
Advancements and Prospects of Genome-Wide Association Studies (GWAS) in Maize


 and
and
Abstract
1. Introduction
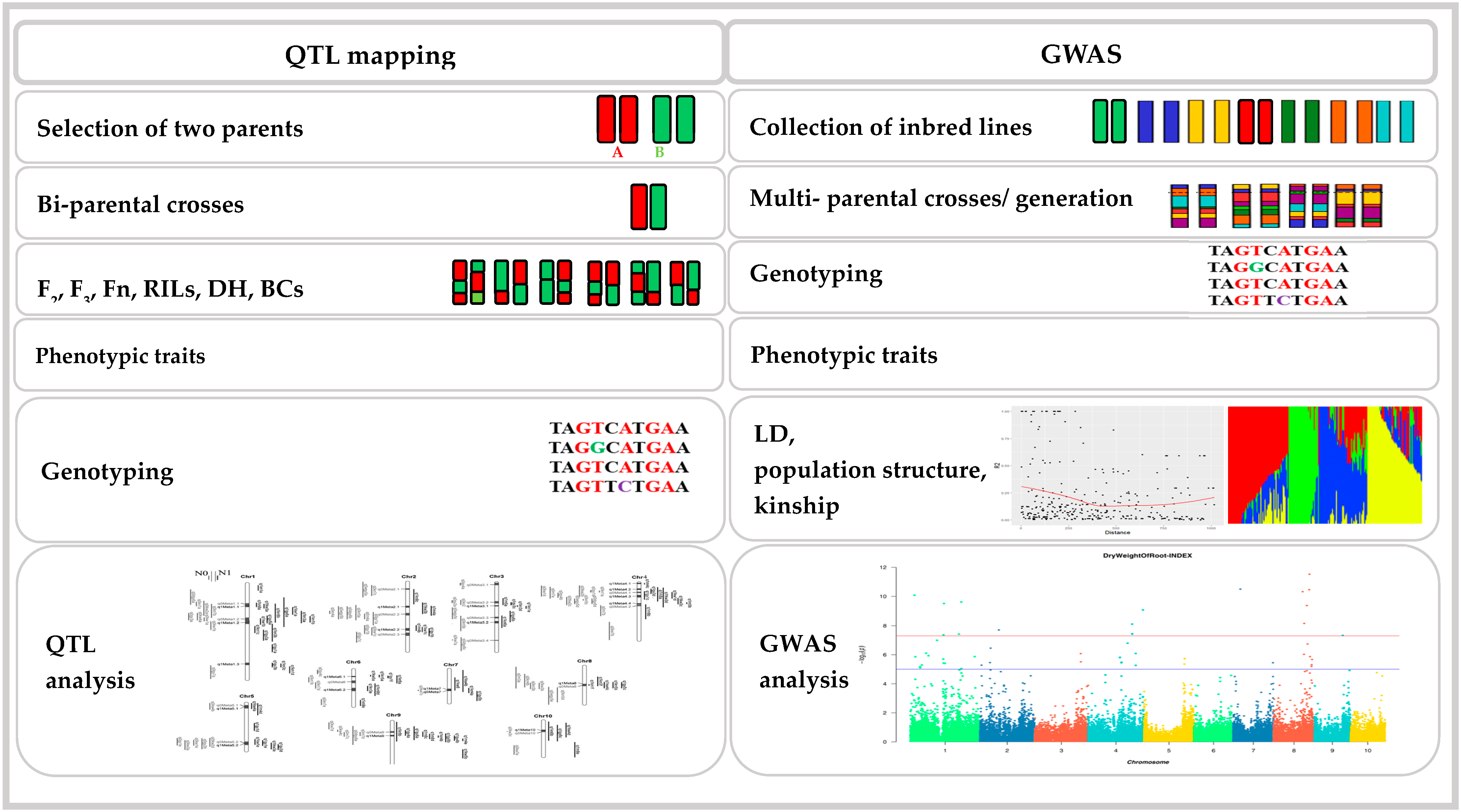
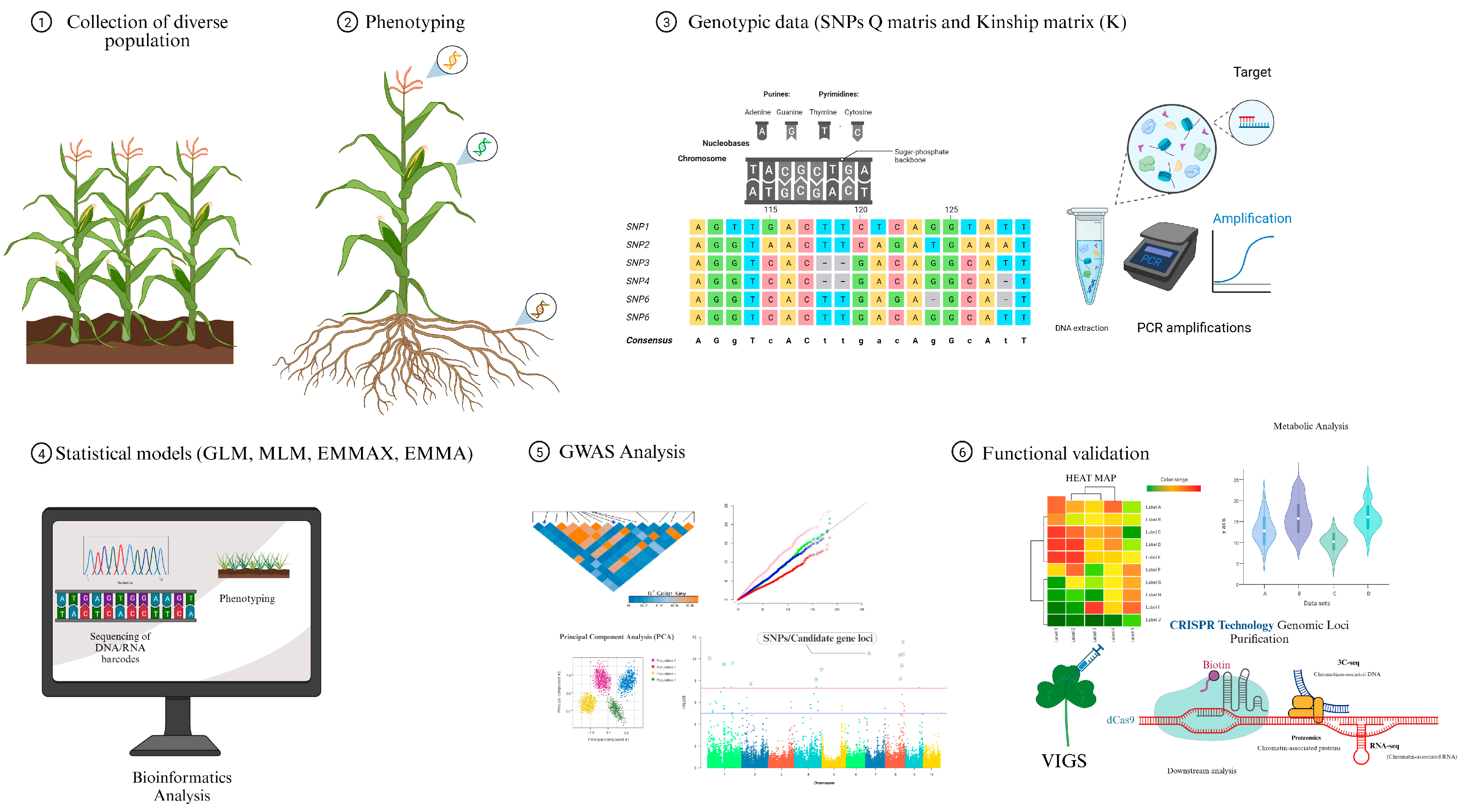
2. GWAS in Maize
3. The Basic GWAS Approach
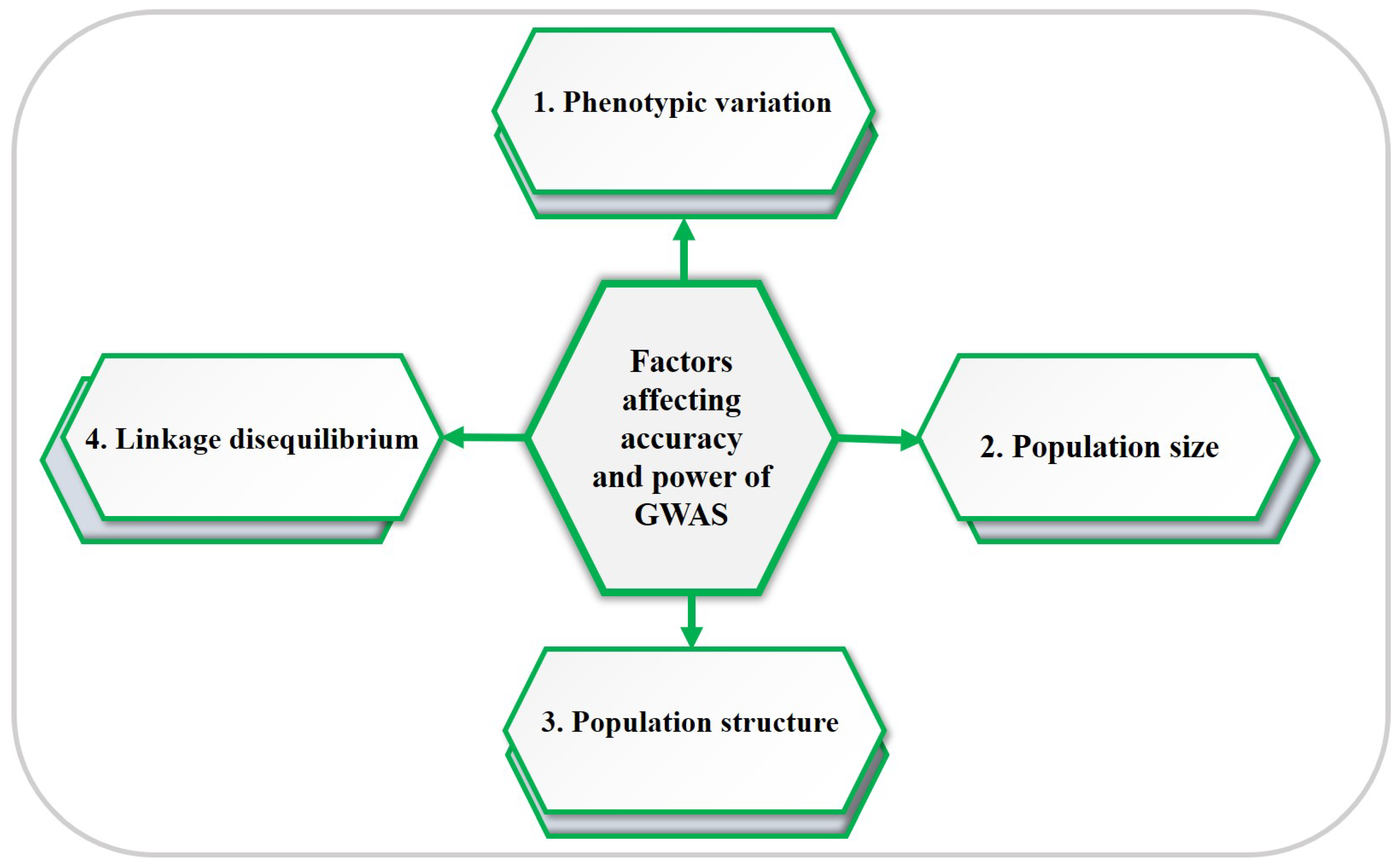
4. Factors Affecting the Accuracy and Statistical Power of GWAS
5. GWAS on Agronomic, Quality, and Quantitative Traits in Maize
6. Factors Affecting Maize Production
7. Identification of SNPs, QTL, and Candidate Genes for Trait Improvement

{kind=link}
{kind=link}
{kind=link}
| Phenotypes Traits | Population | Sample Size | SNPs/QTL/Genes | Chromosomal Location | References |
|---|---|---|---|---|---|
| Ear traits (ear length, diameter, kernel length and width, cob diameter) | Inbred association population | 292 | 20 SNPs | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [124] |
| Corn earworm resistance | Diverse inbreed lines | 287 | 51 SNPs | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [103] |
| Root architecture traits | Diverse inbred lines | 300 | 19 SNPs | 1, 2, 5, 7, and 8 | [103,148] |
| Gray leaf spot resistance | Diverse inbred lines | 157 | 7 SNPs | 1, 2, 3, 4, 5, 6, 7, and 10 | [149] |
| Leaf angle and leaf orientation | diverse inbred lines | 80 | 33 SNPs | 1, 3, 4, 5, 6, 7, and 9 | [150] |
| Male inflorescence morphology | Nested association mapping population | 942 | 242 SNPs | 1, 4, and 6 | [151] |
| Starch pasting properties | Diverse inbred lines | 230 | 60 QTNs | 1, 2, 3, 4, 5, 6, 7, and 8 | [152] |
| Tocopherol content | Diverse inbred lines | 208 | 32 SNPs and 4 candidate genes | Multiple chromosomes | [153] |
| Stalk lodging resistance | Diverse inbred lines | 257 | 423 QTNs and 63 candidate genes | 1, 2, 3, 5, 6, 8, and 9 | [154] |
| Southern corn rust resistance | Diverse inbred lines | 253 | 7 SNPs | 4, 8, and 10 | [155] |
| Corn ear rot resistance | Diverse inbred lines | 242 | 5 candidate genes | 5, 7, and 10 | [156] |
| Ear rot resistance | Diverse inbred lines | 244 | 8 candidate genes | 1, 2, 3, 5, 7, and 9 | [107] |
| Fumonisin accumulation in kernels | Diverse inbred lines | 270 | 39 SNPs/17 QTL | 3 and 4 | [157] |
| Stalk anatomy and stalk biomass | Diverse inbred lines | 492 | 16 candidate genes | Multiple chromosomes | [35] |
| Plant architecture (plant height, leaf length and width and leaf angle | Diverse inbred lines | 87 | 36 QTL | [158] | |
| 13 seedling traits under low phosphorus stress | Diverse inbred lines | 356 | 551 SNPs | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [159] |
| Plant height | Maize hybrids | 300 | 9 SNPs and 2 candidate genes | 1, 2, 4, 7, 9, and 10 | [138] |
| Tassel architecture | Association panel | 359 | 55 candidate genes/19 QTL | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [160] |
| Popping expansion | Diverse inbred lines | 183 | 4 SNPs | [161] | |
| maize lethal necrosis (MLN) and Maize chlorotic mottle virus (MCMV) | Three double-haploid populations | 965 | 54 SNPs and 40 QTL | 1, 2, 3, 4, 5, 6, 7, 8, and 9 | [162] |
| Goss’s wilt | NAM population | 515 | 10 SNPs and 8 candidate genes | [163] | |
| Salt tolerance | Diverse inbred lines | 150 | 7 SNPs and 8 candidate genes | 1, 3, and 6 | [164] |
| Drought tolerance | Diverse inbred lines | 210 | 26 QTL promising candidate genes | 1, 2, 5, 8, and 10 3 | [165] |
| Thermos tolerance of seed | Diverse inbred lines | 261 | 4 QTL, 17 candidate genes and 42 SNPs | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [116] |
| Grain yield and flowering time | Inbred association panel | 300 | 1549 SNPs and 46 candidate genes | 1, 2, 4, 5, 8, and 10 | [166] |
| Husk tightness | Diverse inbred lines | 508 | 27 candidate genes | 1, 2, 3, 5, 6, 7, 8, and 10 | [146] |
| Kernal row number | Diverse inbred lines | 639 | 49 candidate genes and | 1, 2, 3, 5, 9, and 10 | [167] |
| Agronomic traits | Inbred lines | 513 | 3 SNPs | 4, and 3 | [168] |
| Striga resistance | White maize inbred lines | 132 | 24 SNPs | 1, 3, 4, 5, 7, 8, 9, and 10 | [169] |
| Maize leaf necrosis resistance | Diverse inbred lines | 1400 | 32 SNPs and 9 candidate genes | 1, 3, 4, 7, 9, and 10 | [141] |
| Low nitrogen tolerance | Diverse hybrid lines | 49 | 7 candidate genes | Multiple chromosomes | [170] |
| Agronomic traits | Inbred association lines | 224 | 97 candidate genes and 73573 eQTL | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [171] |
| Seminal root length | Inbred association lines | 209 | 7 candidate genes | - | [144] |
| Low temperature | Diverse inbred lines | 222 | 30 SNPs and 82 candidate genes | Multiple chromosomes | [145] |
| Leaf cuticular conductance | Diverse inbred lines | 468 | 9 SNPs and 7 candidate genes | 1, 4, 7, 8, and 10 | [172] |
| Yield related traits | Double haploid population | 250 | 138 SNPs, 100 QTL, and 52 candidate genes | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [173] |
| Root architecture system | Diverse inbred lines | 380 | 87 SNPs and 77 candidate genes | Multiple chromosomes | [174] |
| Fusarium verticillioides resistance | Maize association population | 230 | 42 SNPs and 25 candidate genes | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [175] |
| Corn leaf blight | Association mapping panel | 419 | 22 SNPs | 1, 6, 7, 8, 10 | [176] |
| Aspergillus flavus resistance in kernels | Diverse inbred lines | 313 | 4 SNPs and 16 candidate genes | 1, 2, 8, and 9 | [104] |
| Fusarium ear rot resistance | Diverse inbred lines | 508 | 34 SNPs | - | [177] |
| Gray leaf spot resistance | Diverse inbred lines | 410 | 22 SNPs | 1, 2, 6, 7, and 8 | [178] |
| Agronomic traits | Elite inbred lines | 350 | 129 SNPs | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [179] |
| Aboveground dry matter | Diverse inbred lines | 412 | 129 | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [84] |
| Stover yield | MAGIC population | 408 | 13 SNPs | - | [180] |
| Root architecture traits | RILs population | 179 | 8 SNPs | 1, 2, 4, and 10 | [181] |
| Grain quality traits | Diverse inbred lines | 248 | 49 SNPs and 29 candidate genes | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [75] |
| Root hair length | Diverse inbred lines | 281 | 11 | 1, 2, 4, 5, 6, and 10 | [182] |
| Cold tolerance | Diverse inbred lines | 80 | 4 SNPs and 12 QTL, 1 gene | 3 | [183] |
| Heavy metal stress | Double haploid lines | 187 | 15 QTL and 4 genes | 1, 2, 4, 7, and 10 | [184] |
| Heat tolerance | Double haploid lines | 662 | 46 SNPs | 1, 2, 3, 6, 7, and 8 | [185] |
| Cadmium toxicity | Diverse inbred lines | 513 | 12 SNPs and 1 candidate genes | 2 | [186] |
| Seedling germination traits | MAGIC population | 420 | 28 SNPs | 2, 4, 5, 6, 7, 8, and 9 | [187] |
| Grain yield and related traits | Inbred association panel | 309 | 22 SNPs | - | [188] |
| Accumulation of micronutrients (Fe, Zn, Cu, Mn) | Diverse inbred lines | 305 | 36 SNPs and 11 candidate genes | 2, 3, 4, 6, and 8 | [189] |
| Salt tolerance | Inbred association panel | 305 | 120 candidate genes | - | [73] |
| Kernel moisture and dehydration rate | Diverse inbred lines | 132 | 334 QTNs | 2, 3, 4, 5, 8, and 9 | [190] |
| Root traits | Diverse inbred lines | 319 | 559 SNPs | Multiple chromosomes | [191] |
| Leaf angel | Diverse inbred lines | 285 | 96 SNPs | 1, 2, 3, 4, 5, 6, 7, 9, and 10 | [192] |
| Root system architecture | Diverse inbred lines | 421 | 63 SNPs and 189 candidate genes | 1, 2, 3, 4, 5, 6, 7, 9, and 10 | [137] |
| Grain yield quality traits | Association mapping population | 410 | 42 SNPs | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [193] |
| Yield related traits | Diverse inbred lines | 291 | 59 SNPs and 66 candidate genes | 1, 2, 3, 4, 6, 7, 8, 9, and 10 | [79] |
| Grain yield and other traits | Diverse inbred lines | 169 | 40 SNPs and 6 candidate genes | 1, 2, 8, and 10 | [194] |
| Metaxylem vessel brace roots | Association mapping panel | 508 | 9 SNPs and 5 candidate genes | 2, 4, 7, 8, and 10 | [195] |
| Brace root | Association mapping panel | 508 | 6SNPs and 27 candidate genes | 3, 4, 5, 8, 9, and 10 | [196] |
| Kernal related traits | Association panel | 205 | 139 SNPs and 15 candidate genes | 1, 2, 3, 5, 6, 7, and 9 | [80] |
| Seed germination traits | Diverse inbred lines | 321 | 58 SNPs | 1, 4, 5, 6, 8, 9, and 10 | [197] |
| Stalk lodging resistance | Diverse inbred lines | 248 | 85 SNPs | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [198] |
| Drought and heat resistance | Diverse inbred lines | 162 | 117 SNPs and 20 candidate genes | 1, 2, 5, and 7 | [199] |
| Heat resistance | Diverse inbred lines | 375 | 14 SNPs | 1, 2, 4, 5, and 9 | [200] |
| Rough dwarf disease resistance | Diverse inbred lines | 292 | 22 SNPs | 1, 3, 4, 7, and 8 | [109] |
| Alkaline stress resistance | Association panel | 200 | 9 SNPs | 3, 4, 5, 6, and 9 | [201] |
| Stalk sugar content and agronomic traits | Diverse inbred lines | 188 | 92 SNPs | 1, 3, 4, 6, 7, 8, and 10 | [202] |
| Quality traits and starch pasting | Diverse inbred lines | 292 | 48 SNPs 37 candidate genes | 1, 3, 4, 5, 6, 7, 8, 9, and 10 | [83] |
| Chlorophyll content | Diverse inbred lines | 378 | 19 SNPs | 2, 4, 5, 6, and 10 | [203] |
| Chlorophyll content | Diverse inbred lines | 290 | 140 QTNs and 11 key genes | - | [204] |
| Ear diameter | Multiple parent population | 162 | 11 SNPs and 3 QTL | 1, 2, 3, 6, 8, and 9 | [205] |
| Stalk strength | Diverse inbred lines | 345 | 94 QTL and 241 SNPs | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [206] |
| Chilling tolerant | Diverse inbred lines | 190 | 26 SNPs and 37 candidate genes | 4, 6, 8, and 9 | [119] |
| Striga resistance | Diverse inbred lines | 141 | 22 SNPs | 1, 3, 4, 5, 6, 7, 8, 9, and 10 | [123] |
| Root hair length | Association panel | 200 | 88 QTL | 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 | [207] |
| Total root length | Diverse inbred lines | 280 | 38 candidate genes | 1, 2, 3, 4, 6, 7, 8, and 9 | [208] |
| Root morphology and phosphorus acquisition | Diverse inbred lines | 561 | 7 SNPs | 8 | [209] |
| Leaf streak resistance | Diverse inbred lines | 200 | 11 SNPs | 1, 2, 5, 7, 8 and 9 | [210] |
| Drought resistance | Association panel | 379 | 15 candidate genes | 1, 3, 4, 5, 6, 8, and 9 | [211] |
8. Pervasive Pleiotropy in Maize GWAS Studies
9. Benefits and Limitations of GWAS
9.1. Benefits of GWAS
- Integration of genotype and phenotype: GWAS is a potent method for seamlessly integrating genotype and phenotype data, enhancing our understanding of complex traits.
- Identification of causal and predictive factors: It has the capability to pinpoint both causal and predictive factors associated with specific traits, allowing for in-depth genetic analysis.
- Applicability: GWAS can be conducted on breeding populations as well as natural populations, broadening its utility.
- Discovery of novel associations: It has successfully uncovered novel associations between genetic variants and traits, expanding the scope of genetic research.
- Pathway independence: Unlike QTL mapping, GWAS does not require prior knowledge of the biological pathways related to the studied traits, enabling the discovery of new biological mechanisms.
- Candidate gene discovery: GWAS can identify previously unidentified candidate genes, contributing to the expansion of genetic knowledge.
- Collaboration promotion: GWAS encourages collaborative consortia, facilitating the recruitment of a sufficient number of participants for robust analyses and fostering continued collaboration.
- Ancestry data: It provides ancestry data for each subject, aiding in matching case and control subjects, ensuring the reliability of the analysis.
- Structural variant consideration: GWAS takes into account two types of structural variants sequence variation and copy number, yielding more comprehensive and reliable data.
- Complex trait understanding: It is well suited for unraveling the genetic contributors to complex traits, where an individual’s genes may have a minor influence.
- Data availability: GWAS data is often made publicly available, facilitating the discovery of new trait association and promoting transparency.
- Ethnic diversity: GWAS can shed light on ethnic differences in complex traits, contributing to a more comprehensive understanding of genetic diversity.
9.2. Limitations of GWAS
- Significance threshold: A major limitation is the need for a stringent significance threshold to account for multiple test burdens, potentially missing important associations. Statisticians are strict about this, but if you can prove it with biological evidence, it is not a problem.
- Low-frequency variant analysis: GWAS is not appropriate for studying low-frequency and rare variants. When this happens, a parental population needs to be constructed to detect this rare variant.
- Replication and population size: Findings must be replicated in independent samples from diverse populations, necessitating large and diverse study populations.
- Association vs. causation: GWAS identifies associations but does not pinpoint causal variants and genes. Candidate gene selection and its biological validation are necessary.
- Specific site identification: It may identify specific genetic sites rather than entire genes, and many identified variants are not directly linked to protein-coding regions.
- Missing heritability: GWAS cannot elucidate all genetic determinants of complex traits, leaving much of the heritability unaccounted for.
- Molecular biology insights: Findings related to GWAS variations do not necessarily reveal the underlying molecular biology of traits. Biological validation is necessary.
- Ongoing challenges: While technology, computing, methodology, population stratification, and whole genome sequencing (WGS) may address some limitations, challenges persist in achieving a comprehensive understanding of complex traits.
10. GWAS Interpretation with OMICS
11. Conclusions and Future Prospects in Maize
12. Provoking Questions in GWAS
Author Contributions
Funding
Conflicts of Interest
References
- Blümmel, M.; Grings, E.; Erenstein, O. Potential for dual-purpose maize varieties to meet changing maize demands: Synthesis. Field Crops Res. 2013, 153, 107–112. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Ceasar, S.A.; Duraipandiyan, V.; Ignacimuthu, S. Efficient plant regeneration from shoot apex explants of maize (Zea mays) and analysis of genetic fidelity of regenerated plants by ISSR markers. Plant Cell Tissue Organ Cult. 2014, 119, 183–196. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide association studies in maize: Praise and stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Lobell, D.B.; Schlenker, W.; Costa-Roberts, J. Climate trends and global crop production since 1980. Science 2011, 333, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.-B.; Chu, L.-Y.; Jaleel, C.A.; Manivannan, P.; Panneerselvam, R.; Shao, M.-A. Understanding water deficit stress-induced changes in the basic metabolism of higher plants–biotechnologically and sustainably improving agriculture and the ecoenvironment in arid regions of the globe. Crit. Rev. Biotechnol. 2009, 29, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Plant salt tolerance. Trends Plant Sci. 2001, 6, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.R.G.; Nadarajah, K. QTL and candidate genes: Techniques and advancement in abiotic stress resistance breeding of major cereals. Int. J. Mol. Sci. 2022, 24, 6. [Google Scholar] [CrossRef]
- Almeida, G.D.; Nair, S.; Borém, A.; Cairns, J.; Trachsel, S.; Ribaut, J.-M.; Bänziger, M.; Prasanna, B.M.; Crossa, J.; Babu, R. Molecular mapping across three populations reveals a QTL hotspot region on chromosome 3 for secondary traits associated with drought tolerance in tropical maize. Mol. Breed. 2014, 34, 701–715. [Google Scholar] [CrossRef]
- Dolferus, R.; Thavamanikumar, S.; Sangma, H.; Kleven, S.; Wallace, X.; Forrest, K.; Rebetzke, G.; Hayden, M.; Borg, L.; Smith, A. Determining the genetic architecture of reproductive stage drought tolerance in wheat using a correlated trait and correlated marker effect model. G3 Genes Genomes Genet. 2019, 9, 473–489. [Google Scholar] [CrossRef]
- Uga, Y.; Kitomi, Y.; Yamamoto, E.; Kanno, N.; Kawai, S.; Mizubayashi, T.; Fukuoka, S. A QTL for root growth angle on rice chromosome 7 is involved in the genetic pathway of DEEPER ROOTING 1. Rice 2015, 8, 8. [Google Scholar] [CrossRef]
- Zhao, L.; Lei, J.; Huang, Y.; Zhu, S.; Chen, H.; Huang, R.; Peng, Z.; Tu, Q.; Shen, X.; Yan, S. Mapping quantitative trait loci for heat tolerance at anthesis in rice using chromosomal segment substitution lines. Breed. Sci. 2016, 66, 358–366. [Google Scholar] [CrossRef]
- Xu, Y.; Li, P.; Yang, Z.; Xu, C. Genetic mapping of quantitative trait loci in crops. Crop J. 2017, 5, 175–184. [Google Scholar] [CrossRef]
- Ibrahim, A.K.; Zhang, L.; Niyitanga, S.; Afzal, M.Z.; Xu, Y.; Zhang, L.; Zhang, L.; Qi, J. Principles and approaches of association mapping in plant breeding. Trop. Plant Biol. 2020, 13, 212–224. [Google Scholar] [CrossRef]
- Alqudah, A.M.; Sallam, A.; Baenziger, P.S.; Börner, A. GWAS: Fast-forwarding gene identification and characterization in temperate cereals: Lessons from barley—A review. J. Adv. Res. 2020, 22, 119–135. [Google Scholar] [CrossRef]
- Shikha, K.; Shahi, J.; Vinayan, M.; Zaidi, P.; Singh, A.; Sinha, B. Genome-wide association mapping in maize: Status and prospects. 3 Biotech 2021, 11, 244. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A. The b73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.H.; Gouker, F.E.; Crowell, C.R.; Evans, L.; DiFazio, S.P.; Smart, C.D.; Smart, L.B. Joint linkage and association mapping of complex traits in shrub willow (Salix purpurea L.). Ann. Bot. 2019, 124, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Challa, S.; Neelapu, N.R. Genome-wide association studies (GWAS) for abiotic stress tolerance in plants. In Biochemical, Physiological and Molecular Avenues for Combating Abiotic Stress Tolerance in Plants; Elsevier: Amsterdam, The Netherlands, 2018; pp. 135–150. [Google Scholar]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Yan, J. Crop genome-wide association study: A harvest of biological relevance. Plant J. 2019, 97, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.; Yu, J. Association mapping of genetic resources: Achievements and future perspectives. In Genomics of Plant Genetic Resources; Springer: Berlin/Heidelberg, Germany, 2014; pp. 207–235. [Google Scholar]
- Varshney, R.K.; Ribaut, J.-M.; Buckler, E.S.; Tuberosa, R.; Rafalski, J.A.; Langridge, P. Can genomics boost productivity of orphan crops? Nat. Biotechnol. 2012, 30, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Association mapping in crop plants: Opportunities and challenges. Adv. Genet. 2014, 85, 109–147. [Google Scholar]
- Gupta, P.K.; Rustgi, S.; Kulwal, P.L. Linkage disequilibrium and association studies in higher plants: Present status and future prospects. Plant Mol. Biol. 2005, 57, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Han, B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Ingvarsson, P.K.; Street, N.R. Association genetics of complex traits in plants. New Phytol. 2011, 189, 909–922. [Google Scholar]
- Kulwal, P.L. Association mapping and genomic selection—Where does sorghum stand? In The Sorghum Genome; Springer: Berlin/Heidelberg, Germany, 2016; pp. 137–148. [Google Scholar]
- Borevitz, J.O.; Hazen, S.P.; Michael, T.P.; Morris, G.P.; Baxter, I.R.; Hu, T.T.; Chen, H.; Werner, J.D.; Nordborg, M.; Salt, D.E.; et al. Genome-wide patterns of single-feature polymorphism in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2007, 104, 12057–12062. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Plagnol, V.; Hu, T.T.; Toomajian, C.; Clark, R.M.; Ossowski, S.; Ecker, J.R.; Weigel, D.; Nordborg, M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2007, 39, 1151–1155. [Google Scholar] [CrossRef]
- Todesco, M.; Balasubramanian, S.; Hu, T.T.; Traw, M.B.; Horton, M.; Epple, P.; Kuhns, C.; Sureshkumar, S.; Schwartz, C.; Lanz, C. Natural allelic variation underlying a major fitness trade-off in Arabidopsis thaliana. Nature 2010, 465, 632–636. [Google Scholar] [CrossRef]
- Kleessen, S.; Laitinen, R.; Fusari, C.M.; Antonio, C.; Sulpice, R.; Fernie, A.R.; Stitt, M.; Nikoloski, Z. Metabolic efficiency underpins performance trade-offs in growth of Arabidopsis thaliana. Nat. Commun. 2014, 5, 3537. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Chen, R.; Deng, Y.; Ding, Y.; Guo, J.; Qiu, J.; Wang, B.; Wang, C.; Xie, Y.; Zhang, Z.; Chen, J.; et al. Rice functional genomics: Decades’ efforts and roads ahead. Sci. China Life Sci. 2021, 65, 33–92. [Google Scholar] [CrossRef]
- Cooper, J.S.; Rice, B.R.; Shenstone, E.M.; Lipka, A.E.; Jamann, T.M. Genome-wide analysis and prediction of resistance to goss’s wilt in maize. Plant Genome 2019, 12, 180045. [Google Scholar] [CrossRef]
- Mazaheri, M.; Heckwolf, M.; Vaillancourt, B.; Gage, J.L.; Burdo, B.; Heckwolf, S.; Barry, K.; Lipzen, A.; Ribeiro, C.B.; Kono, T.J.; et al. Genome-wide association analysis of stalk biomass and anatomical traits in maize. BMC Plant Biol. 2019, 19, 45. [Google Scholar] [CrossRef] [PubMed]
- Beló, A.; Zheng, P.; Luck, S.; Shen, B.; Meyer, D.J.; Li, B.; Tingey, S.; Rafalski, A. Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol. Genet. Genom. 2008, 279, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S., IV. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Hindu, V.; Palacios-Rojas, N.; Babu, R.; Suwarno, W.B.; Rashid, Z.; Usha, R.; Saykhedkar, G.R.; Nair, S.K. Identification and validation of genomic regions influencing kernel zinc and iron in maize. Theor. Appl. Genet. 2018, 131, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef]
- Malenica, N.; Dunić, J.A.; Vukadinović, L.; Cesar, V.; Šimić, D. Genetic approaches to enhance multiple stress tolerance in maize. Genes 2021, 12, 1760. [Google Scholar] [CrossRef]
- Comadran, J.; Kilian, B.; Russell, J.; Ramsay, L.; Stein, N.; Ganal, M.; Shaw, P.; Bayer, M.; Thomas, W.; Marshall, D.; et al. Natural variation in a homolog of Antirrhinum centroradialis contributed to spring growth habit and environmental adaptation in cultivated barley. Nat. Genet. 2012, 44, 1388–1392. [Google Scholar] [CrossRef]
- Berhe, M.; Dossa, K.; You, J.; Mboup, P.A.; Diallo, I.N.; Diouf, D.; Zhang, X.; Wang, L. Genome-wide association study and its applications in the non-model crop sesamum indicum. BMC Plant Biol. 2021, 21, 283. [Google Scholar] [CrossRef]
- Wei, X.; Liu, K.; Zhang, Y.; Feng, Q.; Wang, L.; Zhao, Y.; Li, D.; Zhao, Q.; Zhu, X.; Zhu, X.; et al. Genetic discovery for oil production and quality in sesame. Nat. Commun. 2015, 6, 8609. [Google Scholar] [CrossRef]
- Wen, Y.-J.; Zhang, H.; Ni, Y.-L.; Huang, B.; Zhang, J.; Feng, J.-Y.; Wang, S.-B.; Dunwell, J.M.; Zhang, Y.-M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef]
- Fang, C.; Luo, J. Metabolic GWAS-based dissection of genetic bases underlying the diversity of plant metabolism. Plant J. 2019, 97, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Kostova, D.; Bulut, M.; Fernie, A.R. Genome-wide association studies: Assessing trait characteristics in model and crop plants. Cell. Mol. Life Sci. 2021, 78, 5743–5754. [Google Scholar] [CrossRef] [PubMed]
- Schaid, D.J.; Chen, W.; Larson, N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Lu, Y.; Yang, X.; Huang, J.; Zhou, Y.; Ali, F.; Wen, W.; Liu, J.; Li, J.; Yan, J. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet. 2014, 10, e1004573. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.; Daly, M.J.; Lander, E.S. Genetic mapping in human disease. Science 2008, 322, 881–888. [Google Scholar] [CrossRef]
- Burghardt, L.T.; Young, N.D.; Tiffin, P. A guide to genome-wide association mapping in plants. Curr. Protoc. Plant Biol. 2017, 2, 22–38. [Google Scholar] [CrossRef]
- Marchini, J.; Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010, 11, 499–511. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Young, A.I. Solving the missing heritability problem. PLoS Genet. 2019, 15, e1008222. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Z.; Bao, Z.; Li, H.; Lyu, Y.; Zan, Y.; Wu, Y.; Cheng, L.; Fang, Y.; Wu, K.; et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 2022, 606, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Brachi, B.; Morris, G.P.; O Borevitz, J. Genome-wide association studies in plants: The missing heritability is in the field. Genome Biol. 2011, 12, 232. [Google Scholar] [CrossRef] [PubMed]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; De Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Primers 2021, 1, 59. [Google Scholar] [CrossRef]
- Wang, H.; Xu, C.; Liu, X.; Guo, Z.; Xu, X.; Wang, S.; Xie, C.; Li, W.-X.; Zou, C.; Xu, Y. Development of a multiple-hybrid population for genome-wide association studies: Theoretical consideration and genetic mapping of flowering traits in maize. Sci. Rep. 2017, 7, 40239. [Google Scholar] [CrossRef]
- Sanchez, D.L.; Santana, A.S.; Morais, P.I.C.; Peterlini, E.; De La Fuente, G.; Castellano, M.J.; Blanco, M.; Lübberstedt, T. Phenotypic and genome-wide association analyses for nitrogen use efficiency related traits in maize (Zea mays L.) exotic introgression lines. Front. Plant Sci. 2023, 14, 1270166. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, X.; Wang, J.; Li, M.; Wang, Q.; Tian, F.; Su, Z.; Pan, Y.; Liu, D.; Lipka, A.E.; et al. GAPIT version 2: An enhanced integrated tool for genomic association and prediction. Plant Genome 2016, 9. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z. GAPIT version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 2020, 19, 629–640. [Google Scholar] [CrossRef]
- Dwiningsih, S.Y.; Dwiningsih, Y.; Al-Kahtani, J. Genome-wide association study of complex traits in maize detects genomic regions and genes for increasing grain yield and grain quality. Adv. Sustain. Sci. Eng. Technol. 2022, 4, 0220209. [Google Scholar]
- Hamazaki, K.; Kajiya-Kanegae, H.; Yamasaki, M.; Ebana, K.; Yabe, S.; Nakagawa, H.; Iwata, H. Choosing the optimal population for a genome-wide association study: A simulation of whole-genome sequences from rice. Plant Genome 2020, 13, e20005. [Google Scholar] [CrossRef]
- Kumar, J.; Pratap, A.; Solanki, R.K.; Gupta, D.S.; Goyal, A.; Chaturvedi, S.K.; Nadarajan, N.; Kumar, S. Genomic resources for improving food legume crops. J. Agric. Sci. 2012, 150, 289–318. [Google Scholar] [CrossRef]
- Soto-Cerda, B.; Cloutier, S. Association mapping in plant genomes. In Genetic Diversity in Plants; Caliskan, M., Ed.; InTech Open: London, UK, 2012; ISBN 978-953-51-0185-7. [Google Scholar]
- Ersoz, E.S.; Yu, J.; Buckler, E.S. Applications of linkage disequilibrium and association mapping in crop plants. In Genomics-Assisted Crop Improvement: Vol. 1: Genomics Approaches and Platforms; Springer: Berlin/Heidelberg, Germany, 2007; pp. 97–119. [Google Scholar]
- Ersoz, E.S.; Yu, J.; Buckler, E.S. Applications of linkage disequilibrium and association mapping in maize. In Molecular Genetic Approaches to Maize Improvement; Springer: Berlin/Heidelberg, Germany, 2009; pp. 173–195. [Google Scholar]
- Myles, S.; Peiffer, J.; Brown, P.J.; Ersoz, E.S.; Zhang, Z.; Costich, D.E.; Buckler, E.S. Association mapping: Critical considerations shift from genotyping to experimental design. Plant Cell 2009, 21, 2194–2202. [Google Scholar] [CrossRef]
- Sved, J.A.; Cameron, E.C.; Gilchrist, A.S. Estimating effective population size from linkage disequilibrium between unlinked loci: Theory and application to fruit fly outbreak populations. PLoS ONE 2013, 8, e69078. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, M.; Chen, J.; Qing, C.; He, S.; Zou, C.; Yuan, G.; Yang, C.; Peng, H.; Pan, G.; et al. GWAS and wgcna uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor. Appl. Genet. 2021, 134, 3305–3318. [Google Scholar] [CrossRef]
- Tang, J.D.; Perkins, A.; Williams, W.P.; Warburton, M.L. Using genome-wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genom. 2015, 16, 673. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yuan, F.; Huang, Y.; Zhao, Y.; Jia, X.; Zhu, L.; Guo, J. Genome-wide association studies of grain quality traits in maize. Sci. Rep. 2021, 11, 9797. [Google Scholar] [CrossRef]
- Li, H.; Peng, Z.; Yang, X.; Wang, W.; Fu, J.; Wang, J.; Han, Y.; Chai, Y.; Guo, T.; Yang, N.; et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 2013, 45, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Buckler, E.S.; Holland, J.B.; Bradbury, P.J.; Acharya, C.B.; Brown, P.J.; Browne, C.; Ersoz, E.; Flint-Garcia, S.; Garcia, A.; Glaubitz, J.C.; et al. The genetic architecture of maize flowering time. Science 2009, 325, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Li, D.; Li, X.; Gao, Y.; Li, W.; Li, H.; Liu, J.; Liu, H.; Chen, W.; Luo, J.; et al. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 2014, 5, 3438. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Meng, Z.; Yue, R.; Lu, S.; Li, W.; Li, W.; Meng, H.; Sun, Q. Genome wide association analysis for yield related traits in maize. BMC Plant Biol. 2022, 22, 449. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Wu, Y.; Hu, D.; Li, T.; Liang, H.; Ye, F.; Xue, J.; Xu, S. Genome-wide association analysis for candidate genes contributing to kernel-related traits in maize. Front. Plant Sci. 2022, 13, 872292. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, K.; Hu, X.; Liu, Z.; Wu, Y.; Huang, C. Genome-wide association analysis of forage quality in maize mature stalk. BMC Plant Biol. 2016, 16, 227. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, H.; Hu, X.; Liu, Z.; Wu, Y.; Huang, C. Genome-wide association study reveals the genetic basis of stalk cell wall components in maize. PLoS ONE 2016, 11, e0158906. [Google Scholar] [CrossRef]
- Guo, X.; Ge, Z.; Wang, M.; Zhao, M.; Pei, Y.; Song, X. Genome-wide association study of quality traits and starch pasting properties of maize kernels. BMC Genom. 2023, 24, 59. [Google Scholar] [CrossRef]
- Lu, X.; Wang, J.; Wang, Y.; Wen, W.; Zhang, Y.; Du, J.; Zhao, Y.; Guo, X. Genome-wide association study of maize aboveground dry matter accumulation at seedling stage. Front. Genet. 2021, 11, 571236. [Google Scholar] [CrossRef]
- Zhang, X.; Warburton, M.L.; Setter, T.; Liu, H.; Xue, Y.; Yang, N.; Yan, J.; Xiao, Y. Genome-wide association studies of drought-related metabolic changes in maize using an enlarged SNP panel. Theor. Appl. Genet. 2016, 129, 1449–1463. [Google Scholar] [CrossRef]
- Li, P.; Cao, W.; Fang, H.; Xu, S.; Yin, S.; Zhang, Y.; Lin, D.; Wang, J.; Chen, Y.; Xu, C.; et al. Transcriptomic profiling of the maize (Zea mays L.) leaf response to abiotic stresses at the seedling stage. Front. Plant Sci. 2017, 8, 290. [Google Scholar] [CrossRef]
- Yasir, M.; Kanwal, H.H.; Hussain, Q.; Riaz, M.W.; Sajjad, M.; Rong, J.; Jiang, Y. Status and prospects of genome-wide association studies in cotton. Front. Plant Sci. 2022, 13, 1019347. [Google Scholar] [CrossRef] [PubMed]
- Badu-Apraku, B.; Akinwale, R. Cultivar evaluation and trait analysis of tropical early maturing maize under Striga-infested and Striga-free environments. Field Crops Res. 2011, 121, 186–194. [Google Scholar] [CrossRef]
- Balint-Kurti, P.J.; Johal, G.S. Maize disease resistance. In Handbook of Maize: Its Biology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 229–250. [Google Scholar]
- Bennetzen, J.L.; Hake, S.C. Handbook of Maize: Its Biology; Springer Science & Business Media: New York, NY, USA, 2008. [Google Scholar]
- McMullen, M.D.; Frey, M.; Degenhardt, J. Genetics and biochemistry of insect resistance in maize. In Handbook of Maize: Its Biology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 271–289. [Google Scholar]
- Deryng, D.; Conway, D.; Ramankutty, N.; Price, J.; Warren, R. Global crop yield response to extreme heat stress under multiple climate change futures. Environ. Res. Lett. 2014, 9, 034011. [Google Scholar] [CrossRef]
- Farooq, M.; Aziz, T.; Wahid, A.; Lee, D.-J.; Siddique, K.H.M. Chilling tolerance in maize: Agronomic and physiological approaches. Crop Pasture Sci. 2009, 60, 501–516. [Google Scholar] [CrossRef]
- Gunes, A.; Inal, A.; Alpaslan, M.; Cicek, N.; Guneri, E.; Eraslan, F.; Guzelordu, T. Effects of exogenously applied salicylic acid on the induction of multiple stress tolerance and mineral nutrition in maize (Zea mays L.). Arch. Agron. Soil Sci. 2005, 51, 687–695. [Google Scholar] [CrossRef]
- Harrison, M.T.; Tardieu, F.; Dong, Z.; Messina, C.D.; Hammer, G.L. Characterizing drought stress and trait influence on maize yield under current and future conditions. Glob. Change Biol. 2014, 20, 867–878. [Google Scholar] [CrossRef]
- Ribaut, J.-M.; Betran, J.; Monneveux, P.; Setter, T. Drought tolerance in maize. In Handbook of Maize: Its Biology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 311–344. [Google Scholar]
- Subbaiah, C.C.; Sachs, M.M. Responses to oxygen deprivation and potential for enhanced flooding tolerance in maize. In Handbook of Maize: Its Biology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 345–365. [Google Scholar]
- Zaidi, P.; Yadav, M.; Maniselvan, P.; Khan, R.; Shadakshari, T.; Singh, R.; Pal, D. Morpho-physiological traits associated with cold stress tolerance in tropical maize (Zea mays L.). Maydica 2010, 55, 201–208. [Google Scholar]
- Prasad, A.; Senthil-Kumar, M.; Prasad, M. Complex molecular mechanisms determine fitness of plants to biotic and abiotic stresses. J. Plant Biochem. Biotechnol. 2021, 30, 633–635. [Google Scholar] [CrossRef]
- Jakhar, D.S.; Singh, R. Biotic stress response in maize (Zea mays L.). J. Biotechnol. Crop Sci. 2015, 4, 47–51. [Google Scholar]
- Shrestha, V.; Awale, M.; Karn, A. Genome wide association study (GWAS) on disease resistance in maize. In Disease Resistance in Crop Plants: Molecular, Genetic and Genomic Perspectives; Springer: Berlin/Heidelberg, Germany, 2019; pp. 113–130. [Google Scholar]
- Kump, K.L.; Bradbury, P.J.; Wisser, R.J.; Buckler, E.S.; Belcher, A.R.; A Oropeza-Rosas, M.; Zwonitzer, J.C.; Kresovich, S.; McMullen, M.D.; Ware, D.; et al. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 2011, 43, 163–168. [Google Scholar] [CrossRef]
- Warburton, M.L.; Womack, E.D.; Tang, J.D.; Thrash, A.; Smith, J.S.; Xu, W.; Murray, S.C.; Williams, W.P. Genome-wide association and metabolic pathway analysis of corn earworm resistance in maize. Plant Genome 2018, 11, 170069. [Google Scholar] [CrossRef]
- Han, G.; Li, C.; Xiang, F.; Zhao, Q.; Zhao, Y.; Cai, R.; Cheng, B.; Wang, X.; Tao, F. Genome-wide association study leads to novel genetic insights into resistance to Aspergillus flavus in maize kernels. BMC Plant Biol. 2020, 20, 206. [Google Scholar] [CrossRef]
- Rizzardi, D.A.; Peterlini, E.; Scapim, C.A.; Pinto, R.J.B.; Faria, M.V.; Contreras-Soto, R.I. Genome wide association study identifies SNPs associated with northern corn leaf blight caused by Exserohilum turcicum in tropical maize germplasm (Zea mays L.). Euphytica 2022, 218, 40. [Google Scholar] [CrossRef]
- Gowda, M.; Das, B.; Makumbi, D.; Babu, R.; Semagn, K.; Mahuku, G.; Olsen, M.S.; Bright, J.M.; Beyene, Y.; Prasanna, B.M. Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 2015, 128, 1957–1968. [Google Scholar] [CrossRef]
- Han, S.; Miedaner, T.; Utz, H.F.; Schipprack, W.; Schrag, T.A.; Melchinger, A.E. Genomic prediction and GWAS of Gibberella ear rot resistance traits in dent and flint lines of a public maize breeding program. Euphytica 2018, 214, 6. [Google Scholar] [CrossRef]
- Mahuku, G.; Chen, J.; Shrestha, R.; Narro, L.A.; Guerrero, K.V.O.; Arcos, A.L.; Xu, Y. Combined linkage and association mapping identifies a major QTL (qrtsc8-1), conferring tar spot complex resistance in maize. Theor. Appl. Genet. 2016, 129, 1217–1229. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, S.; Pei, Y.; Jiang, X.; Jaqueth, J.S.; Li, B.; Han, J.; Jeffers, D.; Wang, J.; Song, X. Identification of genetic loci associated with rough dwarf disease resistance in maize by integrating GWAS and linkage mapping. Plant Sci. 2022, 315, 111100. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Liu, S.; Ferjani, A.; Li, J.; Yan, J.; Yang, X.; Qin, F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Leipner, J.; Stamp, P.; Guerra-Peraza, O. Low temperature stress in maize (Zea mays L.) induces genes involved in photosynthesis and signal transduction as studied by suppression subtractive hybridization. Plant Physiol. Biochem. 2009, 47, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, H.; Li, W.; Mu, C.; Zhang, F.; Wang, L.; Meng, Z. Genome-wide analysis and environmental response profiling of the fk506-binding protein gene family in maize (Zea mays L.). Gene 2012, 498, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, L.; Xiao, B.; Li, D.; Xing, X.; Kong, X.; Li, D. Enhanced tolerance to low temperature in tobacco by over-expression of a new maize protein phosphatase 2c, ZmPP2C2. J. Plant Physiol. 2010, 167, 1307–1315. [Google Scholar] [CrossRef]
- Kong, X.; Pan, J.; Zhang, M.; Xing, X.; Zhou, Y.; Liu, Y.; Li, D.; Li, D. ZmMKK4, a novel group c mitogen-activated protein kinase kinase in maize (Zea mays), confers salt and cold tolerance in transgenic arabidopsis. Plant Cell Environ. 2011, 34, 1291–1303. [Google Scholar] [CrossRef]
- Luo, B.; Ma, P.; Nie, Z.; Zhang, X.; He, X.; Ding, X.; Feng, X.; Lu, Q.; Ren, Z.; Lin, H.; et al. Metabolite profiling and genome-wide association studies reveal response mechanisms of phosphorus deficiency in maize seedling. Plant J. 2019, 97, 947–969. [Google Scholar] [CrossRef]
- Gao, J.; Wang, S.; Zhou, Z.; Wang, S.; Dong, C.; Mu, C.; Song, Y.; Ma, P.; Li, C.; Wang, Z.; et al. Linkage mapping and genome-wide association reveal candidate genes conferring thermotolerance of seed-set in maize. J. Exp. Bot. 2019, 70, 4849–4864. [Google Scholar] [CrossRef]
- Liu, P.; Zhu, Y.; Liu, H.; Liang, Z.; Zhang, M.; Zou, C.; Yuan, G.; Gao, S.; Pan, G.; Shen, Y.; et al. A combination of a genome-wide association study and a transcriptome analysis reveals circRNAs as new regulators involved in the response to salt stress in maize. Int. J. Mol. Sci. 2022, 23, 9755. [Google Scholar] [CrossRef]
- Strigens, A.; Freitag, N.M.; Gilbert, X.; Grieder, C.; Riedelsheimer, C.; Schrag, T.A.; Messmer, R.; Melchinger, A.E. Association mapping for chilling tolerance in elite flint and dent maize inbred lines evaluated in growth chamber and field experiments. Plant Cell Environ. 2013, 36, 1871–1887. [Google Scholar] [CrossRef]
- Ma, Y.; Yao, L.; Zhang, L.; Su, A.; Wang, R.; Song, W.; Li, Z.; Zhao, J. Genome-wide association analysis of chilling-tolerant germination in a new maize association mapping panel. Food Energy Secur. 2023, 12, e445. [Google Scholar] [CrossRef]
- Zhou, G.; Hao, D.; Xue, L.; Chen, G.; Lu, H.; Zhang, Z.; Shi, M.; Huang, X.; Mao, Y. Genome-wide association study of kernel moisture content at harvest stage in maize. Breed. Sci. 2018, 68, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Luo, J.; Qi, C.; Ruan, Y.; Li, J.; Zhang, A.; Yang, X.; He, Y. Genome-wide association study (GWAS) reveals the genetic architecture of four husk traits in maize. BMC Genom. 2016, 17, 946. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Association mapping in plants in the post-GWAS genomics era. Adv. Genet. 2019, 104, 75–154. [Google Scholar] [PubMed]
- Okunlola, G.; Badu-Apraku, B.; Ariyo, O.; Agre, P.; Offernedo, Q.; Ayo-Vaughan, M. Genome-wide association studies of striga resistance in extra-early maturing quality protein maize inbred lines. G3 Genes Genomes Genet. 2023, 13, jkac237. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-M.; Shao, X.-Y.; Pei, Y.-H.; Guo, X.-M.; Li, J.; Song, X.-Y.; Zhao, M.-A. Genetic diversity and genome-wide association study of major ear quantitative traits using high-density SNPs in maize. Front. Plant Sci. 2018, 9, 966. [Google Scholar] [CrossRef] [PubMed]
- Nordborg, M.; Weigel, D. Next-generation genetics in plants. Nature 2008, 456, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Bin Safdar, L.; Andleeb, T.; Latif, S.; Umer, M.J.; Tang, M.; Li, X.; Liu, S.; Quraishi, U.M. Quraishi. Genome-wide association study and QTL meta-analysis identified novel genomic loci controlling potassium use efficiency and agronomic traits in bread wheat. Front. Plant Sci. 2020, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Atwell, S.; Huang, Y.S.; Vilhjálmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.T.; et al. Genome-wide association study of 107 phenotypes in arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Lin, M.; Lin, Z.; Wang, P.; Yang, Q.; Ye, Z.; Shen, C.; Li, J.; Zhang, L.; et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017, 49, 579–587. [Google Scholar] [CrossRef]
- Du, X.; Huang, G.; He, S.; Yang, Z.; Sun, G.; Ma, X.; Li, N.; Zhang, X.; Sun, J.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated a genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X.; et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef]
- Tieman, D.; Zhu, G.; Resende, M.F.R., Jr.; Lin, T.; Nguyen, C.; Bies, D.; Rambla, J.L.; Beltran, K.S.O.; Taylor, M.; Zhang, B.; et al. A chemical genetic roadmap to improved tomato flavor. Science 2017, 355, 391–394. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261.e12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; He, X.; Wang, Y.; Ma, X.; Yin, D. Genome-wide association study of major agronomic traits related to domestication in peanut. Front. Plant Sci. 2017, 8, 1611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Su, W.; Tao, R.; Zhang, W.; Chen, J.; Wu, P.; Yan, C.; Jia, Y.; Larkin, R.M.; Lavelle, D.; et al. RNA sequencing provides insights into the evolution of lettuce and the regulation of flavonoid biosynthesis. Nat. Commun. 2017, 8, 2264. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Zhou, Z.; Wang, Q.; Guo, J.; Zhao, P.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, X.; et al. Genome-wide association study of 12 agronomic traits in peach. Nat. Commun. 2016, 7, 13246. [Google Scholar] [CrossRef]
- Wu, B.; Ren, W.; Zhao, L.; Li, Q.; Sun, J.; Chen, F.; Pan, Q. Genome-wide association study of root system architecture in maize. Genes 2022, 13, 181. [Google Scholar] [CrossRef]
- Zhang, Y.; Wan, J.; He, L.; Lan, H.; Li, L. Genome-wide association analysis of plant height using the maize F1 population. Plants 2019, 8, 432. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Zhou, J.; Chen, J.; Zhu, L.; Zhao, Y.; Huang, Y. The genetic architecture of zinc and iron content in maize grains as revealed by QTL mapping and meta-analysis. Breed. Sci. 2013, 63, 317–324. [Google Scholar] [CrossRef]
- Qin, H.; Cai, Y.; Liu, Z.; Wang, G.; Wang, J.; Guo, Y.; Wang, H. Identification of QTL for zinc and iron concentration in maize kernel and cob. Euphytica 2012, 187, 345–358. [Google Scholar] [CrossRef]
- Nyaga, C.; Gowda, M.; Beyene, Y.; Muriithi, W.T.; Makumbi, D.; Olsen, M.S.; Suresh, L.M.; Bright, J.M.; Das, B.; Prasanna, B.M. Genome-wide analyses and prediction of resistance to MLN in large tropical maize germplasm. Genes 2019, 11, 16. [Google Scholar] [CrossRef]
- Olukolu, B.A.; Wang, G.-F.; Vontimitta, V.; Venkata, B.P.; Marla, S.; Ji, J.; Gachomo, E.; Chu, K.; Negeri, A.; Benson, J.; et al. A genome-wide association study of the maize hypersensitive defense response identifies genes that cluster in related pathways. PLoS Genet. 2014, 10, e1004562. [Google Scholar] [CrossRef]
- Kaeppler, S.; de Leon, N.; Foerster, J.M.; Muttoni, G. Modifying Flowering Time in Maize; University of Wisconsin: Madison, WI, USA, 2020. [Google Scholar]
- Guo, J.; Li, C.; Zhang, X.; Li, Y.; Zhang, D.; Shi, Y.; Song, Y.; Li, Y.; Yang, D.; Wang, T. Transcriptome and GWAS analyses reveal candidate gene for seminal root length of maize seedlings under drought stress. Plant Sci. 2020, 292, 110380. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, J.; Xu, Q.; Wang, D.; Di, H.; Huang, J.; Yang, X.; Wang, Z.; Zhang, L.; Dong, L.; et al. Identification of candidate tolerance genes to low-temperature during maize germination by GWAS and RNA-seq approaches. BMC Plant Biol. 2020, 20, 333. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, H.; Ni, P.; Yu, S.; Dong, H.; Zhang, A.; Cao, H.; Zhang, L.; Ruan, Y.; Cui, Z. Genome-wide association study dissects the genetic architecture of maize husk tightness. Front. Plant Sci. 2020, 11, 861. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Zhao, H. Molecular and functional dissection of liguleless1 (lg1) in plants. Front. Plant Sci. 2023, 14, 1190004. [Google Scholar] [CrossRef]
- Sanchez, D.L.; Liu, S.; Ibrahim, R.; Blanco, M.; Lübberstedt, T. Genome-wide association studies of doubled haploid exotic introgression lines for root system architecture traits in maize (Zea mays L.). Plant Sci. 2018, 268, 30–38. [Google Scholar] [CrossRef]
- Kuki, M.C.; Scapim, C.A.; Rossi, E.S.; Mangolin, C.A.; do Amaral Júnior, A.T.; Pinto, R.J.B. Genome wide association study for gray leaf spot resistance in tropical maize core. PLoS ONE 2018, 13, e0199539. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, M.; Zhang, Z.; Wang, Z.; Wu, N.; Song, Y.; Wang, P. Screening and verification of genes associated with leaf angle and leaf orientation value in inbred maize lines. PLoS ONE 2018, 13, e0208386. [Google Scholar] [CrossRef] [PubMed]
- Gage, J.L.; White, M.R.; Edwards, J.W.; Kaeppler, S.; de Leon, N. Selection signatures underlying dramatic male inflorescence transformation during modern hybrid maize breeding. Genetics 2018, 210, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, T.; Zhou, Y.; Yin, S.; Li, P.; Liu, J.; Xu, S.; Yang, Z.; Xu, C. Genome-wide association mapping of starch pasting properties in maize using single-locus and multi-locus models. Front. Plant Sci. 2018, 9, 1311. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, S.; Fan, Y.; Liu, N.; Zhan, W.; Liu, H.; Xiao, Y.; Li, K.; Pan, Q.; Li, W.; et al. Beyond pathways: Genetic dissection of tocopherol content in maize kernels by combining linkage and association analyses. Plant Biotechnol. J. 2018, 16, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, P.; Zhang, X.; Zheng, Q.; Chen, M.; Ge, F.; Li, Z.; Sun, W.; Guan, Z.; Liang, T.; et al. Multi-locus genome-wide association study reveals the genetic architecture of stalk lodging resistance-related traits in maize. Front. Plant Sci. 2018, 9, 611. [Google Scholar] [CrossRef]
- Zhou, G.; Hao, D.; Mao, Y.; Zhu, Q.; Chen, G.; Lu, H.; Shi, M.; Huang, X.; Zhang, Z.; Zhao, J.; et al. Identification of genetic loci conferring partial resistance to southern corn rust through a genome-wide association study. Eur. J. Plant Pathol. 2018, 150, 1083–1090. [Google Scholar] [CrossRef]
- de Jong, G.; Pamplona, A.K.A.; Von Pinho, R.G.; Balestre, M. Genome-wide association analysis of ear rot resistance caused by fusarium verticillioides in maize. Genomics 2018, 110, 291–303. [Google Scholar] [CrossRef]
- Samayoa, L.F.; Cao, A.; Santiago, R.; Malvar, R.A.; Butrón, A. Genome-wide association analysis for fumonisin content in maize kernels. BMC Plant Biol. 2019, 19, 166. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, H.; Bo, C.; Dai, W.; Zhang, X.; Cai, R.; Gu, L.; Ma, Q.; Jiang, H.; Zhu, J.; et al. Genome-wide association study of maize plant architecture using f 1 populations. Plant Mol. Biol. 2019, 99, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yuan, Y.; Liao, Z.; Jiang, Y.; Wang, Q.; Zhang, L.; Gao, S.; Wu, F.; Li, M.; Xie, W.; et al. Genome-wide association study of 13 traits in maize seedlings under low phosphorus stress. Plant Genome 2019, 12, 190039. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Guan, Z.; Zhang, X.; Zhang, Y.; Ma, L.; Yao, Y.; Peng, H.; Zhang, Q.; Zhang, B.; et al. Combination of multi-locus genome-wide association study and QTL mapping reveals genetic basis of tassel architecture in maize. Mol. Genet. Genom. 2019, 294, 1421–1440. [Google Scholar] [CrossRef] [PubMed]
- Senhorinho, H.J.C.; Coan, M.M.D.; Marino, T.P.; Kuki, M.C.; Pinto, R.J.B.; Scapim, C.A.; Holland, J.B. Genomic-wide association study of popping expansion in tropical popcorn and field corn germplasm. Crop Sci. 2019, 59, 2007–2019. [Google Scholar] [CrossRef]
- Sitonik, C.; Suresh, L.M.; Beyene, Y.; Olsen, M.S.; Makumbi, D.; Oliver, K.; Das, B.; Bright, J.M.; Mugo, S.; Crossa, J.; et al. Genetic architecture of maize chlorotic mottle virus and maize lethal necrosis through GWAS, linkage analysis and genomic prediction in tropical maize germplasm. Theor. Appl. Genet. 2019, 132, 2381–2399. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Li, G.; Brohammer, A.B.; Jarquin, D.; Hirsch, C.N.; Alfano, J.R.; Lorenz, A.J. Genome-wide association and gene co-expression network analyses reveal complex genetics of resistance to Goss’s wilt of maize. G3 Genes Genomes Genet. 2019, 9, 3139–3152. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Feng, Y.; Chen, Q.; Zhao, F.; Zhou, S.; Ding, Y.; Song, X.; Li, P.; Wang, B. Genome-wide association analysis of salt tolerance QTLs with SNP markers in maize (Zea mays L.). Genes Genom. 2019, 41, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, B.; Liang, X.; Zhou, Y.; Song, J.; Yang, J.; Yong, H.; Weng, J.; Zhang, D.; Li, M.; et al. Genome-wide association study and genomic prediction analyses of drought stress tolerance in china in a collection of off-PVP maize inbred lines. Mol. Breed. 2019, 39, 113. [Google Scholar] [CrossRef]
- Yuan, Y.; Cairns, J.E.; Babu, R.; Gowda, M.; Makumbi, D.; Magorokosho, C.; Zhang, A.; Liu, Y.; Wang, N.; Hao, Z.; et al. Genome-wide association mapping and genomic prediction analyses reveal the genetic architecture of grain yield and flowering time under drought and heat stress conditions in maize. Front. Plant Sci. 2019, 9, 1919. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Chen, L.; Li, Y.-X.; Li, C.; Shi, Y.; Zhang, D.; Li, Y.; Wang, T. Genome-wide association studies and whole-genome prediction reveal the genetic architecture of KRN in maize. BMC Plant Biol. 2020, 20, 490. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hao, Z.; Zhao, Y.; Yang, R. A fast-linear mixed model for genome-wide haplotype association analysis: Application to agronomic traits in maize. BMC Genom. 2020, 21, 151. [Google Scholar] [CrossRef] [PubMed]
- Adewale, S.A.; Badu-Apraku, B.; Akinwale, R.O.; Paterne, A.A.; Gedil, M.; Garcia-Oliveira, A.L. Genome-wide association study of Striga resistance in early maturing white tropical maize inbred lines. BMC Plant Biol. 2020, 20, 203. [Google Scholar] [CrossRef]
- Galli, G.; Alves, F.C.; Morosini, J.S.; Fritsche-Neto, R. On the usefulness of parental lines GWAS for predicting low heritability traits in tropical maize hybrids. PLoS ONE 2020, 15, e0228724. [Google Scholar] [CrossRef]
- Liu, S.; Li, C.; Wang, H.; Wang, S.; Yang, S.; Liu, X.; Yan, J.; Li, B.; Beatty, M.; Zastrow-Hayes, G.; et al. Mapping regulatory variants controlling gene expression in drought response and tolerance in maize. Genome Biol. 2020, 21, 163. [Google Scholar] [CrossRef]
- Lin, M.; Matschi, S.; Vasquez, M.; Chamness, J.; Kaczmar, N.; Baseggio, M.; Miller, M.; Stewart, E.L.; Qiao, P.; Scanlon, M.J.; et al. Genome-wide association study for maize leaf cuticular conductance identifies candidate genes involved in the regulation of cuticle development. G3 Genes Genomes Genet. 2020, 10, 1671–1683. [Google Scholar] [CrossRef]
- Zhang, X.; Guan, Z.; Li, Z.; Liu, P.; Ma, L.; Zhang, Y.; Pan, L.; He, S.; Zhang, Y.; Ge, F.; et al. A combination of linkage mapping and GWAS brings new elements on the genetic basis of yield-related traits in maize across multiple environments. Theor. Appl. Genet. 2020, 133, 2881–2895. [Google Scholar] [CrossRef]
- Zheng, Z.; Hey, S.; Jubery, T.; Liu, H.; Yang, Y.; Coffey, L.; Miao, C.; Sigmon, B.; Schnable, J.C.; Hochholdinger, F.; et al. Shared genetic control of root system architecture between Zea mays and sorghum bicolor. Plant Physiol. 2020, 182, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Stagnati, L.; Rahjoo, V.; Samayoa, L.F.; Holland, J.B.; Borrelli, V.M.G.; Busconi, M.; Lanubile, A.; Marocco, A. A genome-wide association study to understand the effect of Fusarium verticillioides infection on seedlings of a maize diversity panel. G3 Genes Genomes Genet. 2020, 10, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Rashid, Z.; Sofi, M.; Harlapur, S.I.; Kachapur, R.M.; Dar, Z.A.; Singh, P.K.; Zaidi, P.H.; Vivek, B.S.; Nair, S.K. Genome-wide association studies in tropical maize germplasm reveal novel and known genomic regions for resistance to northern corn leaf blight. Sci. Rep. 2020, 10, 21949. [Google Scholar] [CrossRef]
- Yao, L.; Li, Y.; Ma, C.; Tong, L.; Du, F.; Xu, M. Combined genome-wide association study and transcriptome analysis reveal candidate genes for resistance to Fusarium ear rot in maize. J. Integr. Plant Biol. 2020, 62, 1535–1551. [Google Scholar] [CrossRef] [PubMed]
- Kibe, M.; Nair, S.K.; Das, B.; Bright, J.M.; Makumbi, D.; Kinyua, J.; Suresh, L.M.; Beyene, Y.; Olsen, M.S.; Prasanna, B.M.; et al. Genetic dissection of resistance to gray leaf spot by combining genome-wide association, linkage mapping, and genomic prediction in tropical maize germplasm. Front. Plant Sci. 2020, 11, 572027. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, Y. Genetic architecture affecting maize agronomic traits identified by variance heterogeneity association mapping. Genomics 2021, 113, 1681–1688. [Google Scholar] [CrossRef]
- López-Malvar, A.; Butron, A.; Malvar, R.A.; McQueen-Mason, S.J.; Faas, L.; Gómez, L.D.; Revilla, P.; Figueroa-Garrido, D.J.; Santiago, R. Association mapping for maize stover yield and saccharification efficiency using a multiparent advanced generation intercross (MAGIC) population. Sci. Rep. 2021, 11, 3425. [Google Scholar] [CrossRef]
- Moussa, A.A.; Mandozai, A.; Jin, Y.; Qu, J.; Zhang, Q.; Zhao, H.; Anwari, G.; Khalifa, M.A.S.; Lamboro, A.; Noman, M. Genome-wide association screening and verification of potential genes associated with root architectural traits in maize (Zea mays L.) at multiple seedling stages. BMC Genom. 2021, 22, 558. [Google Scholar] [CrossRef]
- Liu, L.; Jiang, L.-G.; Luo, J.-H.; Xia, A.-A.; Chen, L.-Q.; He, Y. Genome-wide association study reveals the genetic architecture of root hair length in maize. BMC Genom. 2021, 22, 664. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, Z.; Xi, Y.; Yang, Z.; Xiao, Z.; Guan, S.; Qu, J.; Wang, P.; Zhao, R. Identification and functional verification of cold tolerance genes in spring maize seedlings based on a genome-wide association study and quantitative trait locus mapping. Front. Plant Sci. 2021, 12, 776972. [Google Scholar] [CrossRef]
- Hou, F.; Zhou, X.; Liu, P.; Yuan, G.; Zou, C.; Lübberstedt, T.; Pan, G.; Ma, L.; Shen, Y. Genetic dissection of maize seedling traits in an ibm syn10 dh population under the combined stress of lead and cadmium. Mol. Genet. Genom. 2021, 296, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Longmei, N.; Gill, G.K.; Zaidi, P.H.; Kumar, R.; Nair, S.K.; Hindu, V.; Vinayan, M.T.; Vikal, Y. Genome wide association mapping for heat tolerance in sub-tropical maize. BMC Genom. 2021, 22, 154. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Luo, M.; Zhang, Y.; Guo, H.; Li, J.; Song, W.; Zhang, R.; Feng, Z.; Kong, M.; Li, H.; et al. Natural variations in the P-type ATPase heavy metal transporter gene ZmHMA3 control cadmium accumulation in maize grains. J. Exp. Bot. 2021, 72, 6230–6246. [Google Scholar] [CrossRef] [PubMed]
- Rida, S.; Maafi, O.; López-Malvar, A.; Revilla, P.; Riache, M.; Djemel, A. Genetics of germination and seedling traits under drought stress in a magic population of maize. Plants 2021, 10, 1786. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Cao, Y. Genetic dissection of grain yield of maize and yield-related traits through association mapping and genomic prediction. Front. Plant Sci. 2021, 12, 690059. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Qing, C.; Zhang, M.; Zou, C.; Pan, G.; Shen, Y. GWAS with a PCA uncovers candidate genes for accumulations of microelements in maize seedlings. Physiol. Plant. 2021, 172, 2170–2180. [Google Scholar] [CrossRef]
- Li, S.; Zhang, C.; Yang, D.; Lu, M.; Qian, Y.; Jin, F.; Liu, X.; Wang, Y.; Liu, W.; Li, X. Detection of QTNs for kernel moisture concentration and kernel dehydration rate before physiological maturity in maize using multi-locus GWAS. Sci. Rep. 2021, 11, 1764. [Google Scholar] [CrossRef]
- Li, P.; Yang, X.; Wang, H.; Pan, T.; Wang, Y.; Xu, Y.; Xu, C.; Yang, Z. Genetic control of root plasticity in response to salt stress in maize. Theor. Appl. Genet. 2021, 134, 1475–1492. [Google Scholar] [CrossRef]
- Peng, B.; Zhao, X.; Wang, Y.; Li, C.; Li, Y.; Zhang, D.; Shi, Y.; Song, Y.; Wang, L.; Li, Y.; et al. Genome-wide association studies of leaf angle in maize. Mol. Breed. 2021, 41, 50. [Google Scholar] [CrossRef]
- Ndlovu, N.; Spillane, C.; McKeown, P.C.; Cairns, J.E.; Das, B.; Gowda, M. Genome-wide association studies of grain yield and quality traits under optimum and low-nitrogen stress in tropical maize (Zea mays L.). Theor. Appl. Genet. 2022, 135, 4351–4370. [Google Scholar] [CrossRef]
- Bhadmus, O.A.; Badu-Apraku, B.; Adeyemo, O.A.; Agre, P.A.; Queen, O.N.; Ogunkanmi, A.L. Genome-wide association analysis reveals genetic architecture and candidate genes associated with grain yield and other traits under low soil nitrogen in early-maturing white quality protein maize inbred lines. Genes 2022, 13, 826. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, M.; Yu, S.; Li, X.; Zhang, A.; Cui, Z.; Dong, X.; Fan, J.; Zhang, L.; Li, C.; et al. A genome-wide association study dissects the genetic architecture of the metaxylem vessel number in maize brace roots. Front. Plant Sci. 2022, 13, 847234. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Chen, S.; Cui, Z.; Lin, J.; Liu, M.; Jin, Y.; Zhang, A.; Gao, Y.; Cao, H.; Ruan, Y. Genome-wide association study reveals the genetic basis of brace root angle and diameter in maize. Front. Genet. 2022, 13, 963852. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liang, Y.; Liu, M.; Zhang, Q.; Wang, Z.; Fan, J.; Ruan, Y.; Zhang, A.; Dong, X.; Yue, J.; et al. Genome-wide association studies provide insights into the genetic architecture of seed germination traits in maize. Front. Plant Sci. 2022, 13, 930438. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zheng, Y.; Jiao, F.; Wang, M.; Zhang, J.; Zhang, Z.; Huang, Y.; Jia, X.; Zhu, L.; Zhao, Y.; et al. Identification of quantitative trait loci for related traits of stalk lodging resistance using genome-wide association studies in maize (Zea mays L.). BMC Genom. Data 2022, 23, 76. [Google Scholar] [CrossRef] [PubMed]
- Osuman, A.S.; Badu-Apraku, B.; Karikari, B.; Ifie, B.E.; Tongoona, P.; Danquah, E.Y. Genome-wide association study reveals genetic architecture and candidate genes for yield and related traits under terminal drought, combined heat and drought in tropical maize germplasm. Genes 2022, 13, 349. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Khalid, M.; Ghafoor, A.; Shah, M.K.N.; Raja, G.K.; Rana, R.M.; Mahmood, T.; Thompson, A.M. SNP-based genome-wide association mapping of pollen viability under heat stress in tropical Zea mays L. Inbred lines. Front. Genet. 2022, 13, 819849. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jia, Y.; Zhou, R.; Liu, L.; Cao, M.; Zhou, Y.; Wang, Z.; Di, H. GWAS and RNA-seq analysis uncover candidate genes associated with alkaline stress tolerance in maize (Zea mays L.) seedlings. Front. Plant Sci. 2022, 13, 963874. [Google Scholar] [CrossRef]
- Chen, J.; Cao, J.; Bian, Y.; Zhang, H.; Li, X.; Wu, Z.; Guo, G.; Lv, G. Identification of genetic variations and candidate genes responsible for stalk sugar content and agronomic traits in fresh corn via GWAS across multiple environments. Int. J. Mol. Sci. 2022, 23, 13490. [Google Scholar] [CrossRef]
- Jin, Y.; Li, D.; Liu, M.; Cui, Z.; Sun, D.; Li, C.; Zhang, A.; Cao, H.; Ruan, Y. Genome-wide association study identified novel SNPs associated with chlorophyll content in maize. Genes 2023, 14, 1010. [Google Scholar] [CrossRef]
- Xiong, X.; Li, J.; Su, P.; Duan, H.; Sun, L.; Xu, S.; Sun, Y.; Zhao, H.; Chen, X.; Ding, D.; et al. Genetic dissection of maize (Zea mays L.) chlorophyll content using multi-locus genome-wide association studies. BMC Genom. 2023, 24, 384. [Google Scholar] [CrossRef]
- Jiang, F.; Liu, L.; Li, Z.; Bi, Y.; Yin, X.; Guo, R.; Wang, J.; Zhang, Y.; Shaw, R.K.; Fan, X. Identification of candidate QTLs and genes for ear diameter by multi-parent population in maize. Genes 2023, 14, 1305. [Google Scholar] [CrossRef]
- Xu, S.; Tang, X.; Zhang, X.; Wang, H.; Ji, W.; Xu, C.; Yang, Z.; Li, P. Genome-wide association study identifies novel candidate loci or genes affecting stalk strength in maize. Crop J. 2023, 11, 220–227. [Google Scholar] [CrossRef]
- Xuhui, L.; Siqi, L.; Weiwei, C.; Hang, Z.; Huanzhang, L.; Danwen, F.; Lina, F.; Junteng, F.; Yuanqiang, H.; Xiangbo, Z.; et al. Genome-wide association study of root hair length in maize. Trop. Plant Biol. 2023, 16, 67–74. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Yang, M.; Liu, R.; Zhang, X.; Jia, Z.; Li, P. Natural variation in zmnac087 contributes to total root length regulation in maize seedlings under salt stress. BMC Plant Biol. 2023, 23, 392. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.A.G.; Tinoco, S.M.d.S.; de Souza, V.F.; Negri, B.F.; Gault, C.M.; Pastina, M.M.; Magalhaes, J.V.; Guimarães, L.J.M.; de Barros, E.G.; Buckler, E.S.; et al. Genome-wide association study for root morphology and phosphorus acquisition efficiency in diverse maize panels. Int. J. Mol. Sci. 2023, 24, 6233. [Google Scholar] [CrossRef]
- Ruiz, M.; Rossi, E.A.; Bonamico, N.C.; Balzarini, M.G. Genome-wide association study for bacterial leaf streak resistance in maize. Agron. J. 2023, 115, 1051–1058. [Google Scholar] [CrossRef]
- Chen, S.; Dang, D.; Liu, Y.; Ji, S.; Zheng, H.; Zhao, C.; Dong, X.; Li, C.; Guan, Y.; Zhang, A.; et al. Genome-wide association study presents insights into the genetic architecture of drought tolerance in maize seedlings under field water-deficit conditions. Front. Plant Sci. 2023, 14, 1165582. [Google Scholar] [CrossRef]
- Balestre, M.; Von Pinho, R.G.; de Souza Junior, C.L.; de Sousa Bueno Filho, J.S. Bayesian mapping of multiple traits in maize: The importance of pleiotropic effects in studying the inheritance of quantitative traits. Theor. Appl. Genet. 2012, 125, 479–493. [Google Scholar] [CrossRef]
- Shikov, A.E.; Skitchenko, R.K.; Predeus, A.V.; Barbitoff, Y.A. Phenome-wide functional dissection of pleiotropic effects highlights key molecular pathways for human complex traits. Sci. Rep. 2020, 10, 1037. [Google Scholar] [CrossRef]
- Singh, B.; Wani, S.H.; Kukreja, S.; Kumar, V.; Goutam, U. Genome-wide association studies (GWAS) for agronomic traits in maize. In Maize Improvement: Current Advances in Yield, Quality, and Stress Tolerance under Changing Climatic Scenarios; Springer: Berlin/Heidelberg, Germany, 2023; pp. 83–98. [Google Scholar]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Khatun, M.; Monir, M.; Lou, X.; Zhu, J.; Xu, H. Genome-wide association studies revealed complex genetic architecture and breeding perspective of maize ear traits. BMC Plant Biol. 2022, 22, 537. [Google Scholar] [CrossRef]
- Akiyama, M. Multi-omics study for interpretation of genome-wide association study. J. Hum. Genet. 2021, 66, 3–10. [Google Scholar] [CrossRef]
- Narkhede, G.W.; Kiranmayee, K.U. Maize improvement using recent omics approaches. In Maize Improvement: Current Advances in Yield, Quality, and Stress Tolerance under Changing Climatic Scenarios; Springer: Berlin/Heidelberg, Germany, 2023; pp. 289–302. [Google Scholar]
- Yang, J.; Yeh, C.-T.; Ramamurthy, R.K.; Qi, X.; Fernando, R.L.; Dekkers, J.C.M.; Garrick, D.J.; Nettleton, D.; Schnable, P.S. Empirical comparisons of different statistical models to identify and validate kernel row number-associated variants from structured multi-parent mapping populations of maize. G3 Genes Genomes Genet. 2018, 8, 3567–3575. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, Z.; Chern, M.; Yin, J.; Yang, C.; Ran, L.; Cheng, M.; He, M.; Wang, K.; Wang, J.; et al. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell 2017, 170, 114–126.e15. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.-C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahito, J.H.; Zhang, H.; Gishkori, Z.G.N.; Ma, C.; Wang, Z.; Ding, D.; Zhang, X.; Tang, J. Advancements and Prospects of Genome-Wide Association Studies (GWAS) in Maize. Int. J. Mol. Sci. 2024, 25, 1918. https://doi.org/10.3390/ijms25031918
Sahito JH, Zhang H, Gishkori ZGN, Ma C, Wang Z, Ding D, Zhang X, Tang J. Advancements and Prospects of Genome-Wide Association Studies (GWAS) in Maize. International Journal of Molecular Sciences. 2024; 25(3):1918. https://doi.org/10.3390/ijms25031918
Chicago/Turabian StyleSahito, Javed Hussain, Hao Zhang, Zeeshan Ghulam Nabi Gishkori, Chenhui Ma, Zhihao Wang, Dong Ding, Xuehai Zhang, and Jihua Tang. 2024. "Advancements and Prospects of Genome-Wide Association Studies (GWAS) in Maize" International Journal of Molecular Sciences 25, no. 3: 1918. https://doi.org/10.3390/ijms25031918
APA StyleSahito, J. H., Zhang, H., Gishkori, Z. G. N., Ma, C., Wang, Z., Ding, D., Zhang, X., & Tang, J. (2024). Advancements and Prospects of Genome-Wide Association Studies (GWAS) in Maize. International Journal of Molecular Sciences, 25(3), 1918. https://doi.org/10.3390/ijms25031918