online: https://www.science.org/content/article/dopamine-may-have-given-humans-our-social-edge-over-other-apes (accessed on 1 October 2023).
- Xenias, H.S.; Ibáñez-Sandoval, O.; Koós, T.; Tepper, J.M. Are Striatal Tyrosine Hydroxylase Interneurons Dopaminergic? J. Neurosci. 2015, 35, 6584–6599. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F.; Armentero, M.-T. Animal Models of Parkinson’s Disease. FEBS J. 2012, 279, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, P.; Zampese, E.; Surmeier, D.J. Selective Neuronal Vulnerability in Parkinson’s Disease. Prog. Brain Res. 2020, 252, 61–89. [Google Scholar]
- Surmeier, D.J.; Zhai, S.; Cui, Q.; Simmons, D.V. Rethinking the network determinants of motor disability in Parkinson’s disease. Front. Synaptic. Neurosci. 2023, 15, 1186484. [Google Scholar] [CrossRef] [PubMed]
- Rommelfanger, K.S.; Wichmann, T. Extrastriatal Dopaminergic Circuits of the Basal Ganglia. Front. Neuroanat. 2010, 4, 139. [Google Scholar] [CrossRef] [PubMed]
- Diederich, N.J.; Surmeier, D.J.; Uchihara, T.; Grillner, S.; Goetz, C.G. Parkinson’s disease: Is it a consequence of human brain evolution? Mov. Disord. 2019, 34, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Ubeda-Bañon, I.; Saiz-Sanchez, D.; Flores-Cuadrado, A.; Rioja-Corroto, E.; Gonzalez-Rodriguez, M.; Villar-Conde, S.; Astillero-Lopez, V.; Cabello-de la Rosa, J.P.; Gallardo-Alcañiz, M.J.; Vaamonde-Gamo, J.; et al. The Human Olfactory System in Two Proteinopathies: Alzheimer’s and Parkinson’s Diseases. Transl. Neurodegener. 2020, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Orimo, S.; Uchihara, T.; Kanazawa, T.; Itoh, Y.; Wakabayashi, K.; Kakita, A.; Takahashi, H. Unmyelinated Axons Are More Vulnerable to Degeneration than Myelinated Axons of the Cardiac Nerve in Parkinson’s Disease. Neuropathol. Appl. Neurobiol. 2011, 37, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Holmes, C.; Sullivan, P.; Lopez, G.; Gelsomino, J.; Moore, S.; Isonaka, R.; Wu, T.; Sharabi, Y. Cardiac Noradrenergic Deficiency Revealed by 18F-Dopamine Positron Emission Tomography Identifies Preclinical Central Lewy Body Diseases. J. Clin. Investig. 2023, 134, e172460. [Google Scholar] [CrossRef]
- Han, Y.; Wu, D.; Wang, Y.; Xie, J.; Zhang, Z. Skin Alpha-Synuclein Deposit Patterns: A Predictor of Parkinson’s Disease Subtypes. EBioMedicine 2022, 80, 104076. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuys, R. Structure and Organization of Fibre Systems. In The Central Nervous System of Vertebrates; Ten Donkelaar, H.J., Nicholson, C.N.R., Eds.; Springer: New York, NY, USA, 1999; Volume 1, pp. 113–157. [Google Scholar]
- Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson’s Disease. Cells 2020, 9, 2045. [Google Scholar] [CrossRef]
- Sokoloff, L.; Reivich, M.; Kennedy, C.; Des Rosiers, M.H.; Patlak, C.S.; Pettigrew, K.D.; Sakurada, O.; Shinohara, M. The [14C]deoxyglucose Method for the Measurement of Local Cerebral Glucose Utilization: Theory, Procedure, and Normal Values in the Conscious and Anesthetized Albino Rat. J. Neurochem. 1977, 28, 897–916. [Google Scholar] [CrossRef]
- Schröter, N.; Blazhenets, G.; Frings, L.; Jost, W.H.; Weiller, C.; Rijntjes, M.; Meyer, P.T.; Brumberg, J. Nigral Glucose Metabolism as a Diagnostic Marker of Neurodegenerative Parkinsonian Syndromes. NPJ Park. Dis. 2022, 8, 123. [Google Scholar] [CrossRef]
- Surmeier, D.J. From Causes of Selective Neuronal Vulnerability in Parkinson’s Disease to a Phase III Clinical Trial. C. David Marsden Award Lecture. 20th International Congress of Parkinson’s Disease and Movement Disorders. Available online: https://www.youtube.com/watch?v=h1WGbC2vMtQ (accessed on 1 October 2023).
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Liu, C.; Kershberg, L.; Wang, J.; Schneeberger, S.; Kaeser, P.S. Dopamine Secretion Is Mediated by Sparse Active Zone-like Release Sites. Cell 2018, 172, 706–718.e15. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Goel, P.; Kaeser, P.S. Spatial and Temporal Scales of Dopamine Transmission. Nat. Rev. Neurosci. 2021, 22, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Andersen, J.K. Dopaminergic Neurons. Int. J. Biochem. Cell Biol. 2005, 37, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Bolam, J.P.; Pissadaki, E.K. Living on the Edge with Too Many Mouths to Feed: Why Dopamine Neurons Die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Pissadaki, E.K.; Bolam, J.P. The Energy Cost of Action Potential Propagation in Dopamine Neurons: Clues to Susceptibility in Parkinson’s Disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Arbuthnott, G.W.; Wickens, J. Space, Time and Dopamine. Trends Neurosci. 2007, 30, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Vandecasteele, M.; Glowinski, J.; Venance, L. Electrical Synapses between Dopaminergic Neurons of the Substantia Nigra Pars Compacta. J. Neurosci. 2005, 25, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Joshua, M.; Adler, A.; Prut, Y.; Vaadia, E.; Wickens, J.R.; Bergman, H. Synchronization of Midbrain Dopaminergic Neurons Is Enhanced by Rewarding Events. Neuron 2009, 62, 695–704. [Google Scholar] [CrossRef]
- Eshel, N.; Tian, J.; Bukwich, M.; Uchida, N. Dopamine Neurons Share Common Response Function for Reward Prediction Error. Nat. Neurosci. 2016, 19, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Toyoshima, O.; Kunimatsu, J.; Yamada, H.; Matsumoto, M. Tonic Firing Mode of Midbrain Dopamine Neurons Continuously Tracks Reward Values Changing Moment-by-Moment. eLife 2021, 10, e63166. [Google Scholar] [CrossRef] [PubMed]
- Hamid, A.A.; Frank, M.J.; Moore, C.I. Wave-like Dopamine Dynamics as a Mechanism for Spatiotemporal Credit Assignment. Cell 2021, 184, 2733–2749.e16. [Google Scholar] [CrossRef] [PubMed]
- Travagli, R.A.; Gillis, R.A.; Rossiter, C.D.; Vicini, S. Glutamate and GABA-Mediated Synaptic Currents in Neurons of the Rat Dorsal Motor Nucleus of the Vagus. Am. J. Physiol. 1991, 260, G531–G536. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson’s Disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef]
- Um, K.B.; Hahn, S.; Kim, S.W.; Lee, Y.J.; Birnbaumer, L.; Kim, H.J.; Park, M.K. TRPC3 and NALCN Channels Drive Pacemaking in Substantia Nigra Dopaminergic Neurons. eLife 2021, 10, e70920. [Google Scholar] [CrossRef]
- Zampese, E.; Wokosin, D.L.; Gonzalez-Rodriguez, P.; Guzman, J.N.; Tkatch, T.; Kondapalli, J.; Surmeier, W.C.; D’Alessandro, K.B.; De Stefani, D.; Rizzuto, R.; et al. Ca2+ Channels Couple Spiking to Mitochondrial Metabolism in Substantia Nigra Dopaminergic Neurons. Sci. Adv. 2022, 8, eabp8701. [Google Scholar] [CrossRef]
- Tubert, C.; Zampese, E.; Pancani, T.; Tkatch, T.; Surmeier, D.J. Feed-Forward Metabotropic Signaling by Cav1 Ca2+ Channels Supports Pacemaking in Pedunculopontine Cholinergic Neurons. Neurobiol. Dis. 2023, 188, 106328. [Google Scholar] [CrossRef]
- Yamada, T.; McGeer, P.L.; Baimbridge, K.G.; McGeer, E.G. Relative Sparing in Parkinson’s Disease of Substantia Nigra Dopamine Neurons Containing Calbindin-D28K. Brain Res. 1990, 526, 303–307. [Google Scholar] [CrossRef]
- Inoue, K.-I.; Miyachi, S.; Nishi, K.; Okado, H.; Nagai, Y.; Minamimoto, T.; Nambu, A.; Takada, M. Recruitment of Calbindin into Nigral Dopamine Neurons Protects against MPTP-Induced Parkinsonism. Mov. Disord. 2019, 34, 200–209. [Google Scholar] [CrossRef]
- Brimblecombe, K.R.; Vietti-Michelina, S.; Platt, N.J.; Kastli, R.; Hnieno, A.; Gracie, C.J.; Cragg, S.J. Calbindin-D28K Limits Dopamine Release in Ventral but Not Dorsal Striatum by Regulating Ca2+ Availability and Dopamine Transporter Function. ACS Chem. Neurosci. 2019, 10, 3419–3426. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Dietz, J.V.; Fox, J.L.; Khalimonchuk, O. Down the Iron Path: Mitochondrial Iron Homeostasis and Beyond. Cells 2021, 10, 2198. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Thomas, R.A.; Ragheb, M.A.; Fallahi, A.; Fon, E.A. Mitochondrial Quality Control in Health and in Parkinson’s Disease. Physiol. Rev. 2022, 102, 1721–1755. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Stotland, A. MitoTimer: A Novel Protein for Monitoring Mitochondrial Turnover in the Heart. J. Mol. Med. 2015, 93, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial Dynamics in Adaptive and Maladaptive Cellular Stress Responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Nagumo, S.; Iwashita, S.; Okamoto, K. The ER Membrane Insertase Get1/2 Is Required for Efficient Mitophagy in Yeast. Biochem. Biophys. Res. Commun. 2018, 503, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-L.; Wang, T.T.; Luby-Phelps, K.; German, D.C. Mitochondria Mass Is Low in Mouse Substantia Nigra Dopamine Neurons: Implications for Parkinson’s Disease. Exp. Neurol. 2007, 203, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Bunney, B.S.; Aghajanian, G.K. D-Amphetamine-Induced Inhibition of Central Dopaminergic Neurons: Direct Effect or Mediated by a Striatonigral Feedback Pathway? Adv. Biochem. Psychopharmacol. 1977, 16, 577–582. [Google Scholar] [PubMed]
- Shepard, P.D.; German, D.C. Electrophysiological and Pharmacological Evidence for the Existence of Distinct Subpopulations of Nigrostriatal Dopaminergic Neuron in the Rat. Neuroscience 1988, 27, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-T.; Huang, N.; Sheng, Z.-H. Programming Axonal Mitochondrial Maintenance and Bioenergetics in Neurodegeneration and Regeneration. Neuron 2022, 110, 1899–1923. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Grady, J.P.; Cosgrave, E.M.; Bennison, E.; Chen, C.; Hepplewhite, P.D.; Morris, C.M. Mitochondrial Dysfunction within the Synapses of Substantia Nigra Neurons in Parkinson’s Disease. NPJ Park. Dis. 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Ilijic, E.; Guzman, J.N.; Surmeier, D.J. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2011, 43, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant Stress Evoked by Pacemaking in Dopaminergic Neurons Is Attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef]
- Guzman, J.N.; Ilijic, E.; Yang, B.; Sanchez-Padilla, J.; Wokosin, D.; Galtieri, D.; Kondapalli, J.; Schumacker, P.T.; Surmeier, D.J. Systemic Isradipine Treatment Diminishes Calcium-Dependent Mitochondrial Oxidant Stress. J. Clin. Investig. 2018, 128, 2266–2280. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Nguyen, J.T.; Lancki, N.; Venuto, C.S.; Oakes, D.; Simuni, T.; Wyse, R.K. Re-Analysis of the STEADY-PD II Trial-Evidence for Slowing the Progression of Parkinson’s Disease. Mov. Disord. 2022, 37, 334–342. [Google Scholar] [CrossRef]
- Venuto, C.S.; Yang, L.; Javidnia, M.; Oakes, D.; James Surmeier, D.; Simuni, T. Isradipine Plasma Pharmacokinetics and Exposure-Response in Early Parkinson’s Disease. Ann. Clin. Transl. Neurol. 2021, 8, 603–612. [Google Scholar] [CrossRef]
- Parkinson Study Group STEADY-PD III Investigators. Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann. Intern. Med. 2020, 172, 591–598. [Google Scholar] [CrossRef]
- Brimblecombe, K.R.; Connor-Robson, N.; Bataille, C.J.R.; Roberts, B.M.; Gracie, C.; O’Connor, B.; Te Water Naude, R.; Karthik, G.; Russell, A.J.; Wade-Martins, R.; et al. Inhibition of Striatal Dopamine Release by the L-Type Calcium Channel Inhibitor Isradipine Co-Varies with Risk Factors for Parkinson’s. Eur. J. Neurosci. 2023. [Google Scholar] [CrossRef]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much More than Mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial Dysfunction and Oxidative Damage in Parkin-Deficient Mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, QLD, Australia, 2018; Chapter 1. [Google Scholar]
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Hoffer, A.; Hoffer, B.; Qi, X. Mitochondria: A Therapeutic Target for Parkinson’s Disease? Int. J. Mol. Sci. 2015, 16, 20704–20730. [Google Scholar] [CrossRef]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-Linked Oxidation in Brain Mitochondria by 1-Methyl-4-Phenyl-Pyridine, a Metabolite of the Neurotoxin, 1-Methyl-4-Phenyl-1,2,5,6-Tetrahydropyridine. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef] [PubMed]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-Inducing Neurotoxin, N-Methyl-4-Phenyl-1,2,3,6 -Tetrahydropyridine: Uptake of the Metabolite N-Methyl-4-Phenylpyridine by Dopamine Neurons Explains Selective Toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of Mitochondrial Complex I Induces Progressive Parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and Propagation of α-Synuclein Aggregation in the Nervous System. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Graybiel, A.M.; Duyckaerts, C.; Javoy-Agid, F. Neuronal Loss in the Pedunculopontine Tegmental Nucleus in Parkinson Disease and in Progressive Supranuclear Palsy. Proc. Natl. Acad. Sci. USA. 1987, 84, 5976–5980. [Google Scholar] [CrossRef] [PubMed]
- Chambers, N.E.; Lanza, K.; Bishop, C. Pedunculopontine Nucleus Degeneration Contributes to Both Motor and Non-Motor Symptoms of Parkinson’s Disease. Front. Pharmacol. 2019, 10, 1494. [Google Scholar] [CrossRef] [PubMed]
- Greenamyre, J.T.; Hastings, T.G. Biomedicine. Parkinson’s--Divergent Causes, Convergent Mechanisms. Science 2004, 304, 1120–1122. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Dopaminergic Neuron-Specific Oxidative Stress Caused by Dopamine Itself. Acta Med. Okayama 2008, 62, 141–150. [Google Scholar]
- Bolton, J.L.; Trush, M.A.; Penning, T.M.; Dryhurst, G.; Monks, T.J. Role of quinones in toxicology. Chem. Res. Toxicol. 2000, 13, 135–160. [Google Scholar] [CrossRef]
- Kleppe, R.; Waheed, Q.; Ruoff, P. DOPA Homeostasis by Dopamine: A Control-Theoretic View. Int. J. Mol. Sci. 2021, 22, 12862. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Zecca, L.; Bellei, C.; Costi, P.; Albertini, A.; Monzani, E.; Casella, L.; Gallorini, M.; Bergamaschi, L.; Moscatelli, A.; Turro, N.J.; et al. New Melanic Pigments in the Human Brain That Accumulate in Aging and Block Environmental Toxic Metals. Proc. Natl. Acad. Sci. USA 2008, 105, 17567–17572. [Google Scholar] [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of Iron, Dopamine and Neuromelanin Pathways in Brain Aging and Parkinson’s Disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- Parent, M.; Parent, A. Substantia Nigra and Parkinson’s Disease: A Brief History of Their Long and Intimate Relationship. Can. J. Neurol. Sci. 2010, 37, 313–319. [Google Scholar] [CrossRef]
- Halliday, G.M.; Ophof, A.; Broe, M.; Jensen, P.H.; Kettle, E.; Fedorow, H.; Cartwright, M.I.; Griffiths, F.M.; Shepherd, C.E.; Double, K.L. Alpha-Synuclein Redistributes to Neuromelanin Lipid in the Substantia Nigra Early in Parkinson’s Disease. Brain 2005, 128, 2654–2664. [Google Scholar] [CrossRef]
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial Iron Metabolism and Its Role in Neurodegeneration. J. Alzheimers. Dis. 2010, 20 (Suppl. S2), S551–S568. [Google Scholar] [CrossRef]
- Zareba, M.; Bober, A.; Korytowski, W.; Zecca, L.; Sarna, T. The Effect of a Synthetic Neuromelanin on Yield of Free Hydroxyl Radicals Generated in Model Systems. Biochim. Biophys. Acta 1995, 1271, 343–348. [Google Scholar] [CrossRef]
- Zecca, L.; Zucca, F.A.; Albertini, A.; Rizzio, E.; Fariello, R.G. A Proposed Dual Role of Neuromelanin in the Pathogenesis of Parkinson’s Disease. Neurology 2006, 67, S8–S11. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Wilms, H.; Geick, S.; Claasen, J.-H.; Brandenburg, L.-O.; Holzknecht, C.; Panizza, M.L.; Zucca, F.A.; Deuschl, G.; Sievers, J.; et al. Human Neuromelanin Induces Neuroinflammation and Neurodegeneration in the Rat Substantia Nigra: Implications for Parkinson’s Disease. Acta Neuropathol. 2008, 116, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Cassidy, C.; Horga, G.; Kang, U.J.; Fahn, S.; Casella, L.; Pezzoli, G.; Langley, J.; Hu, X.P.; Zucca, F.A.; et al. Neuromelanin detection by magnetic resonance imaging (MRI) and its promise as a biomarker for Parkinson’s disease. Npj Park. Dis. 2018, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, S.; Berg, D.; Gasser, T.; Chen, H.; Yao, C.; Postuma, R.B. MDS Task Force on the Definition of Parkinson’s Disease Update of the MDS Research Criteria for Prodromal Parkinson’s Disease. Mov. Disord. 2019, 34, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Belin, A.C.; Westerlund, M. Parkinson’s Disease: A Genetic Perspective. FEBS J. 2008, 275, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental Toxins and Parkinson’s Disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Davis, G.C.; Williams, A.C.; Markey, S.P.; Ebert, M.H.; Caine, E.D.; Reichert, C.M.; Kopin, I.J. Chronic Parkinsonism Secondary to Intravenous Injection of Meperidine Analogues. Psychiatry Res. 1979, 1, 249–254. [Google Scholar] [CrossRef]
- Ullah, I.; Zhao, L.; Hai, Y.; Fahim, M.; Alwayli, D.; Wang, X.; Li, H. Metal Elements and Pesticides as Risk Factors for Parkinson’s Disease—A Review. Toxicol. Rep. 2021, 8, 607–616. [Google Scholar] [CrossRef]
- Goldman, S.M.; Weaver, F.M.; Stroupe, K.T.; Cao, L.; Gonzalez, B.; Colletta, K.; Brown, E.G.; Tanner, C.M. Risk of Parkinson Disease Among Service Members at Marine Corps Base Camp Lejeune. JAMA Neurol. 2023, 80, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition Metals, Ferritin, Glutathione, and Ascorbic Acid in Parkinsonian Brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and Oxidized Glutathione in the Substantia Nigra of Patients with Parkinson’s Disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.L.; Teismann, P. Glutathione--a Review on Its Role and Significance in Parkinson’s Disease. FASEB J. 2009, 23, 3263–3272. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Monoranu, C.; Strobel, S.; Iordache, T.; Sian-Hülsmann, J. Iron as the Concert Master in the Pathogenic Orchestra Playing in Sporadic Parkinson’s Disease. J. Neural Transm. 2021, 128, 1577–1598. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Ben-Shachar, D.; Riederer, P. The Possible Role of Iron in the Etiopathology of Parkinson’s Disease. Mov. Disord. 1993, 8, 1–12. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased Nigral Iron Content in Postmortem Parkinsonian Brain. Lancet 1987, 2, 1219–1220. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B. Increased Iron (III) and Total Iron Content in Post Mortem Substantia Nigra of Parkinsonian Brain. J. Neural Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Zhuang, Q.-Q.; Zhu, L.-B.; Zhu, H.; Li, T.; Li, R.; Chen, S.-F.; Huang, C.-P.; Zhang, X.; Zhu, J.-H. Meta-Analysis of Brain Iron Levels of Parkinson’s Disease Patients Determined by Postmortem and MRI Measurements. Sci. Rep. 2016, 6, 36669. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a Newly Characterized Form of Cell Death in Parkinson’s Disease That Is Regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.; Kumar Sreelatha, A.A.; Pihlstrom, L.; Domenighetti, C.; Schulte, C.; Sugier, P.-E.; Radivojkov-Blagojevic, M.; Lichtner, P.; Mohamed, O.; Portugal, B.; et al. Genome-Wide Association and Meta-Analysis of Age at Onset in Parkinson Disease: Evidence From the COURAGE-PD Consortium. Neurology 2022, 99, e698–e710. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Lima, T.; Li, T.Y.; Mottis, A.; Auwerx, J. Pleiotropic Effects of Mitochondria in Aging. Nat Aging 2022, 2, 199–213. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.; Ye, K.; Picard, M.; Gu, Z. Independent Impacts of Aging on Mitochondrial DNA Quantity and Quality in Humans. BMC Genomics 2017, 18, 890. [Google Scholar] [CrossRef]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The Role of Iron and Copper Molecules in the Neuronal Vulnerability of Locus Coeruleus and Substantia Nigra during Aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef]
- Venkateshappa, C.; Harish, G.; Mythri, R.B.; Mahadevan, A.; Bharath, M.M.S.; Shankar, S.K. Increased Oxidative Damage and Decreased Antioxidant Function in Aging Human Substantia Nigra Compared to Striatum: Implications for Parkinson’s Disease. Neurochem. Res. 2012, 37, 358–369. [Google Scholar] [CrossRef]
- Finger, C.E.; Moreno-Gonzalez, I.; Gutierrez, A.; Moruno-Manchon, J.F.; McCullough, L.D. Age-Related Immune Alterations and Cerebrovascular Inflammation. Mol. Psychiatry 2022, 27, 803–818. [Google Scholar] [CrossRef]
- Cramb, K.M.L.; Beccano-Kelly, D.; Cragg, S.J.; Wade-Martins, R. Impaired Dopamine Release in Parkinson’s Disease. Brain 2023, 146, 3117–3132. [Google Scholar] [CrossRef]
- Chen, H.; Huang, X.; Guo, X.; Mailman, R.B.; Park, Y.; Kamel, F.; Umbach, D.M.; Xu, Q.; Hollenbeck, A.; Schatzkin, A.; et al. Smoking Duration, Intensity, and Risk of Parkinson Disease. Neurology 2010, 74, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-Analysis of Early Nonmotor Features and Risk Factors for Parkinson Disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef]
- Pathak, D.; Berthet, A.; Nakamura, K. Energy Failure: Does It Contribute to Neurodegeneration? Ann. Neurol. 2013, 74, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Saiki, S.; Hatano, T.; Fujimaki, M.; Ishikawa, K.-I.; Mori, A.; Oji, Y.; Okuzumi, A.; Fukuhara, T.; Koinuma, T.; Imamichi, Y.; et al. Decreased Long-Chain Acylcarnitines from Insufficient β-Oxidation as Potential Early Diagnostic Markers for Parkinson’s Disease. Sci. Rep. 2017, 7, 7328. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.M.; Hernán, M.A.; Willett, W.C.; Ascherio, A. Weight Loss in Parkinson’s Disease. Ann. Neurol. 2003, 53, 676–679. [Google Scholar] [CrossRef]
- Song, S.; Luo, Z.; Li, C.; Huang, X.; Shiroma, E.J.; Simonsick, E.M.; Chen, H. Changes in Body Composition Before and After Parkinson’s Disease Diagnosis. Mov. Disord. 2021, 36, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Sheard, J.M.; Ash, S.; Silburn, P.A.; Kerr, G.K. Prevalence of Malnutrition in Parkinson’s Disease: A Systematic Review. Nutr. Rev. 2011, 69, 520–532. [Google Scholar] [CrossRef]
- Yong, V.W.; Tan, Y.J.; Ng, Y.-D.; Choo, X.Y.; Sugumaran, K.; Chinna, K.; Md Shah, M.N.; Raja Aman, R.R.A.; Moy, F.M.; Mohd Ramli, N.; et al. Progressive and Accelerated Weight and Body Fat Loss in Parkinson’s Disease: A Three-Year Prospective Longitudinal Study. Parkinsonism Relat. Disord. 2020, 77, 28–35. [Google Scholar] [CrossRef]
- Kim, H.J.; Oh, E.S.; Lee, J.H.; Moon, J.S.; Oh, J.E.; Shin, J.W.; Lee, K.J.; Baek, I.C.; Jeong, S.-H.; Song, H.-J.; et al. Relationship between Changes of Body Mass Index (BMI) and Cognitive Decline in Parkinson’s Disease (PD). Arch. Gerontol. Geriatr. 2012, 55, 70–72. [Google Scholar] [CrossRef]
- Park, K.; Oeda, T.; Kohsaka, M.; Tomita, S.; Umemura, A.; Sawada, H. Low Body Mass Index and Life Prognosis in Parkinson’s Disease. Parkinsonism Relat. Disord. 2018, 55, 81–85. [Google Scholar] [CrossRef]
- Andersen, K.B.; Hansen, A.K.; Schacht, A.C.; Horsager, J.; Gottrup, H.; Klit, H.; Danielsen, E.H.; Poston, K.L.; Pavese, N.; Brooks, D.J.; et al. Synaptic Density and Glucose Consumption in Patients with Lewy Body Diseases: An [11 C]UCB-J and [18 F]FDG PET Study. Mov. Disord. 2023, 38, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, P.W.; de Pablo Fernandez, E.; König, A.; Outeiro, T.F.; Jaunmuktane, Z.; Warner, T.T. Type 2 Diabetes and Parkinson’s Disease: A Focused Review of Current Concepts. Mov. Disord. 2023, 38, 162–177. [Google Scholar] [CrossRef]
- Hattingen, E.; Magerkurth, J.; Pilatus, U.; Mozer, A.; Seifried, C.; Steinmetz, H.; Zanella, F.; Hilker, R. Phosphorus and Proton Magnetic Resonance Spectroscopy Demonstrates Mitochondrial Dysfunction in Early and Advanced Parkinson’s Disease. Brain 2009, 132, 3285–3297. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Imamura, H.; Sasaoka, N.; Yamamoto, M.; Uemura, N.; Shudo, T.; Fuchigami, T.; Takahashi, R.; Kakizuka, A. ATP Maintenance via Two Types of ATP Regulators Mitigates Pathological Phenotypes in Mouse Models of Parkinson’s Disease. EBioMedicine 2017, 22, 225–241. [Google Scholar] [CrossRef]
- Tang, B.L. Glucose, Glycolysis, and Neurodegenerative Diseases. J. Cell. Physiol. 2020, 235, 7653–7662. [Google Scholar] [CrossRef]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a Biological Hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Takaine, M.; Imamura, H.; Yoshida, S. High and Stable ATP Levels Prevent Aberrant Intracellular Protein Aggregation in Yeast. eLife 2022, 11, e67659. [Google Scholar] [CrossRef]
- McKenzie, M.; Liolitsa, D.; Akinshina, N.; Campanella, M.; Sisodiya, S.; Hargreaves, I.; Nirmalananthan, N.; Sweeney, M.G.; Abou-Sleiman, P.M.; Wood, N.W.; et al. Mitochondrial ND5 Gene Variation Associated with Encephalomyopathy and Mitochondrial ATP Consumption. J. Biol. Chem. 2007, 282, 36845–36852. [Google Scholar] [CrossRef]
- Dunn, L.; Allen, G.F.; Mamais, A.; Ling, H.; Li, A.; Duberley, K.E.; Hargreaves, I.P.; Pope, S.; Holton, J.L.; Lees, A.; et al. Dysregulation of Glucose Metabolism Is an Early Event in Sporadic Parkinson’s Disease. Neurobiol. Aging 2014, 35, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.A.; Sivakumar, K.; Tabakovic, E.E.; Oya, M.; Aldridge, G.M.; Zhang, Q.; Simmering, J.E.; Narayanan, N.S. Glycolysis-Enhancing α1-Adrenergic Antagonists Modify Cognitive Symptoms Related to Parkinson’s Disease. NPJ Park. Dis. 2023, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.A.; Jinnah, H.A.; Kamatani, N. Shortage of Cellular ATP as a Cause of Diseases and Strategies to Enhance ATP. Front. Pharmacol. 2019, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Hattori, T.; Kume, A.; Misu, K.; Ito, T.; Koike, Y.; Johnson, T.A.; Kamitsuji, S.; Kamatani, N.; Sobue, G. Improved Parkinsons disease motor score in a single-arm open-label trial of febuxostat and inosine. Medicine 2020, 99, e21576. [Google Scholar] [CrossRef] [PubMed]
- Chopade, P.; Chopade, N.; Zhao, Z.; Mitragotri, S.; Liao, R.; Chandran Suja, V. Alzheimer’s and Parkinson’s disease therapies in the clinic. Bioeng. Transl. Med. 2022, 8, e10367. [Google Scholar] [CrossRef] [PubMed]
- Rascol, O.; Fabbri, M.; Poewe, W. Amantadine in the treatment of Parkinson’s disease and other movement disorders. Lancet Neurol. 2021, 20, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Gudala, K.; Kanukula, R.; Bansal, D. Reduced Risk of Parkinson’s Disease in Users of Calcium Channel Blockers: A Meta-Analysis. Int. J. Chronic. Dis. 2015, 2015, 697404. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Zhang, Y.; Simmering, J.E.; Schultz, J.L.; Li, Y.; Fernandez-Carasa, I.; Consiglio, A.; Raya, A.; Polgreen, P.M.; Narayanan, N.S.; et al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J. Clin. Investig. 2019, 129, 4539–4549. [Google Scholar] [CrossRef]
- Simmering, J.E.; Welsh, M.J.; Liu, L.; Narayanan, N.S.; Pottegård, A. Association of Glycolysis-Enhancing α-1 Blockers With Risk of Developing Parkinson Disease. JAMA Neurol. 2021, 78, 407–413. [Google Scholar] [CrossRef]
- Kamatani, N.; Hashimoto, M.; Sakurai, K.; Gokita, K.; Yoshihara, J.; Sekine, M.; Mochii, M.; Fukuuchi, T.; Yamaoka, N.; Kaneko, K. Clinical studies on changes in purine compounds in blood and urine by the simultaneous administration of febuxostat and inosine, or by single administration of each. Gout Nucleic Acid Metab. 2017, 41, 171–181. [Google Scholar] [CrossRef][Green Version]
- Song, Y.; Racette, B.A.; Camacho-Soto, A.; Searles Nielsen, S. Biologic targets of prescription medications and risk of neurodegenerative disease in United States Medicare beneficiaries. PLoS ONE 2023, 18, e0285011. [Google Scholar] [CrossRef] [PubMed]
- García-Beltrán, O.; Urrutia, P.J.; Núñez, M.T. On the Chemical and Biological Characteristics of Multifunctional Compounds for the Treatment of Parkinson’s Disease. Antioxidants 2023, 12, 214. [Google Scholar] [CrossRef]
- Kim, S.; Moon, M.; Park, S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J. Endocrinol. 2009, 202, 431–439. [Google Scholar] [CrossRef]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Price, D.L.; Khan, A.; Angers, R.; Cardenas, A.; Prato, M.K.; Bani, M.; Bonhaus, D.W.; Citron, M.; Biere, A.L. In vivo effects of the alpha-synuclein misfolding inhibitor minzasolmin supports clinical development in Parkinson’s disease. NPJ Park. Dis. 2023, 9, 114. [Google Scholar] [CrossRef]
- Pagano, G.; PASADENA Investigators; Prasinezumab Study Group. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Huang, L.Z.; Parameswaran, N.; Bordia, T.; Campos, C.; Perez, X.A. Multiple roles for nicotine in Parkinson’s disease. Biochem. Pharmacol. 2009, 78, 677–685. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
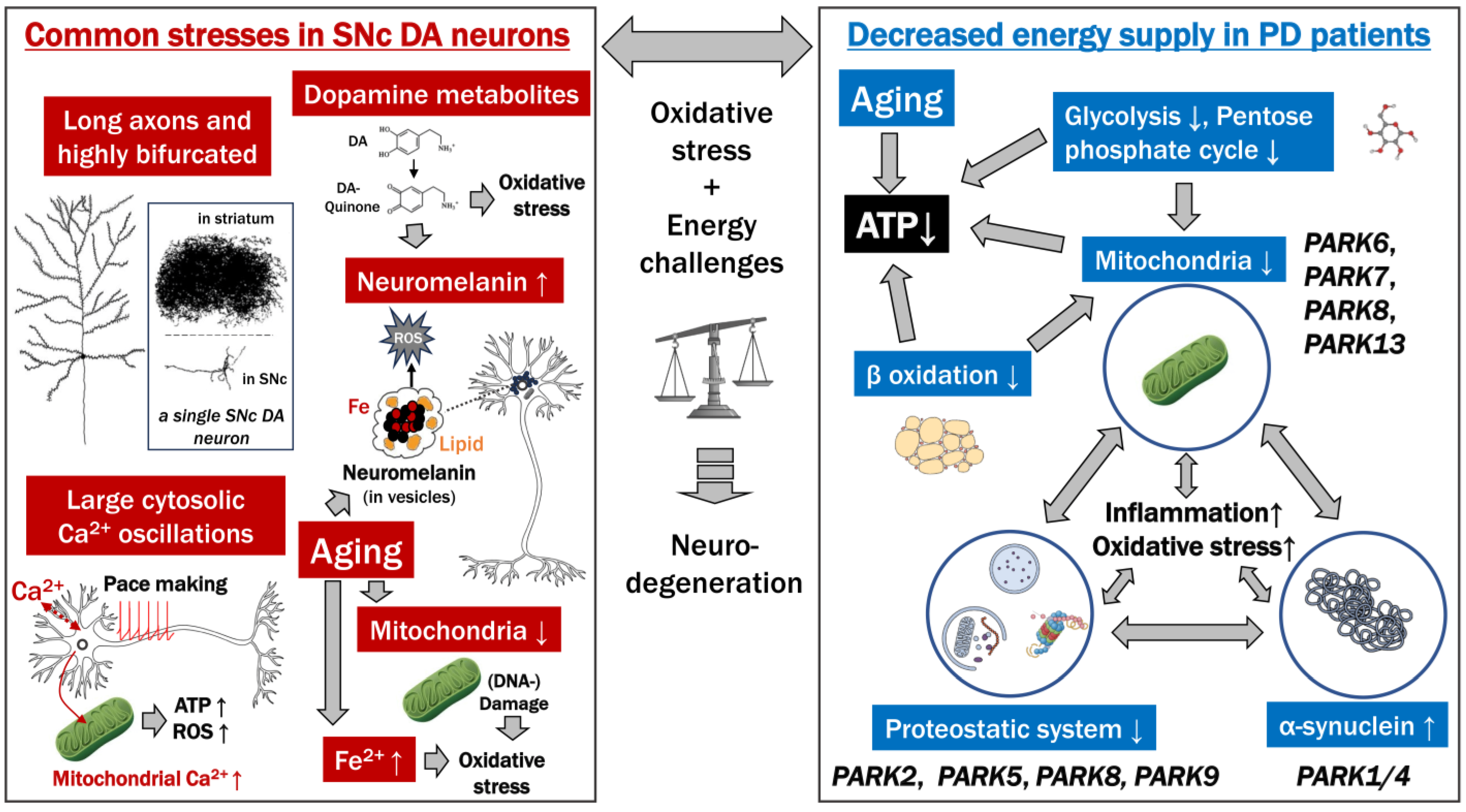
Relatively certain contributing factors
|
Speculative contributing factors
|
| Examples of Models Explaining Neural Vulnerabilities in PD | |
|---|---|
| Reference | Statements Relevant to the Model |
| Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s Disease: Possible Routes by Which Vulnerable Neuronal Types May Be Subject to Neuroinvasion by an Unknown Pathogen. J. Neural Transm. 2003, 110, 517–536 [20]. | [about the common properties of neurons vulnerable to PD] “All of the vulnerable cells belong to the class of projection neurons. Within this class, only some neuronal types, namely those which generate axons that are disproportionately long in relation to their somata, demonstrate a pronounced tendency to develop the lesions”. “An additional feature shared by all of the endangered neuronal types is that their long and thin axons are unmyelinated or only partially myelinated”. [about the energy usage by these neurons] “The maintenance of unmyelinated or incompletely myelinated axons requires prodigious expenditures of energy”. |
| Sulzer, D.; Surmeier, D.J. Neuronal Vulnerability, Pathogenesis, and Parkinson’s Disease. Mov. Disord. 2013, 28, 715–724 [35]. | [about the (common) properties of neurons vulnerable to PD] “There appears to be a small number of risk factors contributing to vulnerability. These include autonomous activity, broad action potentials, low intrinsic calcium buffering capacity, poorly myelinated long highly branched axons and terminal fields, and use of a monoamine neurotransmitter, often with the catecholamine-derived neuromelanin pigment”. “SNc, LC, RN, PPN, and NBM neurons all have unusually long highly branched axons that are unmyelinated or thinly myelinated”. [about the energy usage and metabolic stress of SNc DA neurons related to their calcium pumping] “Calcium entry is energetically expensive because it must be pumped out of the cell against a much steeper electrochemical gradient than any of the other ions”. “One of the ion channels contributing to the basal metabolic stress in SNc DA neurons is the L-type calcium channel”. |
| Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson’s Disease. Cells 2020, 9, 2045 [81]. | [about the properties of neurons, especially SNc DA neurons, that make them vulnerable to PD] “The available data indicates that an extensive axonal branching, autonomous pacemaking, and Cav1 channel-mediated feedforward control of mitochondrial OXPHOS (and the consequent mitochondrial oxidant stress) might be key features determining neuronal vulnerability in PD”. “Unlike most neurons, SN DAergic neurons appear to have a high basal bioenergetic demand. This demand may have its roots in several factors. The most important of these is likely to be the neuron’s massive axonal arbor. This arbor creates an anabolic demand, as it has to be supplied with release-related proteins and lipids largely delivered by axonal transport from the somatic region”. |
| Some Critical Considerations | |
| 1. There may not be good evidence that SNc DA neurons are poorly myelinated. | |
| 2. The glucose consumption of the SNc is not especially high (Sokoloff, L.; et al. J. Neurochem. 1977, 28, 897–916 [82]; Schröter, N.; et al. NPJ Parkinson’s Dis. 2022, 8, 123 [83]), arguing against an explanation of stress in SNc DA neurons due to unusually high energy demands. | |
Arguments that neurodegeneration starts in the cell bodies
|
Arguments that neurodegeneration starts in the axonal arbor
|
| PARK | Gene | Protein | Function |
|---|---|---|---|
| PARK1, PARK4 | SNCA | α-synuclein | Uncertain, but misfolding causes Lewy Bodies |
| PARK2 | PRKN | Parkin, E3 ubiquitin ligase | Mitochondrial |
| PARK5 | UCHL1 | Ubiquitin C-terminal hydrolase L1 | Ubiquitin-proteasome |
| PARK6 | PINK1 | PTEN-induced putative kinase 1 | Mitochondrial |
| PARK7 | DJ-1 | Parkinsonism-associated deglycase | Mitochondrial |
| PARK8 | LRRK2 | Leucine-rich repeat kinase 2 | Lysosomal, mitochondrial, microtubule |
| PARK9 | ATP13A2 | Cation-transporting ATPase 13A2 | Lysosomal |
| PARK11 | GIGYF2 | GRB10 interacting GYF protein 2 | Uncertain |
| PARK13 | HTRA2 | HtrA serine peptidase 2 | Mitochondrial |
| PARK14 | PLA2G6 | Calcium-independent phospholipase A2 enzyme | Cell membrane |
| PARK15 | FBX07 | F-box protein 7 | Mitochondrial |
| PARK17 | VPS35 | Vacuolar protein sorting-associated protein 35 | Retromer and endosomal trafficking |
| PARK18 | EIF4G1 | Eukaryotic translation initiation factor 4 gamma 1 | Transcription |
| PARK19 | DNAJC6 | HSP40 Auxilin | Synaptic vesicle formation and trafficking |
| PARK20 | SYNJ1 | Synaptojanin 1 | Synaptic vesicle formation and trafficking |
| PARK21 | DNAJC13 | Receptor-mediated endocytosis 8 (RME-8) | Synaptic vesicle formation and trafficking |
| PARK23 | VPS13C | Vacuolar protein sorting-associated protein 13C | Mitochondrial |
Reducing the accumulation of intracellular DA
|
Reducing the calcium ion influx
|
Assuring a proper energy supply
|
Antioxidant
|
Inhibition of inflammation
|
Reducing α-synuclein aggregation
|
Why is PD only prevalent in humans?
|
Does increased DA release help protect against PD?
(note: We speculate that the lack of major success in clinical trials that administered nicotine to halt PD progression (reviewed by Quik M.; et al., Biochem. Pharmacol. 2009, 8, 677–685 [213]) may not have sufficiently replicated the DA release regimen of a heavy smoker) |
Why has isradipine not been (very) successful against PD?
|
Can the energy status of SNc DA neurons be improved/protected?
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, H.; Dijkstra, J.M.; Nagatsu, T. Parkinson’s Disease: Cells Succumbing to Lifelong Dopamine-Related Oxidative Stress and Other Bioenergetic Challenges. Int. J. Mol. Sci. 2024, 25, 2009. https://doi.org/10.3390/ijms25042009
Watanabe H, Dijkstra JM, Nagatsu T. Parkinson’s Disease: Cells Succumbing to Lifelong Dopamine-Related Oxidative Stress and Other Bioenergetic Challenges. International Journal of Molecular Sciences. 2024; 25(4):2009. https://doi.org/10.3390/ijms25042009
Chicago/Turabian StyleWatanabe, Hirohisa, Johannes M. Dijkstra, and Toshiharu Nagatsu. 2024. "Parkinson’s Disease: Cells Succumbing to Lifelong Dopamine-Related Oxidative Stress and Other Bioenergetic Challenges" International Journal of Molecular Sciences 25, no. 4: 2009. https://doi.org/10.3390/ijms25042009
APA StyleWatanabe, H., Dijkstra, J. M., & Nagatsu, T. (2024). Parkinson’s Disease: Cells Succumbing to Lifelong Dopamine-Related Oxidative Stress and Other Bioenergetic Challenges. International Journal of Molecular Sciences, 25(4), 2009. https://doi.org/10.3390/ijms25042009