
Serum/Plasma Proteome in Non-Malignant Liver Disease

 ,
, 
Abstract
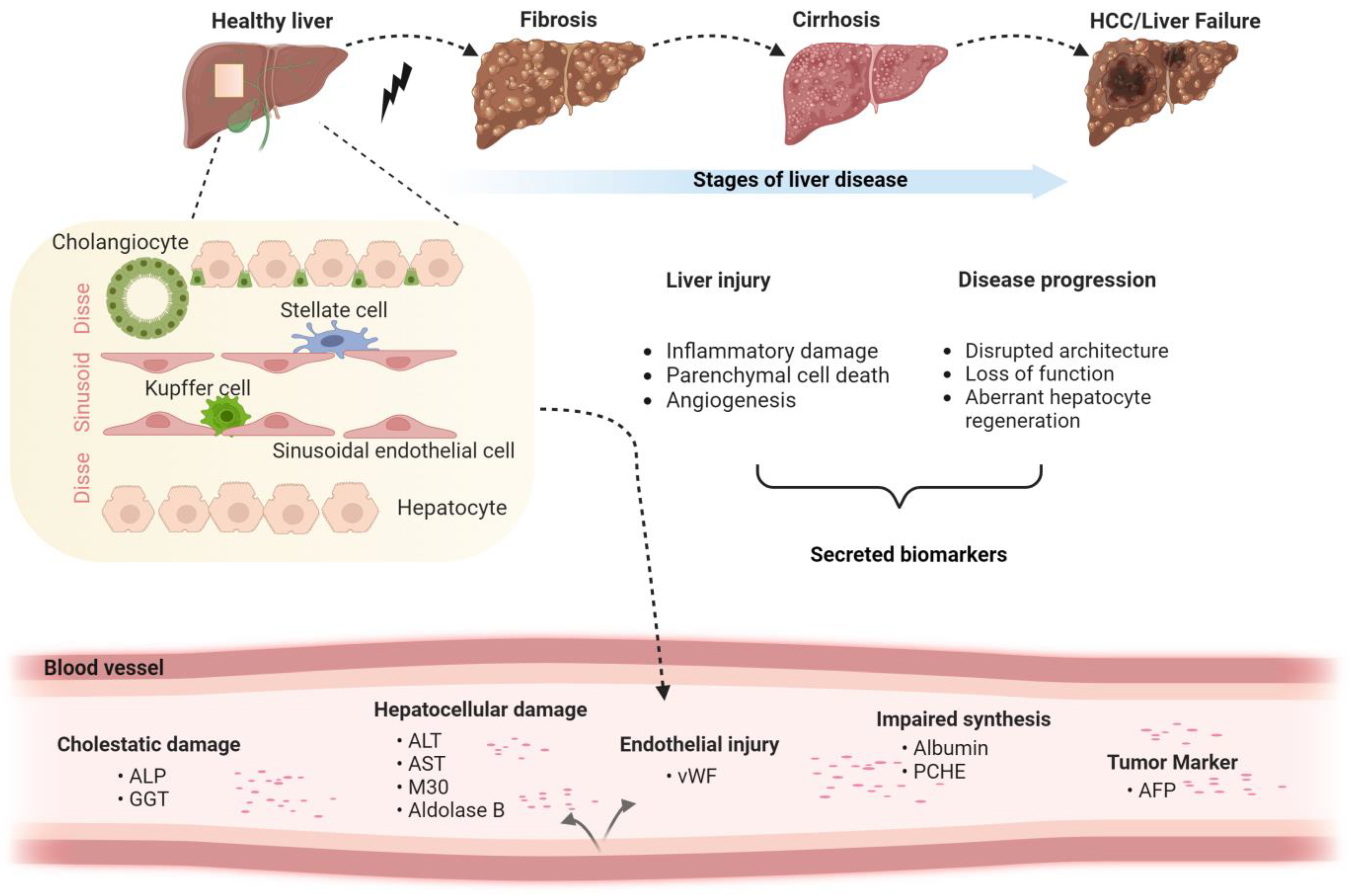
1. Introduction
2. Overview of Traditional Protein Biomarkers and Their Usefulness
2.1. Liver Injury Markers
2.1.1. Aspartate/Alanine Amino Transferase
2.1.2. Soluble Keratin 18 (K18) and Fragmented K18
2.1.3. Aldolase B
2.1.4. Golgi Protein 73 (GP73)
2.2. Biomarkers of Advanced Liver Disease
2.2.1. Von Willebrand Factor (vWF)
2.2.2. Apolipoproteins
2.2.3. Pseudocholinesterase (PCHE)
3. Proteomics as the Next Generation Approach
3.1. Key Methods
3.1.1. Non-Depleted vs. Depleted Proteomics
3.1.2. Mass Spec. vs. Affinity-Based Methods (Olink and Somascan)
4. Data Analysis of Serum Proteomics
4.1. Bioinformatic Methods
4.2. Key Papers
5. Conclusions and Future Directions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pabst, O.; Hornef, M.W.; Schaap, F.G.; Cerovic, V.; Clavel, T.; Bruns, T. Gut-liver axis: Barriers and functional circuits. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 447–461. [Google Scholar] [CrossRef]
- Kuscuoglu, D.; Janciauskiene, S.; Hamesch, K.; Haybaeck, J.; Trautwein, C.; Strnad, P. Liver—Master and servant of serum proteome. J. Hepatol. 2018, 69, 512–524. [Google Scholar] [CrossRef]
- Kwo, P.Y.; Cohen, S.M.; Lim, J.K. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. Am. J. Gastroenterol. 2017, 112, 18–35. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.S.; Taylor, R.J.; Bayliss, S.; Hagstrom, H.; Nasr, P.; Schattenberg, J.M.; Ishigami, M.; Toyoda, H.; Wai-Sun Wong, V.; Peleg, N.; et al. Association Between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 2020, 158, 1611–1625. [Google Scholar] [CrossRef] [PubMed]
- Gurbuz, B.; Guldiken, N.; Reuken, P.; Fu, L.; Remih, K.; Preisinger, C.; Bruha, R.; Lenicek, M.; Petrtyl, J.; Reissing, J.; et al. Biomarkers of hepatocellular synthesis in patients with decompensated cirrhosis. Hepatol. Int. 2023, 17, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Moshage, H. Cytokines and the hepatic acute phase response. J. Pathol. 1997, 181, 257–266. [Google Scholar] [CrossRef]
- Tapper, E.B.; Lok, A.S.F. Use of Liver Imaging and Biopsy in Clinical Practice. N. Engl. J. Med. 2017, 377, 2296–2297. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Thiele, M.; Geyer, P.E.; Rasmussen, D.N.; Webel, H.E.; Santos, A.; Gupta, R.; Meier, F.; Strauss, M.; Kjaergaard, M.; et al. Noninvasive proteomic biomarkers for alcohol-related liver disease. Nat. Med. 2022, 28, 1277–1287. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B.; et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 561–577. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Liu, W.; Xing, S.; Wang, D.; Chen, J.; Sun, L.; Mu, J.; Liu, W.; Xing, B.; et al. Identification of noninvasive diagnostic biomarkers for hepatocellular carcinoma by urinary proteomics. J. Proteom. 2020, 225, 103780. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Liu, X.; Wei, X.; Luo, H.; Li, P.; Shi, M.; Guo, B.; Cui, Y.; Su, Z.; Zeng, J.; et al. Quantitative proteomics identifies a plasma multi-protein model for detection of hepatocellular carcinoma. Sci. Rep. 2020, 10, 15552. [Google Scholar] [CrossRef]
- Ben-Moshe, S.; Itzkovitz, S. Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Kakisaka, K.; Kataoka, K.; Onodera, M.; Suzuki, A.; Endo, K.; Tatemichi, Y.; Kuroda, H.; Ishida, K.; Takikawa, Y. Alpha-fetoprotein: A biomarker for the recruitment of progenitor cells in the liver in patients with acute liver injury or failure. Hepatol. Res. 2015, 45, E12–E20. [Google Scholar] [CrossRef] [PubMed]
- Caraceni, P.; Tufoni, M.; Zaccherini, G.; Riggio, O.; Angeli, P.; Alessandria, C.; Neri, S.; Foschi, F.G.; Levantesi, F.; Airoldi, A.; et al. On-treatment serum albumin level can guide long-term treatment in patients with cirrhosis and uncomplicated ascites. J. Hepatol. 2021, 74, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.; Wu, Q.; Fang, X.; Duan, L.; et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Olsavsky Goyak, K.M.; Laurenzana, E.M.; Omiecinski, C.J. Hepatocyte differentiation. Methods Mol. Biol. 2010, 640, 115–138. [Google Scholar] [CrossRef] [PubMed]
- De Pablo-Moreno, J.A.; Serrano, L.J.; Revuelta, L.; Sanchez, M.J.; Liras, A. The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V. Int. J. Mol. Sci. 2022, 23, 8283. [Google Scholar] [CrossRef]
- Gao, J.; Zuo, B.; He, Y. Liver sinusoidal endothelial cells as potential drivers of liver fibrosis (Review). Mol. Med. Rep. 2024, 29, 40. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Androutsakos, T.; Flessa, C.M.; Kyrou, I.; Siasos, G.; Randeva, H.S.; Kassi, E.; Papavassiliou, A.G. Endothelial Cell Dysfunction and Nonalcoholic Fatty Liver Disease (NAFLD): A Concise Review. Cells 2022, 11, 2511. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Yagmur, E.; Linka, J.; Schumacher, F.; Bruensing, J.; Buendgens, L.; Herbers, U.; Koek, G.H.; Weiskirchen, R.; Trautwein, C.; et al. High Circulating Caspase-Cleaved Keratin 18 Fragments (M30) Indicate Short-Term Mortality in Critically Ill Patients. Dis. Markers 2018, 2018, 8583121. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.A.; Bradwell, A.R.; Thompson, R.A. Quantitation of vitronectin in serum: Evaluation of its usefulness in routine clinical practice. J. Clin. Pathol. 1993, 46, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, W.; Sipe, J.D. Levels of the serum amyloid A protein (SAA) in normal persons of different age groups. Clin. Exp. Immunol. 1979, 35, 96–100. [Google Scholar] [PubMed]
- Ng, C.; Motto, D.G.; Di Paola, J. Diagnostic approach to von Willebrand disease. Blood 2015, 125, 2029–2037. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef]
- Eguchi, A.; Wree, A.; Feldstein, A.E. Biomarkers of liver cell death. J. Hepatol. 2014, 60, 1063–1074. [Google Scholar] [CrossRef]
- Thietart, S.; Rautou, P.E. Extracellular vesicles as biomarkers in liver diseases: A clinician’s point of view. J. Hepatol. 2020, 73, 1507–1525. [Google Scholar] [CrossRef]
- Kew, M.C. Serum aminotransferase concentration as evidence of hepatocellular damage. Lancet 2000, 355, 591–592. [Google Scholar] [CrossRef]
- Kim, W.R.; Flamm, S.L.; Di Bisceglie, A.M.; Bodenheimer, H.C.; Public Policy Committee of the American Association for the Study of Liver, D. Serum activity of alanine aminotransferase (ALT) as an indicator of health and disease. Hepatology 2008, 47, 1363–1370. [Google Scholar] [CrossRef]
- Williams, A.L.; Hoofnagle, J.H. Ratio of serum aspartate to alanine aminotransferase in chronic hepatitis. Relationship to cirrhosis. Gastroenterology 1988, 95, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ma, Y.; Cai, J.; Sun, M.; Zeng, L.; Wu, F.; Zhang, Y.; Hu, M. Serum biomarkers for liver fibrosis. Clin. Chim. Acta 2022, 537, 16–25. [Google Scholar] [CrossRef]
- Branchi, F.; Conti, C.B.; Baccarin, A.; Lampertico, P.; Conte, D.; Fraquelli, M. Non-invasive assessment of liver fibrosis in chronic hepatitis B. World J. Gastroenterol. 2014, 20, 14568–14580. [Google Scholar] [CrossRef] [PubMed]
- Berzigotti, A.; Seijo, S.; Arena, U.; Abraldes, J.G.; Vizzutti, F.; Garcia-Pagan, J.C.; Pinzani, M.; Bosch, J. Elastography, spleen size, and platelet count identify portal hypertension in patients with compensated cirrhosis. Gastroenterology 2013, 144, 102–111.e1. [Google Scholar] [CrossRef] [PubMed]
- Serra-Burriel, M.; Juanola, A.; Serra-Burriel, F.; Thiele, M.; Graupera, I.; Pose, E.; Pera, G.; Grgurevic, I.; Caballeria, L.; Piano, S.; et al. Development, validation, and prognostic evaluation of a risk score for long-term liver-related outcomes in the general population: A multicohort study. Lancet 2023, 402, 988–996. [Google Scholar] [CrossRef]
- Bedogni, G.; Bellentani, S.; Miglioli, L.; Masutti, F.; Passalacqua, M.; Castiglione, A.; Tiribelli, C. The Fatty Liver Index: A simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006, 6, 33. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Sheron, N.; Zelber-Sagi, S.; Carrieri, P.; Dusheiko, G.; Bugianesi, E.; Pryke, R.; Hutchinson, S.J.; Sangro, B.; Martin, N.K.; et al. The EASL-Lancet Liver Commission: Protecting the next generation of Europeans against liver disease complications and premature mortality. Lancet 2022, 399, 61–116. [Google Scholar] [CrossRef]
- Toivola, D.M.; Boor, P.; Alam, C.; Strnad, P. Keratins in health and disease. Curr. Opin. Cell Biol. 2015, 32, 73–81. [Google Scholar] [CrossRef]
- Ku, N.O.; Strnad, P.; Bantel, H.; Omary, M.B. Keratins: Biomarkers and modulators of apoptotic and necrotic cell death in the liver. Hepatology 2016, 64, 966–976. [Google Scholar] [CrossRef]
- Schutte, B.; Henfling, M.; Kolgen, W.; Bouman, M.; Meex, S.; Leers, M.P.; Nap, M.; Bjorklund, V.; Bjorklund, P.; Bjorklund, B.; et al. Keratin 8/18 breakdown and reorganization during apoptosis. Exp. Cell Res. 2004, 297, 11–26. [Google Scholar] [CrossRef]
- Zheng, S.J.; Liu, S.; Liu, M.; McCrae, M.A.; Li, J.F.; Han, Y.P.; Xu, C.H.; Ren, F.; Chen, Y.; Duan, Z.P. Prognostic value of M30/M65 for outcome of hepatitis B virus-related acute-on-chronic liver failure. World J. Gastroenterol. 2014, 20, 2403–2411. [Google Scholar] [CrossRef]
- Vatsalya, V.; Cave, M.C.; Kong, M.; Gobejishvili, L.; Falkner, K.C.; Craycroft, J.; Mitchell, M.; Szabo, G.; McCullough, A.; Dasarathy, S.; et al. Keratin 18 Is a Diagnostic and Prognostic Factor for Acute Alcoholic Hepatitis. Clin. Gastroenterol. Hepatol. 2020, 18, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Dolar, E.; Ulukaya, E.; Akgoz, S.; Keskin, M.; Kiyici, M.; Aker, S.; Yilmaztepe, A.; Gurel, S.; Gulten, M.; et al. Soluble forms of extracellular cytokeratin 18 may differentiate simple steatosis from nonalcoholic steatohepatitis. World J. Gastroenterol. 2007, 13, 837–844. [Google Scholar] [CrossRef]
- Joka, D.; Wahl, K.; Moeller, S.; Schlue, J.; Vaske, B.; Bahr, M.J.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Prospective biopsy-controlled evaluation of cell death biomarkers for prediction of liver fibrosis and nonalcoholic steatohepatitis. Hepatology 2012, 55, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, S.R.; Grove, J.I.; Liebig, S.; Astbury, S.; Vergis, N.; Goldin, R.; Quaglia, A.; Bantel, H.; Guha, I.N.; Thursz, M.R.; et al. In Severe Alcoholic Hepatitis, Serum Keratin-18 Fragments Are Diagnostic, Prognostic, and Theragnostic Biomarkers. Am. J. Gastroenterol. 2020, 115, 1857–1868. [Google Scholar] [CrossRef]
- Mueller, S.; Nahon, P.; Rausch, V.; Peccerella, T.; Silva, I.; Yagmur, E.; Straub, B.K.; Lackner, C.; Seitz, H.K.; Rufat, P.; et al. Caspase-cleaved keratin-18 fragments increase during alcohol withdrawal and predict liver-related death in patients with alcoholic liver disease. Hepatology 2017, 66, 96–107. [Google Scholar] [CrossRef]
- Bell, L.N.; Vuppalanchi, R.; Watkins, P.B.; Bonkovsky, H.L.; Serrano, J.; Fontana, R.J.; Wang, M.; Rochon, J.; Chalasani, N.; Group, U.S.D.-I.L.I.N.R. Serum proteomic profiling in patients with drug-induced liver injury. Aliment. Pharmacol. Ther. 2012, 35, 600–612. [Google Scholar] [CrossRef]
- Asaka, M.; Miyazaki, T.; Hollinger, F.B.; Alpert, E. Human aldolase B serum levels: A marker of liver injury. Hepatology 1984, 4, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Ravindra, K.C.; Vaidya, V.S.; Wang, Z.; Federspiel, J.D.; Virgen-Slane, R.; Everley, R.A.; Grove, J.I.; Stephens, C.; Ocana, M.F.; Robles-Diaz, M.; et al. Tandem mass tag-based quantitative proteomic profiling identifies candidate serum biomarkers of drug-induced liver injury in humans. Nat. Commun. 2023, 14, 1215. [Google Scholar] [CrossRef] [PubMed]
- Morota, K.; Nakagawa, M.; Sekiya, R.; Hemken, P.M.; Sokoll, L.J.; Elliott, D.; Chan, D.W.; Dowell, B.L. A comparative evaluation of Golgi protein-73, fucosylated hemopexin, alpha-fetoprotein, and PIVKA-II in the serum of patients with chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Clin. Chem. Lab. Med. 2011, 49, 711–718. [Google Scholar] [CrossRef]
- Marrero, J.A.; Romano, P.R.; Nikolaeva, O.; Steel, L.; Mehta, A.; Fimmel, C.J.; Comunale, M.A.; D’Amelio, A.; Lok, A.S.; Block, T.M. GP73, a resident Golgi glycoprotein, is a novel serum marker for hepatocellular carcinoma. J. Hepatol. 2005, 43, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, R.; Kladney, R.D.; Havlioglu, N.; Schmitt-Graff, A.; Gusmirovic, I.; Solomon, H.; Luxon, B.A.; Bacon, B.R.; Fimmel, C.J. Disease- and cell-specific expression of GP73 in human liver disease. Am. J. Gastroenterol. 2004, 99, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ma, J.; Chen, P.; Liu, S.; Guo, Y.; Tan, M.; Guo, X.; Feng, Y.; Wang, Q.; Li, W.; et al. Novel serum biomarker of Golgi protein 73 for the diagnosis of clinically significant portal hypertension in patients with compensated cirrhosis. J. Med. Virol. 2024, 96, e29380. [Google Scholar] [CrossRef] [PubMed]
- Drake, R.R.; Schwegler, E.E.; Malik, G.; Diaz, J.; Block, T.; Mehta, A.; Semmes, O.J. Lectin capture strategies combined with mass spectrometry for the discovery of serum glycoprotein biomarkers. Mol. Cell Proteom. 2006, 5, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Q.; Zhang, H.; Zhao, H.; Mao, R.; Li, Z.; Ya, S.; Jia, C.; Bao, Y. Silencing of GP73 inhibits invasion and metastasis via suppression of epithelial-mesenchymal transition in hepatocellular carcinoma. Oncol. Rep. 2017, 37, 1182–1188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vischer, U.M. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J. Thromb. Haemost. 2006, 4, 1186–1193. [Google Scholar] [CrossRef]
- Lisman, T.; Bongers, T.N.; Adelmeijer, J.; Janssen, H.L.; de Maat, M.P.; de Groot, P.G.; Leebeek, F.W. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology 2006, 44, 53–61. [Google Scholar] [CrossRef]
- Hugenholtz, G.C.; Adelmeijer, J.; Meijers, J.C.; Porte, R.J.; Stravitz, R.T.; Lisman, T. An unbalance between von Willebrand factor and ADAMTS13 in acute liver failure: Implications for hemostasis and clinical outcome. Hepatology 2013, 58, 752–761. [Google Scholar] [CrossRef]
- La Mura, V.; Reverter, J.C.; Flores-Arroyo, A.; Raffa, S.; Reverter, E.; Seijo, S.; Abraldes, J.G.; Bosch, J.; Garcia-Pagan, J.C. Von Willebrand factor levels predict clinical outcome in patients with cirrhosis and portal hypertension. Gut 2011, 60, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Ferlitsch, M.; Reiberger, T.; Hoke, M.; Salzl, P.; Schwengerer, B.; Ulbrich, G.; Payer, B.A.; Trauner, M.; Peck-Radosavljevic, M.; Ferlitsch, A. von Willebrand factor as new noninvasive predictor of portal hypertension, decompensation and mortality in patients with liver cirrhosis. Hepatology 2012, 56, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Gyori, G.P.; Pereyra, D.; Rumpf, B.; Hackl, H.; Koditz, C.; Ortmayr, G.; Reiberger, T.; Trauner, M.; Berlakovich, G.A.; Starlinger, P. The von Willebrand Factor Facilitates Model for End-Stage Liver Disease-Independent Risk Stratification on the Waiting List for Liver Transplantation. Hepatology 2020, 72, 584–594. [Google Scholar] [CrossRef]
- Starlinger, P.; Ahn, J.C.; Mullan, A.; Gyoeri, G.P.; Pereyra, D.; Alva-Ruiz, R.; Hackl, H.; Reiberger, T.; Trauner, M.; Santol, J.; et al. The Addition of C-Reactive Protein and von Willebrand Factor to Model for End-Stage Liver Disease-Sodium Improves Prediction of Waitlist Mortality. Hepatology 2021, 74, 1533–1545. [Google Scholar] [CrossRef]
- Dixon, J.L.; Ginsberg, H.N. Hepatic synthesis of lipoproteins and apolipoproteins. Semin. Liver Dis. 1992, 12, 364–372. [Google Scholar] [CrossRef]
- Sundaram, M.; Yao, Z. Intrahepatic role of exchangeable apolipoproteins in lipoprotein assembly and secretion. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1073–1078. [Google Scholar] [CrossRef]
- Green, P.H.; Glickman, R.M.; Saudek, C.D.; Blum, C.B.; Tall, A.R. Human intestinal lipoproteins. Studies in chyluric subjects. J. Clin. Investig. 1979, 64, 233–242. [Google Scholar] [CrossRef]
- Sparks, J.D.; Cianci, J.; Jokinen, J.; Chen, L.S.; Sparks, C.E. Interleukin-6 mediates hepatic hypersecretion of apolipoprotein B. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G980–G989. [Google Scholar] [CrossRef] [PubMed]
- Andus, T.; Bauer, J.; Gerok, W. Effects of cytokines on the liver. Hepatology 1991, 13, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Trieb, M.; Horvath, A.; Birner-Gruenberger, R.; Spindelboeck, W.; Stadlbauer, V.; Taschler, U.; Curcic, S.; Stauber, R.E.; Holzer, M.; Pasterk, L.; et al. Liver disease alters high-density lipoprotein composition, metabolism and function. Biochim. Biophys. Acta 2016, 1861, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Onufer, E.J.; Huang, L.H.; Sprung, R.W.; Davidson, W.S.; Czepielewski, R.S.; Wohltmann, M.; Sorci-Thomas, M.G.; Warner, B.W.; Randolph, G.J. Enterically derived high-density lipoprotein restrains liver injury through the portal vein. Science 2021, 373, eabe6729. [Google Scholar] [CrossRef] [PubMed]
- Trieb, M.; Rainer, F.; Stadlbauer, V.; Douschan, P.; Horvath, A.; Binder, L.; Trakaki, A.; Knuplez, E.; Scharnagl, H.; Stojakovic, T.; et al. HDL-related biomarkers are robust predictors of survival in patients with chronic liver failure. J. Hepatol. 2020, 73, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.; Mihas, A.A.; Abou-Assi, S.G.; Williams, L.M.; Gavis, E.; Pandak, W.M.; Heuman, D.M. High-density lipoprotein cholesterol as an indicator of liver function and prognosis in noncholestatic cirrhotics. Clin. Gastroenterol. Hepatol. 2005, 3, 286–291. [Google Scholar] [CrossRef]
- Miller, M.H.; Walsh, S.V.; Atrih, A.; Huang, J.T.; Ferguson, M.A.; Dillon, J.F. Serum proteome of nonalcoholic fatty liver disease: A multimodal approach to discovery of biomarkers of nonalcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2014, 29, 1839–1847. [Google Scholar] [CrossRef]
- Benner, A.; Lewallen, N.F.; Sadiq, N.M. Biochemistry, Pseudocholinesterase. In StatPearls; Treasure Island (FL) Ineligible Companies: Brielle, NJ, USA, 2023. [Google Scholar]
- Tan, L.; Meng, Y.; Zeng, T.; Wang, Q.; Long, T.; Wu, S.; Guan, X.; Fu, H.; Zheng, W.; Tian, Y.; et al. Clinical diagnostic significance of prealbumin, cholinesterase and retinol binding protein in liver cirrhosis combined with encephalopathy. Br. J. Biomed. Sci. 2019, 76, 24–28. [Google Scholar] [CrossRef]
- Ramachandran, J.; Sajith, K.G.; Priya, S.; Dutta, A.K.; Balasubramanian, K.A. Serum cholinesterase is an excellent biomarker of liver cirrhosis. Trop. Gastroenterol. 2014, 35, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yin, X.; Ma, X.; Guo, X.D.; Jin, B.; Li, H. Assessment of the value of serum cholinesterase as a liver function test for cirrhotic patients. Biomed. Rep. 2013, 1, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Hosp, F.; Mann, M. A Primer on Concepts and Applications of Proteomics in Neuroscience. Neuron 2017, 96, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Altelaar, A.F.; Munoz, J.; Heck, A.J. Next-generation proteomics: Towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2013, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef]
- Klein, C.; Garcia-Rizo, C.; Bisle, B.; Scheffer, B.; Zischka, H.; Pfeiffer, F.; Siedler, F.; Oesterhelt, D. The membrane proteome of Halobacterium salinarum. Proteomics 2005, 5, 180–197. [Google Scholar] [CrossRef]
- Olsen, J.V.; de Godoy, L.M.; Li, G.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell Proteom. 2005, 4, 2010–2021. [Google Scholar] [CrossRef]
- Beck, S.; Michalski, A.; Raether, O.; Lubeck, M.; Kaspar, S.; Goedecke, N.; Baessmann, C.; Hornburg, D.; Meier, F.; Paron, I.; et al. The Impact II, a Very High-Resolution Quadrupole Time-of-Flight Instrument (QTOF) for Deep Shotgun Proteomics. Mol. Cell Proteom. 2015, 14, 2014–2029. [Google Scholar] [CrossRef]
- Rosenberger, G.; Koh, C.C.; Guo, T.; Rost, H.L.; Kouvonen, P.; Collins, B.C.; Heusel, M.; Liu, Y.; Caron, E.; Vichalkovski, A.; et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 2014, 1, 140031. [Google Scholar] [CrossRef]
- Meier, F.; Park, M.A.; Mann, M. Trapped Ion Mobility Spectrometry and Parallel Accumulation-Serial Fragmentation in Proteomics. Mol. Cell Proteom. 2021, 20, 100138. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Martinez-Val, A.; Steigerwald, S.; Ruther, P.; Fort, K.L.; Arrey, T.N.; Harder, A.; Makarov, A.; Olsen, J.V. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell Proteom. 2020, 19, 716–729. [Google Scholar] [CrossRef]
- Hebert, A.S.; Prasad, S.; Belford, M.W.; Bailey, D.J.; McAlister, G.C.; Abbatiello, S.E.; Huguet, R.; Wouters, E.R.; Dunyach, J.J.; Brademan, D.R.; et al. Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal. Chem. 2018, 90, 9529–9537. [Google Scholar] [CrossRef]
- Bache, N.; Geyer, P.E.; Bekker-Jensen, D.B.; Hoerning, O.; Falkenby, L.; Treit, P.V.; Doll, S.; Paron, I.; Muller, J.B.; Meier, F.; et al. A Novel LC System Embeds Analytes in Pre-formed Gradients for Rapid, Ultra-robust Proteomics. Mol. Cell Proteom. 2018, 17, 2284–2296. [Google Scholar] [CrossRef] [PubMed]
- Karayel, O.; Virreira Winter, S.; Padmanabhan, S.; Kuras, Y.I.; Vu, D.T.; Tuncali, I.; Merchant, K.; Wills, A.M.; Scherzer, C.R.; Mann, M. Proteome profiling of cerebrospinal fluid reveals biomarker candidates for Parkinson’s disease. Cell Rep. Med. 2022, 3, 100661. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Wang, Z.; Hartl, J.; Kustatscher, G.; Mulleder, M.; Ralser, M. Mass spectrometry-based high-throughput proteomics and its role in biomedical studies and systems biology. Proteomics 2023, 23, e2200013. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Rodriguez, P.; Selheim, F.; Hernandez-Valladares, M. Mass Spectrometry-Based Proteomics Workflows in Cancer Research: The Relevance of Choosing the Right Steps. Cancers 2023, 15, 555. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell Proteom. 2012, 11, O111–016717. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Szyrwiel, L.; Yu, F.; Teo, G.C.; Rosenberger, G.; Niewienda, A.; Ludwig, D.; Decker, J.; Kaspar-Schoenefeld, S.; Lilley, K.S.; et al. dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 2022, 13, 3944. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Teo, G.C.; Kong, A.T.; Frohlich, K.; Li, G.X.; Demichev, V.; Nesvizhskii, A.I. Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform. Nat. Commun. 2023, 14, 4154. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Somasundaram, K. Targeted Proteomics Comes to the Benchside and the Bedside: Is it Ready for Us? Bioessays 2019, 41, e1800042. [Google Scholar] [CrossRef] [PubMed]
- Kusebauch, U.; Campbell, D.S.; Deutsch, E.W.; Chu, C.S.; Spicer, D.A.; Brusniak, M.Y.; Slagel, J.; Sun, Z.; Stevens, J.; Grimes, B.; et al. Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome. Cell 2016, 166, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef] [PubMed]
- Paul, J.; Veenstra, T.D. Separation of Serum and Plasma Proteins for In-Depth Proteomic Analysis. Separations 2022, 9, 89. [Google Scholar] [CrossRef]
- Ahn, S.B.; Sharma, S.; Mohamedali, A.; Mahboob, S.; Redmond, W.J.; Pascovici, D.; Wu, J.X.; Zaw, T.; Adhikari, S.; Vaibhav, V.; et al. Potential early clinical stage colorectal cancer diagnosis using a proteomics blood test panel. Clin. Proteom. 2019, 16, 34. [Google Scholar] [CrossRef]
- Palstrom, N.B.; Rasmussen, L.M.; Beck, H.C. Affinity Capture Enrichment versus Affinity Depletion: A Comparison of Strategies for Increasing Coverage of Low-Abundant Human Plasma Proteins. Int. J. Mol. Sci. 2020, 21, 5903. [Google Scholar] [CrossRef]
- Wewer Albrechtsen, N.J.; Geyer, P.E.; Doll, S.; Treit, P.V.; Bojsen-Moller, K.N.; Martinussen, C.; Jorgensen, N.B.; Torekov, S.S.; Meier, F.; Niu, L.; et al. Plasma Proteome Profiling Reveals Dynamics of Inflammatory and Lipid Homeostasis Markers after Roux-En-Y Gastric Bypass Surgery. Cell Syst. 2018, 7, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Blume, J.E.; Manning, W.C.; Troiano, G.; Hornburg, D.; Figa, M.; Hesterberg, L.; Platt, T.L.; Zhao, X.; Cuaresma, R.A.; Everley, P.A.; et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat. Commun. 2020, 11, 3662. [Google Scholar] [CrossRef]
- Ferdosi, S.; Tangeysh, B.; Brown, T.R.; Everley, P.A.; Figa, M.; McLean, M.; Elgierari, E.M.; Zhao, X.; Garcia, V.J.; Wang, T.; et al. Engineered nanoparticles enable deep proteomics studies at scale by leveraging tunable nano-bio interactions. Proc. Natl. Acad. Sci. USA 2022, 119, e2106053119. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Walt, D.R. Highly Sensitive and Multiplexed Protein Measurements. Chem. Rev. 2019, 119, 293–321. [Google Scholar] [CrossRef]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Bjorkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Rennel Dickens, E.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, A.; Nong, R.; Darmanis, S.; Hertz, E.; Landegren, U.; Kamali-Moghaddam, M. Protein biomarker validation via proximity ligation assays. Biochim. Biophys. Acta 2014, 1844, 933–939. [Google Scholar] [CrossRef]
- Rohloff, J.C.; Gelinas, A.D.; Jarvis, T.C.; Ochsner, U.A.; Schneider, D.J.; Gold, L.; Janjic, N. Nucleic Acid Ligands With Protein-like Side Chains: Modified Aptamers and Their Use as Diagnostic and Therapeutic Agents. Mol. Ther. Nucleic Acids 2014, 3, e201. [Google Scholar] [CrossRef]
- Brody, E.N.; Willis, M.C.; Smith, J.D.; Jayasena, S.; Zichi, D.; Gold, L. The use of aptamers in large arrays for molecular diagnostics. Mol. Diagn. 1999, 4, 381–388. [Google Scholar] [CrossRef]
- Lim, S.Y.; Lee, J.H.; Welsh, S.J.; Ahn, S.B.; Breen, E.; Khan, A.; Carlino, M.S.; Menzies, A.M.; Kefford, R.F.; Scolyer, R.A.; et al. Evaluation of two high-throughput proteomic technologies for plasma biomarker discovery in immunotherapy-treated melanoma patients. Biomark. Res. 2017, 5, 32. [Google Scholar] [CrossRef]
- Raffield, L.M.; Dang, H.; Pratte, K.A.; Jacobson, S.; Gillenwater, L.A.; Ampleford, E.; Barjaktarevic, I.; Basta, P.; Clish, C.B.; Comellas, A.P.; et al. Comparison of Proteomic Assessment Methods in Multiple Cohort Studies. Proteomics 2020, 20, e1900278. [Google Scholar] [CrossRef]
- Finkernagel, F.; Reinartz, S.; Schuldner, M.; Malz, A.; Jansen, J.M.; Wagner, U.; Worzfeld, T.; Graumann, J.; von Strandmann, E.P.; Muller, R. Dual-platform affinity proteomics identifies links between the recurrence of ovarian carcinoma and proteins released into the tumor microenvironment. Theranostics 2019, 9, 6601–6617. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Kemp, T.J.; Pfeiffer, R.M.; Biancotto, A.; Williams, M.; Munuo, S.; Purdue, M.P.; Hsing, A.W.; Pinto, L.; McCoy, J.P.; et al. Evaluation of multiplexed cytokine and inflammation marker measurements: A methodologic study. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Valikangas, T.; Suomi, T.; Elo, L.L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Webb-Robertson, B.J.; Wiberg, H.K.; Matzke, M.M.; Brown, J.N.; Wang, J.; McDermott, J.E.; Smith, R.D.; Rodland, K.D.; Metz, T.O.; Pounds, J.G.; et al. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J. Proteome Res. 2015, 14, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dongre, A. Proper imputation of missing values in proteomics datasets for differential expression analysis. Brief. Bioinform. 2021, 22, bbaa112. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Hill, E.G.; Schwacke, J.H.; Comte-Walters, S.; Slate, E.H.; Oberg, A.L.; Eckel-Passow, J.E.; Therneau, T.M.; Schey, K.L. A statistical model for iTRAQ data analysis. J. Proteome Res. 2008, 7, 3091–3101. [Google Scholar] [CrossRef]
- Herbrich, S.M.; Cole, R.N.; West, K.P., Jr.; Schulze, K.; Yager, J.D.; Groopman, J.D.; Christian, P.; Wu, L.; O’Meally, R.N.; May, D.H.; et al. Statistical inference from multiple iTRAQ experiments without using common reference standards. J. Proteome Res. 2013, 12, 594–604. [Google Scholar] [CrossRef]
- Choi, H.; Nesvizhskii, A.I. False discovery rates and related statistical concepts in mass spectrometry-based proteomics. J. Proteome Res. 2008, 7, 47–50. [Google Scholar] [CrossRef]
- Malik, R.; Dulla, K.; Nigg, E.A.; Korner, R. From proteome lists to biological impact--tools and strategies for the analysis of large MS data sets. Proteomics 2010, 10, 1270–1283. [Google Scholar] [CrossRef]
- Lavallee-Adam, M.; Rauniyar, N.; McClatchy, D.B.; Yates, J.R., 3rd. PSEA-Quant: A protein set enrichment analysis on label-free and label-based protein quantification data. J. Proteome Res. 2014, 13, 5496–5509. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]
- Chatr-aryamontri, A.; Ceol, A.; Palazzi, L.M.; Nardelli, G.; Schneider, M.V.; Castagnoli, L.; Cesareni, G. MINT: The Molecular INTeraction database. Nucleic Acids Res. 2007, 35, D572–D574. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Sveinbjornsson, G.; Ulfarsson, M.O.; Thorolfsdottir, R.B.; Jonsson, B.A.; Einarsson, E.; Gunnlaugsson, G.; Rognvaldsson, S.; Arnar, D.O.; Baldvinsson, M.; Bjarnason, R.G.; et al. Multiomics study of nonalcoholic fatty liver disease. Nat. Genet. 2022, 54, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Govaere, O.; Cockell, S.; Tiniakos, D.; Queen, R.; Younes, R.; Vacca, M.; Alexander, L.; Ravaioli, F.; Palmer, J.; Petta, S.; et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020, 12, eaba4448. [Google Scholar] [CrossRef] [PubMed]
- Govaere, O.; Hasoon, M.; Alexander, L.; Cockell, S.; Tiniakos, D.; Ekstedt, M.; Schattenberg, J.M.; Boursier, J.; Bugianesi, E.; Ratziu, V.; et al. A proteo-transcriptomic map of non-alcoholic fatty liver disease signatures. Nat. Metab. 2023, 5, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Corey, K.E.; Pitts, R.; Lai, M.; Loureiro, J.; Masia, R.; Osganian, S.A.; Gustafson, J.L.; Hutter, M.M.; Gee, D.W.; Meireles, O.R.; et al. ADAMTSL2 protein and a soluble biomarker signature identify at-risk non-alcoholic steatohepatitis and fibrosis in adults with NAFLD. J. Hepatol. 2022, 76, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Abozaid, Y.J.; Ayada, I.; van Kleef, L.A.; Vallerga, C.L.; Pan, Q.; Brouwer, W.P.; Ikram, M.A.; Van Meurs, J.; de Knegt, R.J.; Ghanbari, M. Plasma proteomic signature of fatty liver disease: The Rotterdam Study. Hepatology 2023, 78, 284–294. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Biomarkers | MW (kDa) | Normal Serum Values * | Pros | Cons |
|---|---|---|---|---|
| Hepatocyte injury markers | ||||
| M30 | 30 | 48.3 to 217.1 U/L a | Sensitive to hepatocellular injury | Not widely available |
| ALT | 54.6 | 10 to 40 U/L | Widely available | Less useful in advanced liver disease |
| AST | 46.1 | 10 to 59 U/L | Widely available | Not very specific |
| Aldolase B | 160 | 1.0 to 7.5 U/L | Liver-specific | Not widely available |
| Secreted hepatocellular proteins | ||||
| PCHE | 342 | 4.9 to 11.9 U/mL | Widely available | False low in some patients |
| ALB | 66.5 | 32 to 52 g/L | Widely available | Long half-life, non-specific |
| Apolipoprotein A-1 | 28.96 | 0.94–1.99 g/L | Not widely available | |
| Vn | 75 | 239 to 711 mg/L b | Not widely available | |
| SAA | 11.4 to 12.5 | 20 mg/mL c | Not widely available | |
| Other | ||||
| AFP | ~70 | <15 ng/mL | Widely available | Some tumors are AFP-negative |
| vWF | 500–20,000 | 50 to 200 IU/dL d | Widely available | Complex biology |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, L.; Guldiken, N.; Remih, K.; Karl, A.S.; Preisinger, C.; Strnad, P. Serum/Plasma Proteome in Non-Malignant Liver Disease. Int. J. Mol. Sci. 2024, 25, 2008. https://doi.org/10.3390/ijms25042008
Fu L, Guldiken N, Remih K, Karl AS, Preisinger C, Strnad P. Serum/Plasma Proteome in Non-Malignant Liver Disease. International Journal of Molecular Sciences. 2024; 25(4):2008. https://doi.org/10.3390/ijms25042008
Chicago/Turabian StyleFu, Lei, Nurdan Guldiken, Katharina Remih, Anna Sophie Karl, Christian Preisinger, and Pavel Strnad. 2024. "Serum/Plasma Proteome in Non-Malignant Liver Disease" International Journal of Molecular Sciences 25, no. 4: 2008. https://doi.org/10.3390/ijms25042008
APA StyleFu, L., Guldiken, N., Remih, K., Karl, A. S., Preisinger, C., & Strnad, P. (2024). Serum/Plasma Proteome in Non-Malignant Liver Disease. International Journal of Molecular Sciences, 25(4), 2008. https://doi.org/10.3390/ijms25042008