
Pathogenic Impact of Fatty Acid-Binding Proteins in Parkinson’s Disease—Potential Biomarkers and Therapeutic Targets


Abstract
:1. Introduction
2. Physiological Function of FABP and Involvement in Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FABP Subfamily | Tissue Distribution | Expressed Cells | Ref |
|---|---|---|---|
| FABP1 (Liver FABP) | Liver, intestine, kidney, pancreas | Hepatocytes, enterocytes | [33,34,35] |
| FABP2 (Intestinal FABP) | Intestine | Enterocytes | [36,37,38] |
| FABP3 (Heart FABP) | Heart, skeletal muscle, brain | Cardiomyocytes, myocytes, neurons | [18,39,40,41,42,43] |
| FABP4 (Adipocyte FABP) | Adipose tissue, macrophages | Adipocytes, macrophages | [44,45,46,47] |
| FABP5 (Epidermal FABP) | Epidermis, brain, adipose tissue | Keratinocytes, adipocytes, glial cells, neurons | [18,43,48,49,50] |
| FABP6 (Ileal FABP) | Intestine | Enterocytes | [51,52,53,54] |
| FABP7 (Brain FABP) | Brain, eye, kidney, mammary gland | Neural stem cells, oligodendrocytes, astrocytes, ependymal cells | [18,41,43,55,56] |
| FABP8 (Myelin FABP) | Myelin-forming cells in the peripheral nervous system | Schwann cells, oligodendrocytes | [43,56] |
| FABP9 (Testis FABP) | Testis | Salivary gland, mammary gland | [57,58] |
3. Pathology of Parkinson’s Disease and Current Issues
4. Biochemistry and Pathology of Tyrosine Hydroxylase
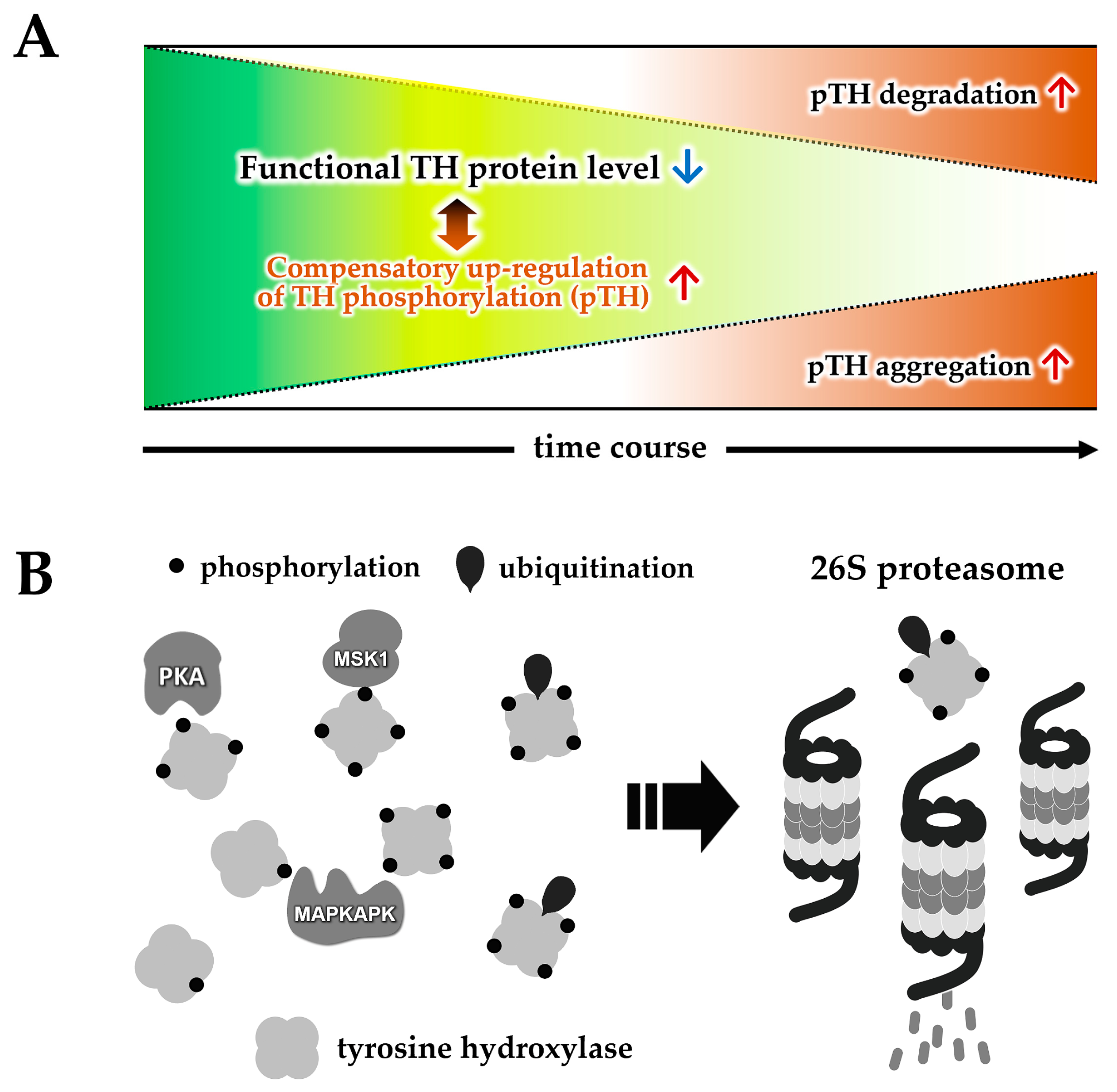
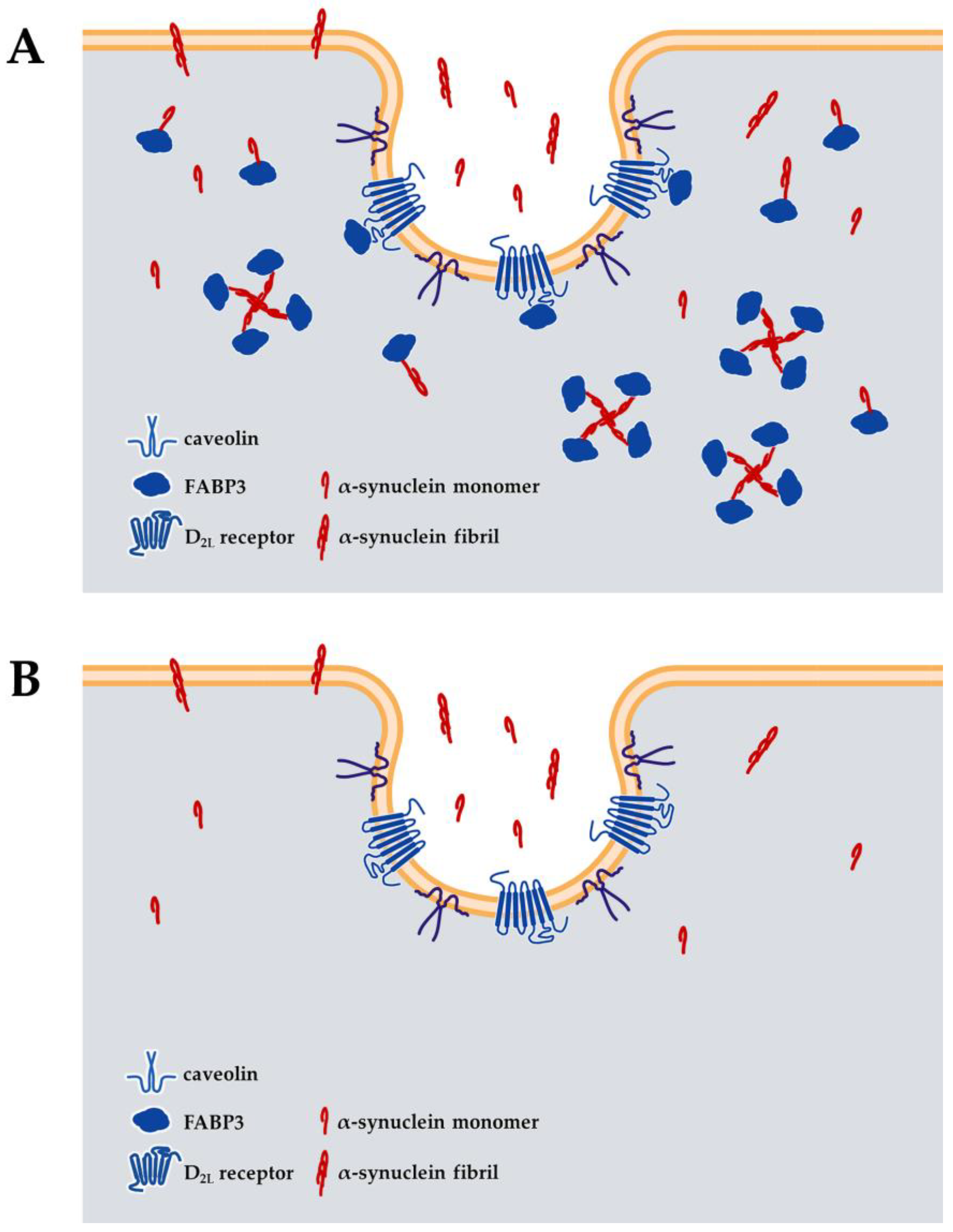
5. Impact of FABP on the Loss of TH Protein and α-Synuclein Aggregation
6. Pathogenic Impact of Fatty Acid-Binding Proteins on Mitochondrial Homeostasis
7. Therapeutic Potential of FABP-Targeting Drugs for Parkinson’s Disease
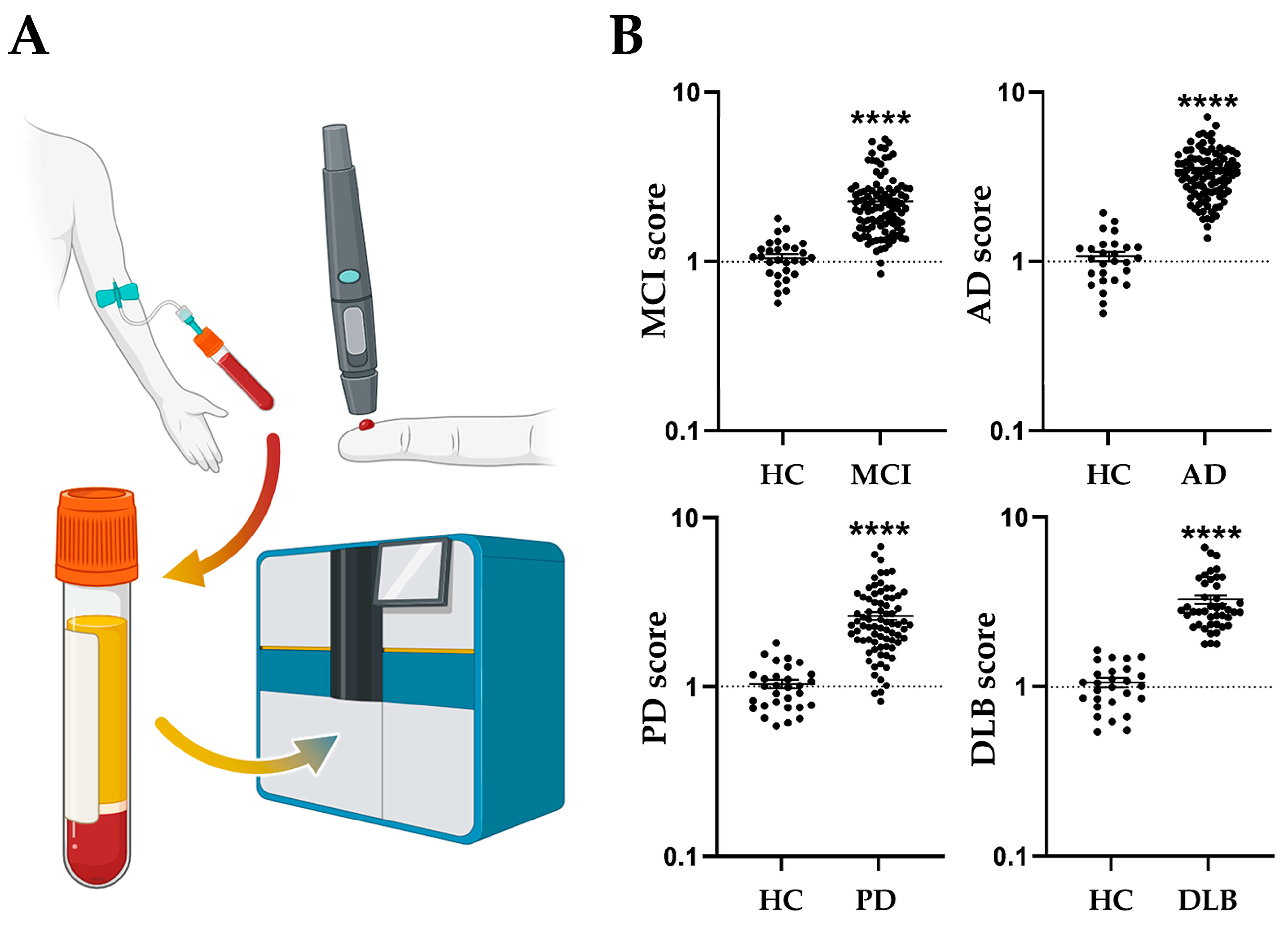
8. Diagnostic Potential of FABPs as a Prodromal Biomarker for Parkinson’s Diseases
9. Conclusions
10. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zimmermann-Ivol, C.G.; Burkhard, P.R.; Le Floch-Rohr, J.; Allard, L.; Hochstrasser, D.F.; Sanchez, J.C. Fatty acid binding protein as a serum marker for the early diagnosis of stroke: A pilot study. Mol. Cell. Proteom. 2004, 3, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Steinacker, P.; Bahn, E.; Bibl, M.; Brechlin, P.; Schlossmacher, M.G.; Locascio, J.J.; Wiltfang, J.; Kretzschmar, H.A.; Poser, S.; et al. Serum heart-type fatty acid-binding protein and cerebrospinal fluid tau: Marker candidates for dementia with Lewy bodies. Neurodegener. Dis. 2007, 4, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Wada-Isoe, K.; Imamura, K.; Kitamaya, M.; Kowa, H.; Nakashima, K. Serum heart-fatty acid binding protein levels in patients with Lewy body disease. J. Neurol. Sci. 2008, 266, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; Veerhuis, R.; De Vente, J.; Verhey, F.R.; Vreeling, F.; van Boxtel, M.P.; Glatz, J.F.; Pelsers, M.A. Brain-specific fatty acid-binding protein is elevated in serum of patients with dementia-related diseases. Eur. J. Neurol. 2011, 18, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Diolintzi, A.; Storch, J. Fatty acid-binding proteins: Functional understanding and diagnostic implications. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Ockner, R.K.; Manning, J.A.; Poppenhausen, R.B.; Ho, W.K. A binding protein for fatty acids in cytosol of intestinal mucosa, liver, myocardium, and other tissues. Science 1972, 177, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Veerkamp, J.H. Fatty acid transport and fatty acid-binding proteins. Proc. Nutr. Soc. 1995, 54, 23–37. [Google Scholar] [CrossRef]
- Coe, N.R.; Bernlohr, D.A. Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim. Biophys. Acta 1998, 1391, 287–306. [Google Scholar] [CrossRef]
- Storch, J.; Thumser, A.E. The fatty acid transport function of fatty acid-binding proteins. Biochim. Biophys. Acta 2000, 1486, 28–44. [Google Scholar] [CrossRef]
- Chmurzyńska, A. The multigene family of fatty acid-binding proteins (FABPs): Function, structure and polymorphism. J. Appl. Genet. 2006, 47, 39–48. [Google Scholar] [CrossRef]
- Smathers, R.L.; Petersen, D.R. The human fatty acid-binding protein family: Evolutionary divergences and functions. Hum. Genom. 2011, 5, 170–191. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.W.; Veerkamp, J.H. New insights into the structure and function of fatty acid-binding proteins. Cell. Mol. Life Sci. 2002, 59, 1096–1116. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.W.; van Moerkerk, H.T.; Veerkamp, J.H. Ligand specificity and conformational stability of human fatty acid-binding proteins. Int. J. Biochem. Cell Biol. 2001, 33, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Z.; Mita, R.; Beaulieu, M.; Gao, Z.; Godbout, R. Fatty acid binding proteins in brain development and disease. Int. J. Dev. Biol. 2010, 54, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chen, L.; Zhan, Y.; Marquez, K.N.S.; Zhuo, L.; Qi, S.; Zhu, J.; He, Y.; Chen, X.; Zhang, H.; et al. The Biological Functions and Regulatory Mechanisms of Fatty Acid Binding Protein 5 in Various Diseases. Front. Cell Dev. Biol. 2022, 10, 857919. [Google Scholar] [CrossRef]
- Binas, B.; Danneberg, H.; McWhir, J.; Mullins, L.; Clark, A.J. Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization. FASEB J. 1999, 13, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Schaap, F.G.; Binas, B.; Danneberg, H.; van der Vusse, G.J.; Glatz, J.F. Impaired long-chain fatty acid utilization by cardiac myocytes isolated from mice lacking the heart-type fatty acid binding protein gene. Circ. Res. 1999, 85, 329–337. [Google Scholar] [CrossRef]
- Owada, Y.; Yoshimoto, T.; Kondo, H. Spatio-temporally differential expression of genes for three members of fatty acid binding proteins in developing and mature rat brains. J. Chem. Neuroanat. 1996, 12, 113–122. [Google Scholar] [CrossRef]
- Owada, Y. Fatty acid binding protein: Localization and functional significance in the brain. Tohoku J. Exp. Med. 2008, 214, 213–220. [Google Scholar] [CrossRef]
- Hanhoff, T.; Lucke, C.; Spener, F. Insights into binding of fatty acids by fatty acid binding proteins. Mol. Cell. Biochem. 2002, 239, 45–54. [Google Scholar] [CrossRef]
- Armstrong, E.H.; Goswami, D.; Griffin, P.R.; Noy, N.; Ortlund, E.A. Structural basis for ligand regulation of the fatty acid-binding protein 5, peroxisome proliferator-activated receptor beta/delta (FABP5-PPARbeta/delta) signaling pathway. J. Biol. Chem. 2014, 289, 14941–14954. [Google Scholar] [CrossRef] [PubMed]
- Mita, R.; Beaulieu, M.J.; Field, C.; Godbout, R. Brain fatty acid-binding protein and omega-3/omega-6 fatty acids: Mechanistic insight into malignant glioma cell migration. J. Biol. Chem. 2010, 285, 37005–37015. [Google Scholar] [CrossRef] [PubMed]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J. Biol. Chem. 2001, 276, 41958–41962. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Yakunin, E.; Loeb, V.; Kisos, H.; Biala, Y.; Yehuda, S.; Yaari, Y.; Selkoe, D.J.; Sharon, R. Alpha-synuclein neuropathology is controlled by nuclear hormone receptors and enhanced by docosahexaenoic acid in a mouse model for Parkinson’s disease. Brain Pathol. 2012, 22, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Sasaki, S.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Dietary fat intake and risk of Parkinson’s disease: A case-control study in Japan. J. Neurol. Sci. 2010, 288, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Pelsers, M.M.; Hanhoff, T.; Van der Voort, D.; Arts, B.; Peters, M.; Ponds, R.; Honig, A.; Rudzinski, W.; Spener, F.; de Kruijk, J.R.; et al. Brain- and heart-type fatty acid-binding proteins in the brain: Tissue distribution and clinical utility. Clin. Chem. 2004, 50, 1568–1575. [Google Scholar] [CrossRef]
- Basso, M.; Giraudo, S.; Corpillo, D.; Bergamasco, B.; Lopiano, L.; Fasano, M. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteomics 2004, 4, 3943–3952. [Google Scholar] [CrossRef]
- Oizumi, H.; Yamasaki, K.; Suzuki, H.; Hasegawa, T.; Sugimura, Y.; Baba, T.; Fukunaga, K.; Takeda, A. Fatty Acid-Binding Protein 3 Expression in the Brain and Skin in Human Synucleinopathies. Front. Aging Neurosci. 2021, 13, 648982. [Google Scholar] [CrossRef]
- Kawahata, I.; Sekimori, T.; Oizumi, H.; Takeda, A.; Fukunaga, K. Using Fatty Acid-Binding Proteins as Potential Biomarkers to Discriminate between Parkinson’s Disease and Dementia with Lewy Bodies: Exploration of a Novel Technique. Int. J. Mol. Sci. 2023, 24, 13267. [Google Scholar] [CrossRef] [PubMed]
- Backstrom, D.C.; Eriksson Domellof, M.; Linder, J.; Olsson, B.; Ohrfelt, A.; Trupp, M.; Zetterberg, H.; Blennow, K.; Forsgren, L. Cerebrospinal Fluid Patterns and the Risk of Future Dementia in Early, Incident Parkinson Disease. JAMA Neurol. 2015, 72, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Ockner, R.K.; Manning, J.A.; Kane, J.P. Fatty acid binding protein. Isolation from rat liver, characterization, and immunochemical quantification. J. Biol. Chem. 1982, 257, 7872–7878. [Google Scholar] [CrossRef]
- Lowe, J.B.; Boguski, M.S.; Sweetser, D.A.; Elshourbagy, N.A.; Taylor, J.M.; Gordon, J.I. Human liver fatty acid binding protein. Isolation of a full length cDNA and comparative sequence analyses of orthologous and paralogous proteins. J. Biol. Chem. 1985, 260, 3413–3417. [Google Scholar] [CrossRef] [PubMed]
- Sweetser, D.A.; Lowe, J.B.; Gordon, J.I. The nucleotide sequence of the rat liver fatty acid-binding protein gene. Evidence that exon 1 encodes an oligopeptide domain shared by a family of proteins which bind hydrophobic ligands. J. Biol. Chem. 1986, 261, 5553–5561. [Google Scholar] [CrossRef] [PubMed]
- Alpers, D.H.; Strauss, A.W.; Ockner, R.K.; Bass, N.M.; Gordon, J.I. Cloning of a cDNA encoding rat intestinal fatty acid binding protein. Proc. Natl. Acad. Sci. USA 1984, 81, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Sweetser, D.A.; Birkenmeier, E.H.; Klisak, I.J.; Zollman, S.; Sparkes, R.S.; Mohandas, T.; Lusis, A.J.; Gordon, J.I. The human and rodent intestinal fatty acid binding protein genes. A comparative analysis of their structure, expression, and linkage relationships. J. Biol. Chem. 1987, 262, 16060–16071. [Google Scholar] [CrossRef]
- Green, R.P.; Cohn, S.M.; Sacchettini, J.C.; Jackson, K.E.; Gordon, J.I. The mouse intestinal fatty acid binding protein gene: Nucleotide sequence, pattern of developmental and regional expression, and proposed structure of its protein product. DNA Cell Biol. 1992, 11, 31–41. [Google Scholar] [CrossRef]
- Sacchettini, J.C.; Said, B.; Schulz, H.; Gordon, J.I. Rat heart fatty acid-binding protein is highly homologous to the murine adipocyte 422 protein and the P2 protein of peripheral nerve myelin. J. Biol. Chem. 1986, 261, 8218–8223. [Google Scholar] [CrossRef]
- Heuckeroth, R.O.; Birkenmeier, E.H.; Levin, M.S.; Gordon, J.I. Analysis of the tissue-specific expression, developmental regulation, and linkage relationships of a rodent gene encoding heart fatty acid binding protein. J. Biol. Chem. 1987, 262, 9709–9717. [Google Scholar] [CrossRef]
- Kurtz, A.; Zimmer, A.; Schnütgen, F.; Brüning, G.; Spener, F.; Müller, T. The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development 1994, 120, 2637–2649. [Google Scholar] [CrossRef] [PubMed]
- Sellner, P.A.; Chu, W.; Glatz, J.F.; Berman, N.E. Developmental role of fatty acid-binding proteins in mouse brain. Brain Res. Dev. Brain Res. 1995, 89, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Veerkamp, J.H.; Zimmerman, A.W. Fatty acid-binding proteins of nervous tissue. J. Mol. Neurosci. 2001, 16, 133–142; discussion 151–157. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Frank, M.; Green, H. Molecular cloning of mRNA from 3T3 adipocytes. Regulation of mRNA content for glycerophosphate dehydrogenase and other differentiation-dependent proteins during adipocyte development. J. Biol. Chem. 1983, 258, 10083–10089. [Google Scholar] [CrossRef] [PubMed]
- Bernlohr, D.A.; Angus, C.W.; Lane, M.D.; Bolanowski, M.A.; Kelly, T.J., Jr. Expression of specific mRNAs during adipose differentiation: Identification of an mRNA encoding a homologue of myelin P2 protein. Proc. Natl. Acad. Sci. USA 1984, 81, 5468–5472. [Google Scholar] [CrossRef] [PubMed]
- Amri, E.Z.; Bertrand, B.; Ailhaud, G.; Grimaldi, P. Regulation of adipose cell differentiation. I. Fatty acids are inducers of the aP2 gene expression. J. Lipid Res. 1991, 32, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Bernlohr, D.A.; Coe, N.R.; Simpson, M.A.; Hertzel, A.V. Regulation of gene expression in adipose cells by polyunsaturated fatty acids. Adv. Exp. Med. Biol. 1997, 422, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.; Rasmussen, H.H.; Leffers, H.; Honoré, B.; Celis, J.E. Molecular cloning and expression of a novel keratinocyte protein (psoriasis-associated fatty acid-binding protein [PA-FABP]) that is highly up-regulated in psoriatic skin and that shares similarity to fatty acid-binding proteins. J. Investig. Dermatol. 1992, 99, 299–305. [Google Scholar] [CrossRef]
- Krieg, P.; Feil, S.; Fürstenberger, G.; Bowden, G.T. Tumor-specific overexpression of a novel keratinocyte lipid-binding protein. Identification and characterization of a cloned sequence activated during multistage carcinogenesis in mouse skin. J. Biol. Chem. 1993, 268, 17362–17369. [Google Scholar] [CrossRef]
- Liu, Y.; Longo, L.D.; De Leon, M. In situ and immunocytochemical localization of E-FABP mRNA and protein during neuronal migration and differentiation in the rat brain. Brain Res. 2000, 852, 16–27. [Google Scholar] [CrossRef]
- Gong, Y.Z.; Everett, E.T.; Schwartz, D.A.; Norris, J.S.; Wilson, F.A. Molecular cloning, tissue distribution, and expression of a 14-kDa bile acid-binding protein from rat ileal cytosol. Proc. Natl. Acad. Sci. USA 1994, 91, 4741–4745. [Google Scholar] [CrossRef]
- Vodenlich, A.D., Jr.; Gong, Y.Z.; Geoghegan, K.F.; Lin, M.C.; Lanzetti, A.J.; Wilson, F.A. Identification of the 14 kDa bile acid transport protein of rat ileal cytosol as gastrotropin. Biochem. Biophys. Res. Commun. 1991, 177, 1147–1154. [Google Scholar] [CrossRef]
- Walz, D.A.; Wider, M.D.; Snow, J.W.; Dass, C.; Desiderio, D.M. The complete amino acid sequence of porcine gastrotropin, an ileal protein which stimulates gastric acid and pepsinogen secretion. J. Biol. Chem. 1988, 263, 14189–14195. [Google Scholar] [CrossRef] [PubMed]
- Gantz, I.; Nothwehr, S.F.; Lucey, M.; Sacchettini, J.C.; DelValle, J.; Banaszak, L.J.; Naud, M.; Gordon, J.I.; Yamada, T. Gastrotropin: Not an enterooxyntin but a member of a family of cytoplasmic hydrophobic ligand binding proteins. J. Biol. Chem. 1989, 264, 20248–20254. [Google Scholar] [CrossRef]
- Feng, L.; Hatten, M.E.; Heintz, N. Brain lipid-binding protein (BLBP): A novel signaling system in the developing mammalian CNS. Neuron 1994, 12, 895–908. [Google Scholar] [CrossRef]
- Shimizu, F.; Watanabe, T.K.; Shinomiya, H.; Nakamura, Y.; Fujiwara, T. Isolation and expression of a cDNA for human brain fatty acid-binding protein (B-FABP). Biochim. Biophys. Acta 1997, 1354, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Oko, R.; Morales, C.R. A novel testicular protein, with sequence similarities to a family of lipid binding proteins, is a major component of the rat sperm perinuclear theca. Dev. Biol. 1994, 166, 235–245. [Google Scholar] [CrossRef]
- Pouresmaeili, F.; Morales, C.R.; Oko, R. Molecular cloning and structural analysis of the gene encoding PERF 15 protein present in the perinuclear theca of the rat spermatozoa. Biol. Reprod. 1997, 57, 655–659. [Google Scholar] [CrossRef] [PubMed]
- de Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef]
- Ruan, X.; Lin, F.; Wu, D.; Chen, L.; Weng, H.; Yu, J.; Wang, Y.; Chen, Y.; Chen, X.; Ye, Q.; et al. Comparative Efficacy and Safety of Dopamine Agonists in Advanced Parkinson’s Disease With Motor Fluctuations: A Systematic Review and Network Meta-Analysis of Double-Blind Randomized Controlled Trials. Front. Neurosci. 2021, 15, 728083. [Google Scholar] [CrossRef]
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236; discussion 222. [Google Scholar] [CrossRef] [PubMed]
- Phillips, O.; Ghosh, D.; Fernandez, H.H. Parkinson Disease Dementia Management: An Update of Current Evidence and Future Directions. Curr. Treat. Options Neurol. 2023, 25, 93–119. [Google Scholar] [CrossRef]
- Takeda, A.; Baba, T.; Kikuchi, A.; Hasegawa, T.; Sugeno, N.; Konno, M.; Miura, E.; Mori, E. Olfactory dysfunction and dementia in Parkinson’s disease. J. Park. Dis. 2014, 4, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A. How Useful is an Olfactory Test for Diagnosing Alzheimer’s Syndrome? Brain Nerve 2023, 75, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Rey, N.L.; Wesson, D.W.; Brundin, P. The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol. Dis. 2018, 109, 226–248. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Shetty, A.S.; Bhatia, K.P.; Lang, A.E. Dystonia and Parkinson’s disease: What is the relationship? Neurobiol. Dis. 2019, 132, 104462. [Google Scholar] [CrossRef]
- Montague, P.R.; Hyman, S.E.; Cohen, J.D. Computational roles for dopamine in behavioural control. Nature 2004, 431, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Schultz, W. Behavioral theories and the neurophysiology of reward. Annu. Rev. Psychol. 2006, 57, 87–115. [Google Scholar] [CrossRef] [PubMed]
- Berridge, K.C. From prediction error to incentive salience: Mesolimbic computation of reward motivation. Eur. J. Neurosci. 2012, 35, 1124–1143. [Google Scholar] [CrossRef]
- Stern, G. The effects of lesions in the substantia nigra. Brain 1966, 89, 449–478. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, P.F.A. Action Potentials of Muscles in Rigidity and Tremor. Arch. Neurol. Psychiatry 1940, 43, 704–725. [Google Scholar] [CrossRef]
- Nagatsu, T. The catecholamine system in health and disease -Relation to tyrosine 3-monooxygenase and other catecholamine-synthesizing enzymes. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2007, 82, 388–415. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Nagatsu, I. Tyrosine hydroxylase (TH), its cofactor tetrahydrobiopterin (BH4), other catecholamine-related enzymes, and their human genes in relation to the drug and gene therapies of Parkinson’s disease (PD): Historical overview and future prospects. J. Neural Transm. 2016, 123, 1255–1278. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T. Catecholamines and Parkinson’s disease: Tyrosine hydroxylase (TH) over tetrahydrobiopterin (BH4) and GTP cyclohydrolase I (GCH1) to cytokines, neuromelanin, and gene therapy: A historical overview. J. Neural Transm. 2023. ahead of print. [Google Scholar] [CrossRef]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. Tyrosine Hydroxylase. The initial step in norepinephrine biosynthesis. J. Biol. Chem. 1964, 239, 2910–2917. [Google Scholar] [CrossRef]
- Klein, M.O.; Battagello, D.S.; Cardoso, A.R.; Hauser, D.N.; Bittencourt, J.C.; Correa, R.G. Dopamine: Functions, Signaling, and Association with Neurological Diseases. Cell. Mol. Neurobiol. 2019, 39, 31–59. [Google Scholar] [CrossRef]
- Helman, G.; Pappa, M.B.; Pearl, P.L. Widening Phenotypic Spectrum of AADC Deficiency, a Disorder of Dopamine and Serotonin Synthesis. JIMD Rep. 2014, 17, 23–27. [Google Scholar] [CrossRef]
- Atwal, P.S.; Donti, T.R.; Cardon, A.L.; Bacino, C.A.; Sun, Q.; Emrick, L.; Reid Sutton, V.; Elsea, S.H. Aromatic L-amino acid decarboxylase deficiency diagnosed by clinical metabolomic profiling of plasma. Mol. Genet. Metab. 2015, 115, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, H.; Ohye, T.; Takahashi, E.; Seki, N.; Hori, T.; Segawa, M.; Nomura, Y.; Endo, K.; Tanaka, H.; Tsuji, S.; et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat. Genet. 1994, 8, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Segawa, M. Dopa-responsive dystonia. Handb. Clin. Neurol. 2011, 100, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.P.; Hardie, D.G.; Vulliet, P.R. Site-specific phosphorylation of tyrosine hydroxylase after KCl depolarization and nerve growth factor treatment of PC12 cells. J. Biol. Chem. 1990, 265, 22358–22364. [Google Scholar] [CrossRef]
- Campbell, D.G.; Hardie, D.G.; Vulliet, P.R. Identification of four phosphorylation sites in the N-terminal region of tyrosine hydroxylase. J. Biol. Chem. 1986, 261, 10489–10492. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Bobrovskaya, L.; Graham, M.E.; von Nagy-Felsobuki, E.I.; Dickson, P.W. Tyrosine hydroxylase phosphorylation: Regulation and consequences. J. Neurochem. 2004, 91, 1025–1043. [Google Scholar] [CrossRef] [PubMed]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef]
- Leal, R.B.; Sim, A.T.; Goncalves, C.A.; Dunkley, P.R. Tyrosine hydroxylase dephosphorylation by protein phosphatase 2A in bovine adrenal chromaffin cells. Neurochem. Res. 2002, 27, 207–213. [Google Scholar] [CrossRef]
- Bezem, M.T.; Baumann, A.; Skjærven, L.; Meyer, R.; Kursula, P.; Martinez, A.; Flydal, M.I. Stable preparations of tyrosine hydroxylase provide the solution structure of the full-length enzyme. Sci. Rep. 2016, 6, 30390. [Google Scholar] [CrossRef]
- Bueno-Carrasco, M.T.; Cuéllar, J.; Flydal, M.I.; Santiago, C.; Kråkenes, T.A.; Kleppe, R.; López-Blanco, J.R.; Marcilla, M.; Teigen, K.; Alvira, S.; et al. Structural mechanism for tyrosine hydroxylase inhibition by dopamine and reactivation by Ser40 phosphorylation. Nat. Commun. 2022, 13, 74. [Google Scholar] [CrossRef]
- Urano, F.; Hayashi, N.; Arisaka, F.; Kurita, H.; Murata, S.; Ichinose, H. Molecular mechanism for pterin-mediated inactivation of tyrosine hydroxylase: Formation of insoluble aggregates of tyrosine hydroxylase. J. Biochem. 2006, 139, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Jorge-Finnigan, A.; Jung-Kc, K.; Sauter, A.; Horvath, I.; Morozova-Roche, L.A.; Martinez, A. Tyrosine Hydroxylase Binding to Phospholipid Membranes Prompts Its Amyloid Aggregation and Compromises Bilayer Integrity. Sci. Rep. 2016, 6, 39488. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Belizaire, R.; Jenner, P.; Olanow, C.W.; Isacson, O. Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci. Lett. 2002, 326, 155–158. [Google Scholar] [CrossRef]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.Y.; Tang, Z.; Liu, C.W. alpha-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: Understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J. Biol. Chem. 2008, 283, 20288–20298. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Tokuoka, H.; Parvez, H.; Ichinose, H. Accumulation of phosphorylated tyrosine hydroxylase into insoluble protein aggregates by inhibition of an ubiquitin-proteasome system in PC12D cells. J. Neural Transm. 2009, 116, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Yagishita, S.; Hasegawa, K.; Nagatsu, I.; Nagatsu, T.; Ichinose, H. Immunohistochemical analyses of the postmortem human brains from patients with Parkinson’s disease with anti-tyrosine hydroxylase antibodies. Biog. Amines 2009, 23, 1–7. [Google Scholar]
- Kawahata, I.; Ohtaku, S.; Tomioka, Y.; Ichinose, H.; Yamakuni, T. Dopamine or biopterin deficiency potentiates phosphorylation at (40)Ser and ubiquitination of tyrosine hydroxylase to be degraded by the ubiquitin proteasome system. Biochem. Biophys. Res. Commun. 2015, 465, 53–58. [Google Scholar] [CrossRef]
- Døskeland, A.P.; Flatmark, T. Ubiquitination of soluble and membrane-bound tyrosine hydroxylase and degradation of the soluble form. Eur. J. Biochem. 2002, 269, 1561–1569. [Google Scholar] [CrossRef]
- Kawahata, I.; Fukunaga, K. Degradation of Tyrosine Hydroxylase by the Ubiquitin-Proteasome System in the Pathogenesis of Parkinson’s Disease and Dopa-Responsive Dystonia. Int. J. Mol. Sci. 2020, 21, 3779. [Google Scholar] [CrossRef]
- Wu, B.; Liu, Q.; Duan, C.; Li, Y.; Yu, S.; Chan, P.; Ueda, K.; Yang, H. Phosphorylation of alpha-synuclein upregulates tyrosine hydroxylase activity in MN9D cells. Acta Histochem. 2011, 113, 32–35. [Google Scholar] [CrossRef]
- Canerina-Amaro, A.; Pereda, D.; Diaz, M.; Rodriguez-Barreto, D.; Casañas-Sánchez, V.; Heffer, M.; Garcia-Esparcia, P.; Ferrer, I.; Puertas-Avendaño, R.; Marin, R. Differential Aggregation and Phosphorylation of Alpha Synuclein in Membrane Compartments Associated With Parkinson Disease. Front. Neurosci. 2019, 13, 382. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Bousset, L.; Melki, R.; Fukunaga, K. Fatty Acid-Binding Protein 3 is Critical for alpha-Synuclein Uptake and MPP(+)-Induced Mitochondrial Dysfunction in Cultured Dopaminergic Neurons. Int. J. Mol. Sci. 2019, 20, 5358. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Sekimori, T.; Wang, H.; Wang, Y.; Sasaoka, T.; Bousset, L.; Melki, R.; Mizobata, T.; Kawata, Y.; Fukunaga, K. Dopamine D2 Long Receptors Are Critical for Caveolae-Mediated alpha-Synuclein Uptake in Cultured Dopaminergic Neurons. Biomedicines 2021, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Yabuki, Y.; Matsuo, K.; Kawahata, I.; Fukui, N.; Mizobata, T.; Kawata, Y.; Owada, Y.; Shioda, N.; Fukunaga, K. Fatty Acid Binding Protein 3 Enhances the Spreading and Toxicity of alpha-Synuclein in Mouse Brain. Int. J. Mol. Sci. 2020, 21, 2230. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Kawahata, I.; Melki, R.; Bousset, L.; Owada, Y.; Fukunaga, K. Suppression of alpha-synuclein propagation after intrastriatal injection in FABP3 null mice. Brain Res. 2021, 1760, 147383. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Cheng, Z.; Piao, J.; Cui, R.; Li, B. Dopamine Receptors: Is It Possible to Become a Therapeutic Target for Depression? Front. Pharmacol. 2022, 13, 947785. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Fukunaga, K. Differential subcellular localization of two dopamine D2 receptor isoforms in transfected NG108-15 cells. J. Neurochem. 2003, 85, 1064–1074. [Google Scholar] [CrossRef]
- Sharma, M.; Celver, J.; Octeau, J.C.; Kovoor, A. Plasma membrane compartmentalization of D2 dopamine receptors. J. Biol. Chem. 2013, 288, 12554–12568. [Google Scholar] [CrossRef]
- Cho, D.I.; Min, C.; Jung, K.S.; Cheong, S.Y.; Zheng, M.; Cheong, S.J.; Oak, M.H.; Cheong, J.H.; Lee, B.K.; Kim, K.M. The N-terminal region of the dopamine D2 receptor, a rhodopsin-like GPCR, regulates correct integration into the plasma membrane and endocytic routes. Br. J. Pharmacol. 2012, 166, 659–675. [Google Scholar] [CrossRef]
- Farde, L.; Hall, H.; Ehrin, E.; Sedvall, G. Quantitative analysis of D2 dopamine receptor binding in the living human brain by PET. Science 1986, 231, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.C.; Gatti McArthur, S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Fukui, N.; Yamamoto, H.; Miyabe, M.; Aoyama, Y.; Hongo, K.; Mizobata, T.; Kawahata, I.; Yabuki, Y.; Shinoda, Y.; Fukunaga, K.; et al. An alpha-synuclein decoy peptide prevents cytotoxic alpha-synuclein aggregation caused by fatty acid binding protein 3. J. Biol. Chem. 2021, 296, 100663. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grünewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Park. Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Naini, A.B.; Jakowec, M.; Petzinger, G.; Miller, R.; Akram, M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): A technical review of its utility and safety. J. Neurochem. 2001, 76, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.C.; Williams, A.C.; Markey, S.P.; Ebert, M.H.; Caine, E.D.; Reichert, C.M.; Kopin, I.J. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979, 1, 249–254. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Jenner, P.; Marsden, C.D. The actions of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in animals as a model of Parkinson’s disease. J. Neural Transm. Suppl. 1986, 20, 11–39. [Google Scholar]
- Heikkila, R.E.; Nicklas, W.J.; Vyas, I.; Duvoisin, R.C. Dopaminergic toxicity of rotenone and the 1-methyl-4-phenylpyridinium ion after their stereotaxic administration to rats: Implication for the mechanism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity. Neurosci. Lett. 1985, 62, 389–394. [Google Scholar] [CrossRef]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology 2015, 46, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Innos, J.; Hickey, M.A. Using Rotenone to Model Parkinson’s Disease in Mice: A Review of the Role of Pharmacokinetics. Chem. Res. Toxicol. 2021, 34, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- Landau, R.; Halperin, R.; Sullivan, P.; Zibly, Z.; Leibowitz, A.; Goldstein, D.S.; Sharabi, Y. The rat rotenone model reproduces the abnormal pattern of central catecholamine metabolism found in Parkinson’s disease. Dis. Model. Mech. 2022, 15, dmm049082. [Google Scholar] [CrossRef] [PubMed]
- Haga, H.; Yamada, R.; Izumi, H.; Shinoda, Y.; Kawahata, I.; Miyachi, H.; Fukunaga, K. Novel fatty acid-binding protein 3 ligand inhibits dopaminergic neuronal death and improves motor and cognitive impairments in Parkinson’s disease model mice. Pharmacol. Biochem. Behav. 2020, 191, 172891. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fukunaga, K.; Cheng, A.; Wang, Y.; Arimura, N.; Yoshino, H.; Sasaki, T.; Kawahata, I. Novel FABP3 ligand, HY-11-9, ameliorates neuropathological deficits in MPTP-induced Parkinsonism in mice. J. Pharmacol. Sci. 2023, 152, 30–38. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Wang, Y.; Shinoda, Y.; Cheng, A.; Kawahata, I.; Fukunaga, K. Epidermal Fatty Acid-Binding Protein 5 (FABP5) Involvement in Alpha-Synuclein-Induced Mitochondrial Injury under Oxidative Stress. Biomedicines 2021, 9, 110. [Google Scholar] [CrossRef]
- Cheng, A.; Kawahata, I.; Fukunaga, K. Fatty Acid Binding Protein 5 Mediates Cell Death by Psychosine Exposure through Mitochondrial Macropores Formation in Oligodendrocytes. Biomedicines 2020, 8, 635. [Google Scholar] [CrossRef]
- Cheng, A.; Jia, W.; Kawahata, I.; Fukunaga, K. A novel fatty acid-binding protein 5 and 7 inhibitor ameliorates oligodendrocyte injury in multiple sclerosis mouse models. EBioMedicine 2021, 72, 103582. [Google Scholar] [CrossRef]
- Guo, Q.; Kawahata, I.; Degawa, T.; Ikeda-Matsuo, Y.; Sun, M.; Han, F.; Fukunaga, K. Fatty Acid-Binding Proteins Aggravate Cerebral Ischemia-Reperfusion Injury in Mice. Biomedicines 2021, 9, 529. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Kawahata, I.; Cheng, A.; Wang, H.; Jia, W.; Yoshino, H.; Fukunaga, K. Fatty acid-binding proteins 3 and 5 are involved in the initiation of mitochondrial damage in ischemic neurons. Redox Biol. 2023, 59, 102547. [Google Scholar] [CrossRef] [PubMed]
- de Araújo, F.M.; Cuenca-Bermejo, L.; Fernández-Villalba, E.; Costa, S.L.; Silva, V.D.A.; Herrero, M.T. Role of Microgliosis and NLRP3 Inflammasome in Parkinson’s Disease Pathogenesis and Therapy. Cell. Mol. Neurobiol. 2022, 42, 1283–1300. [Google Scholar] [CrossRef] [PubMed]
- MacMahon Copas, A.N.; McComish, S.F.; Fletcher, J.M.; Caldwell, M.A. The Pathogenesis of Parkinson’s Disease: A Complex Interplay Between Astrocytes, Microglia, and T Lymphocytes? Front. Neurol. 2021, 12, 666737. [Google Scholar] [CrossRef]
- Ma, D.; Zhang, M.; Mori, Y.; Yao, C.; Larsen, C.P.; Yamashima, T.; Zhou, L. Cellular localization of epidermal-type and brain-type fatty acid-binding proteins in adult hippocampus and their response to cerebral ischemia. Hippocampus 2010, 20, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Boneva, N.B.; Kaplamadzhiev, D.B.; Sahara, S.; Kikuchi, H.; Pyko, I.V.; Kikuchi, M.; Tonchev, A.B.; Yamashima, T. Expression of fatty acid-binding proteins in adult hippocampal neurogenic niche of postischemic monkeys. Hippocampus 2011, 21, 162–171. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; McTigue, D.M.; Jakeman, L.B. Regional heterogeneity in astrocyte responses following contusive spinal cord injury in mice. J. Comp. Neurol. 2010, 518, 1370–1390. [Google Scholar] [CrossRef]
- Bannerman, P.; Hahn, A.; Soulika, A.; Gallo, V.; Pleasure, D. Astrogliosis in EAE spinal cord: Derivation from radial glia, and relationships to oligodendroglia. Glia 2007, 55, 57–64. [Google Scholar] [CrossRef]
- Cheng, A.; Wang, Y.F.; Shinoda, Y.; Kawahata, I.; Yamamoto, T.; Jia, W.B.; Yamamoto, H.; Mizobata, T.; Kawata, Y.; Fukunaga, K. Fatty acid-binding protein 7 triggers alpha-synuclein oligomerization in glial cells and oligodendrocytes associated with oxidative stress. Acta Pharmacol. Sin. 2022, 43, 552–562. [Google Scholar] [CrossRef]
- Cheng, A.; Kawahata, I.; Wang, Y.; Jia, W.; Wang, H.; Sekimori, T.; Chen, Y.; Suzuki, H.; Takeda, A.; Stefanova, N.; et al. Epsin2, a novel target for multiple system atrophy therapy via alpha-synuclein/FABP7 propagation. Brain 2023, 146, 3172–3180. [Google Scholar] [CrossRef]
- Guo, Q.; Kawahata, I.; Jia, W.; Wang, H.; Cheng, A.; Yabuki, Y.; Shioda, N.; Fukunaga, K. alpha-Synuclein decoy peptide protects mice against alpha-synuclein-induced memory loss. CNS Neurosci. Ther. 2023, 29, 1547–1560. [Google Scholar] [CrossRef]
- Cheng, A.; Jia, W.; Finkelstein, D.I.; Stefanova, N.; Wang, H.; Sasaki, T.; Kawahata, I.; Fukunaga, K. Pharmacological inhibition of FABP7 by MF 6 counteracts cerebellum dysfunction in an experimental multiple system atrophy mouse model. Acta Pharmacol. Sin. 2023. ahead of print. [Google Scholar] [CrossRef]
- Ntetsika, T.; Papathoma, P.E.; Markaki, I. Novel targeted therapies for Parkinson’s disease. Mol. Med. 2021, 27, 17. [Google Scholar] [CrossRef] [PubMed]
- Prasad, E.M.; Hung, S.Y. Current Therapies in Clinical Trials of Parkinson’s Disease: A 2021 Update. Pharmaceuticals 2021, 14, 717. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Cullen, V.; Kahn, I.; Krastins, B.; Outeiro, T.F.; Pepivani, I.; Ng, J.; Schulz-Schaeffer, W.; Kretzschmar, H.A.; McLean, P.J.; et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp. Neurol. 2008, 213, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, X.; Yang, H.; Liu, Y. Review of Metabolomics-Based Biomarker Research for Parkinson’s Disease. Mol. Neurobiol. 2022, 59, 1041–1057. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.L.; Wang, Z.L.; Zhang, F.Y.; Liu, H.X.; Mao, L.H.; Yuan, L. Biomarkers of Parkinson’s Disease: From Basic Research to Clinical Practice. Aging Dis. 2023. ahead of print. [Google Scholar] [CrossRef]
- Tsamourgelis, A.; Swann, P.; Chouliaras, L.; O’Brien, J.T. From protein biomarkers to proteomics in dementia with Lewy Bodies. Ageing Res. Rev. 2023, 83, 101771. [Google Scholar] [CrossRef]
- Baba, T.; Kikuchi, A.; Hirayama, K.; Nishio, Y.; Hosokai, Y.; Kanno, S.; Hasegawa, T.; Sugeno, N.; Konno, M.; Suzuki, K.; et al. Severe olfactory dysfunction is a prodromal symptom of dementia associated with Parkinson’s disease: A 3 year longitudinal study. Brain 2012, 135, 161–169. [Google Scholar] [CrossRef]
- Domellof, M.E.; Lundin, K.F.; Edstrom, M.; Forsgren, L. Olfactory dysfunction and dementia in newly diagnosed patients with Parkinson’s disease. Park. Relat. Disord. 2017, 38, 41–47. [Google Scholar] [CrossRef]
- Fullard, M.E.; Morley, J.F.; Duda, J.E. Olfactory Dysfunction as an Early Biomarker in Parkinson’s Disease. Neurosci. Bull. 2017, 33, 515–525. [Google Scholar] [CrossRef]
- Chiasserini, D.; Biscetti, L.; Eusebi, P.; Salvadori, N.; Frattini, G.; Simoni, S.; De Roeck, N.; Tambasco, N.; Stoops, E.; Vanderstichele, H.; et al. Differential role of CSF fatty acid binding protein 3, alpha-synuclein, and Alzheimer’s disease core biomarkers in Lewy body disorders and Alzheimer’s dementia. Alzheimers Res. Ther. 2017, 9, 52. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Korczyn, A.D. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 2018, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Sepe, F.N.; Chiasserini, D.; Parnetti, L. Role of FABP3 as biomarker in Alzheimer’s disease and synucleinopathies. Future Neurol. 2018, 13, 199–207. [Google Scholar] [CrossRef]
- Sezgin, M.; Bilgic, B.; Tinaz, S.; Emre, M. Parkinson’s Disease Dementia and Lewy Body Disease. Semin. Neurol. 2019, 39, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.; Stefanis, L.; Attems, J. Clinical and neuropathological differences between Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies—Current issues and future directions. J. Neurochem. 2019, 150, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Gaetani, L.; Eusebi, P.; Paciotti, S.; Hansson, O.; El-Agnaf, O.; Mollenhauer, B.; Blennow, K.; Calabresi, P. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 2019, 18, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Triozzi, P.L.; Stirling, E.R.; Song, Q.; Westwood, B.; Kooshki, M.; Forbes, M.E.; Holbrook, B.C.; Cook, K.L.; Alexander-Miller, M.A.; Miller, L.D.; et al. Circulating Immune Bioenergetic, Metabolic, and Genetic Signatures Predict Melanoma Patients’ Response to Anti-PD-1 Immune Checkpoint Blockade. Clin. Cancer Res. 2022, 28, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Priya Rajan, S.A.; Song, Q.; Zhao, Y.; Wan, M.; Aleman, J.; Skardal, A.; Bishop, C.; Atala, A.; Lu, B. 3D scaffold-free microlivers with drug metabolic function generated by lineage-reprogrammed hepatocytes from human fibroblasts. Biomaterials 2021, 269, 120668. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, E.; Trivedi, D.K.; Sarkar, D.; Walton-Doyle, C.; Milne, J.; Kunath, T.; Rijs, A.M.; de Bie, R.M.A.; Goodacre, R.; Silverdale, M.; et al. Metabolomics of sebum reveals lipid dysregulation in Parkinson’s disease. Nat. Commun. 2021, 12, 1592. [Google Scholar] [CrossRef]
- Oizumi, H.; Sugimura, Y.; Totsune, T.; Kawasaki, I.; Ohshiro, S.; Baba, T.; Kimpara, T.; Sakuma, H.; Hasegawa, T.; Kawahata, I.; et al. Plasma sphingolipid abnormalities in neurodegenerative diseases. PLoS ONE 2022, 17, e0279315. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawahata, I.; Fukunaga, K. Pathogenic Impact of Fatty Acid-Binding Proteins in Parkinson’s Disease—Potential Biomarkers and Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 17037. https://doi.org/10.3390/ijms242317037
Kawahata I, Fukunaga K. Pathogenic Impact of Fatty Acid-Binding Proteins in Parkinson’s Disease—Potential Biomarkers and Therapeutic Targets. International Journal of Molecular Sciences. 2023; 24(23):17037. https://doi.org/10.3390/ijms242317037
Chicago/Turabian StyleKawahata, Ichiro, and Kohji Fukunaga. 2023. "Pathogenic Impact of Fatty Acid-Binding Proteins in Parkinson’s Disease—Potential Biomarkers and Therapeutic Targets" International Journal of Molecular Sciences 24, no. 23: 17037. https://doi.org/10.3390/ijms242317037
APA StyleKawahata, I., & Fukunaga, K. (2023). Pathogenic Impact of Fatty Acid-Binding Proteins in Parkinson’s Disease—Potential Biomarkers and Therapeutic Targets. International Journal of Molecular Sciences, 24(23), 17037. https://doi.org/10.3390/ijms242317037