

Stratification of Colorectal Patients Based on Survival Analysis Shows the Value of Consensus Molecular Subtypes and Reveals the CBLL1 Gene as a Biomarker of CMS2 Tumours

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
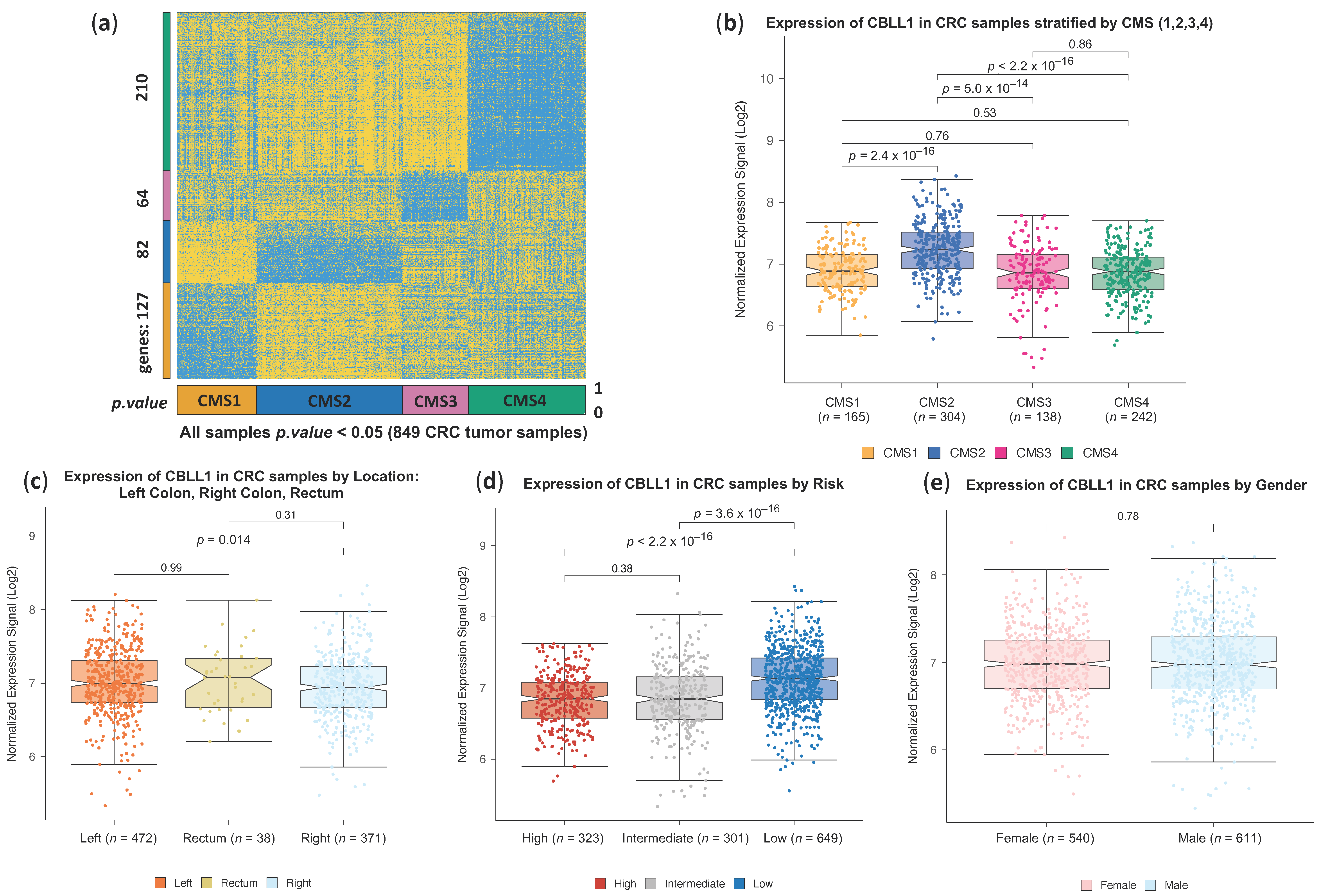
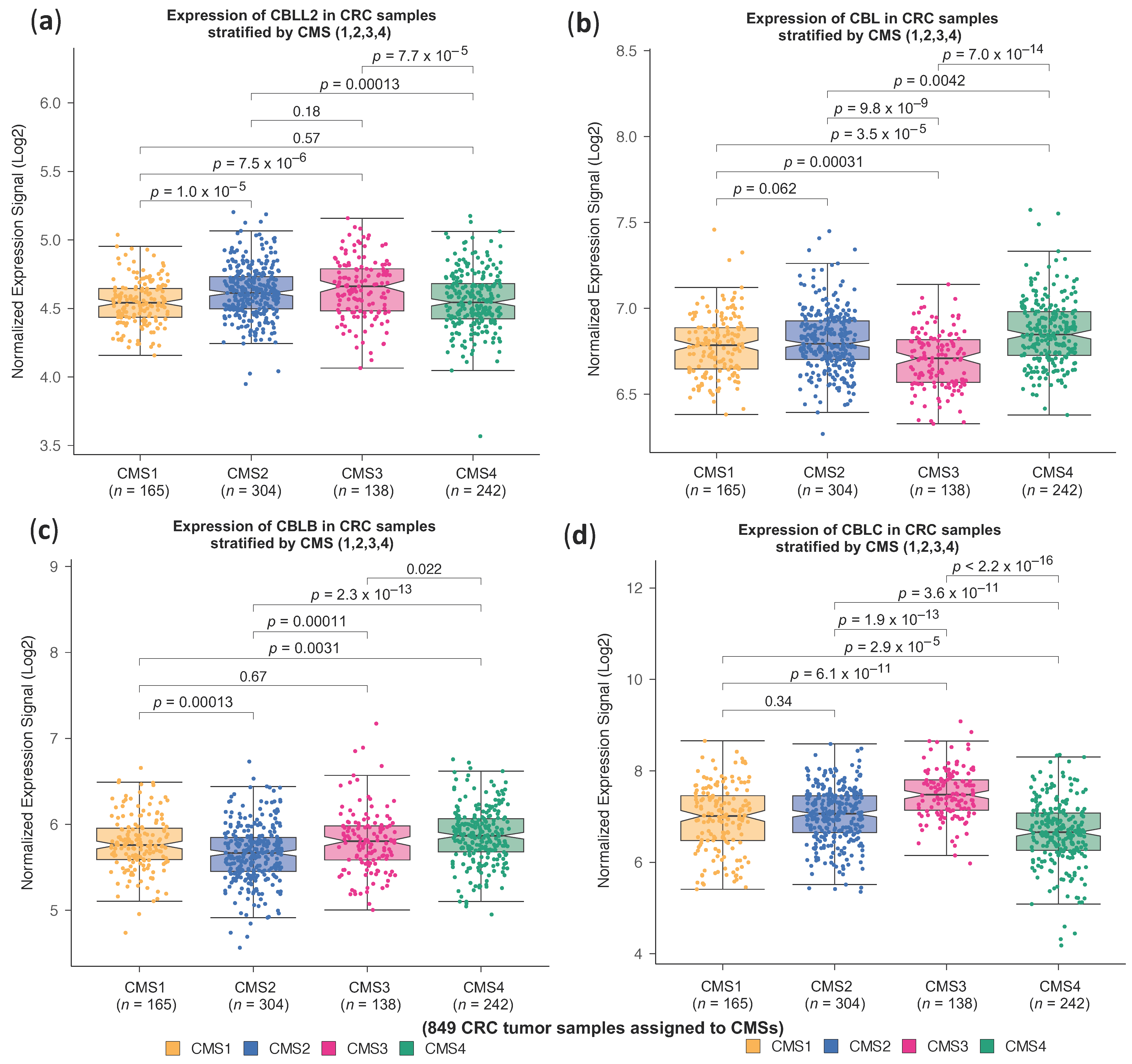
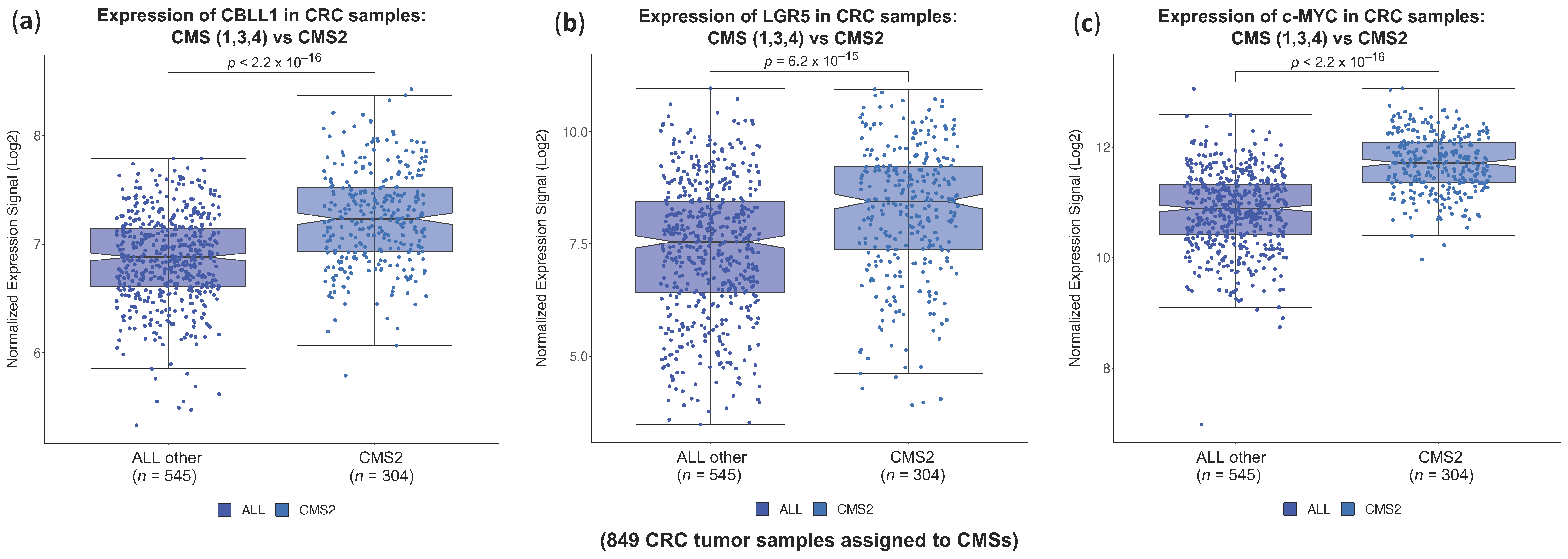
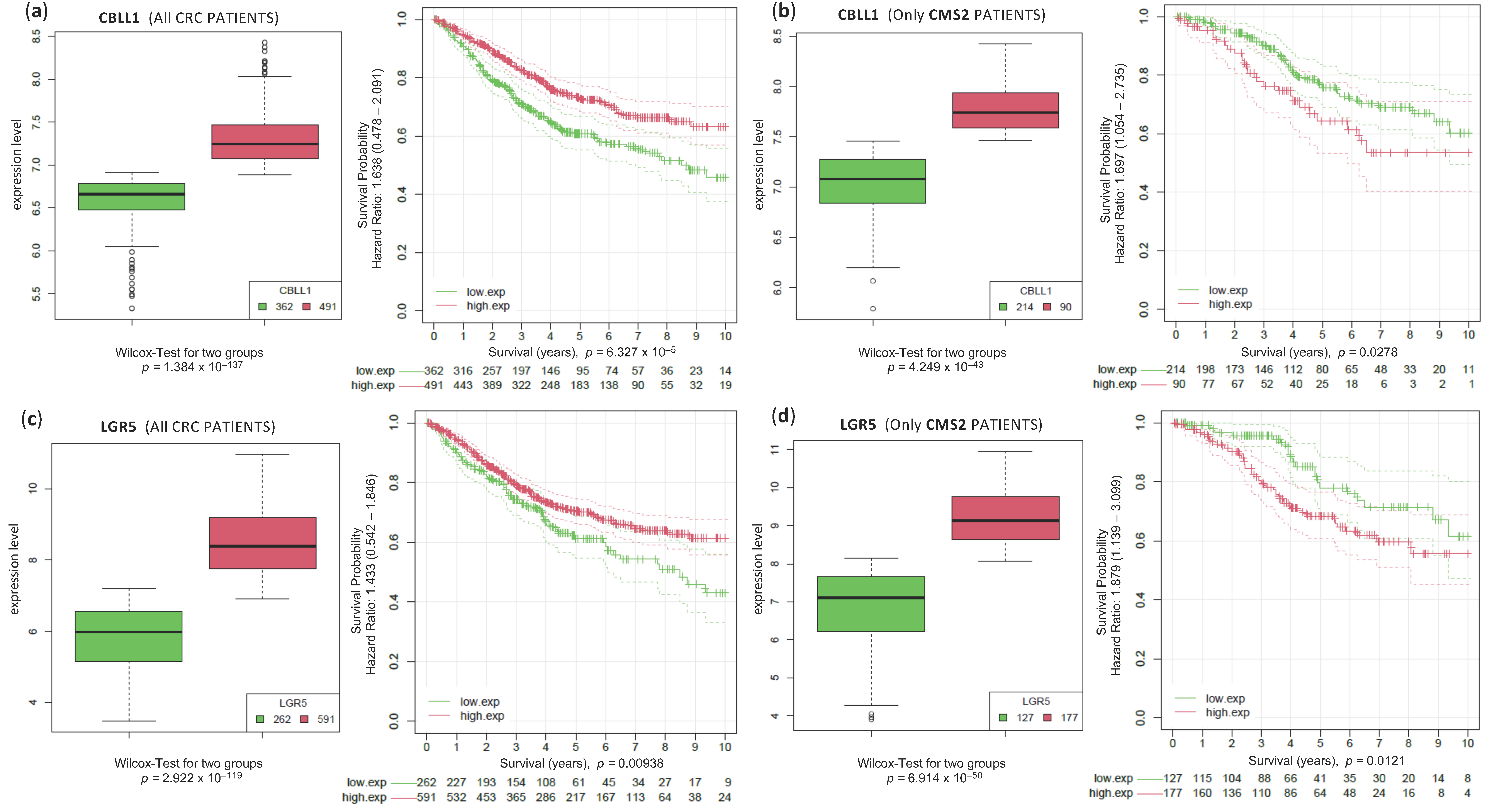
2.1. High CBLL1 Gene Expression Is Specifically Associated with the Consensus Molecular Subtype 2 (CMS2) in Colorectal Cancer Patients
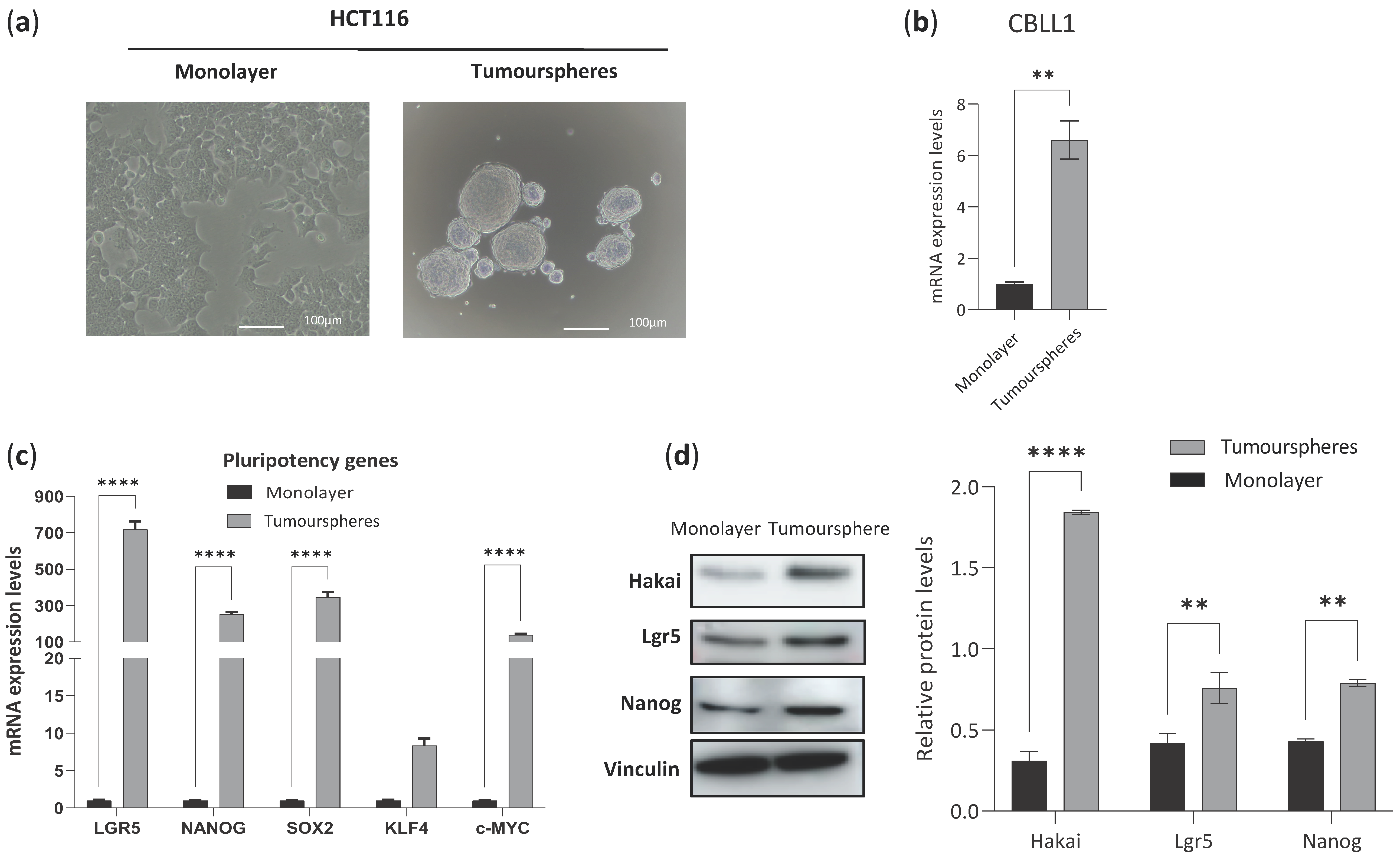
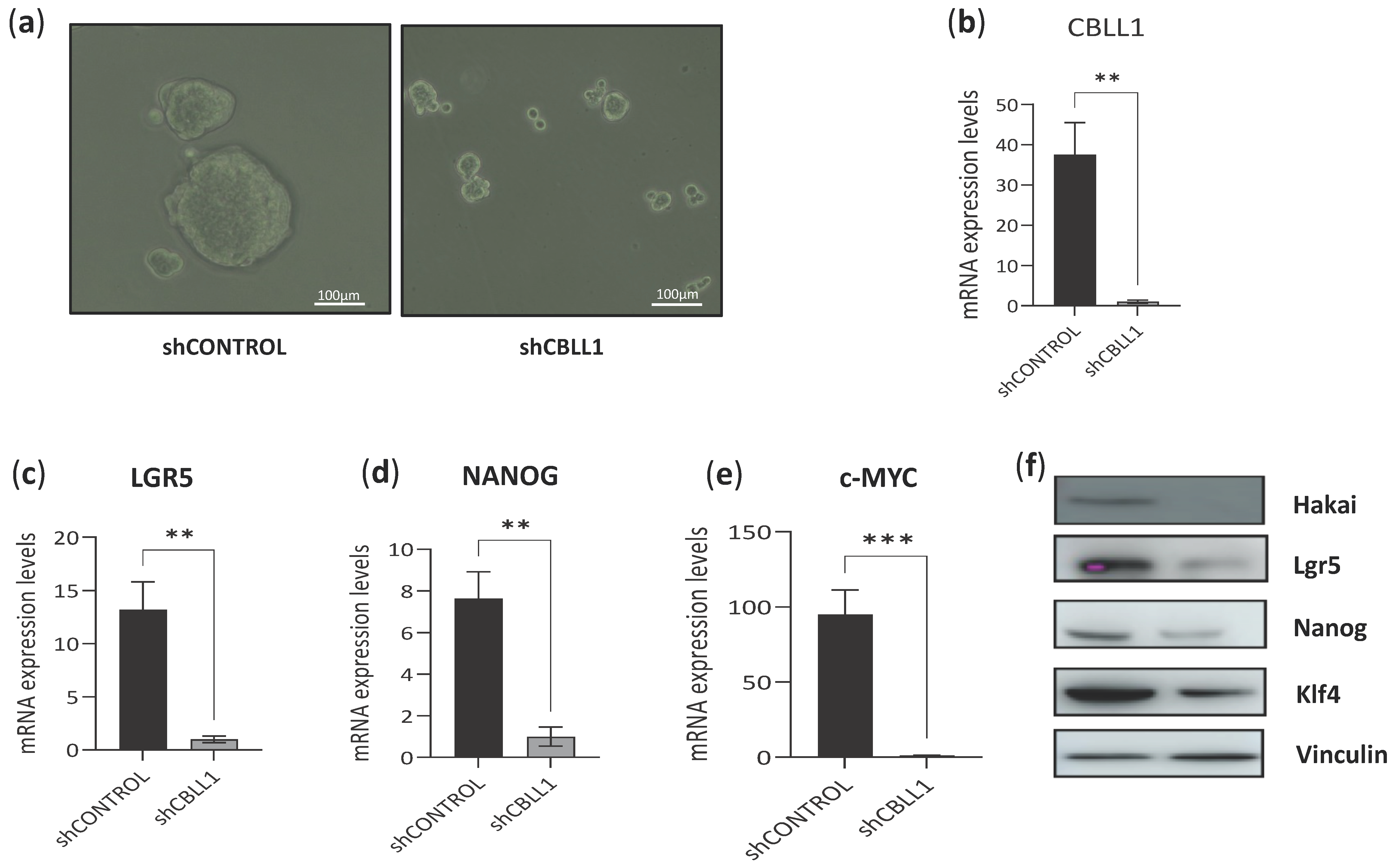
2.2. Impact of Differential Expression of Specific Genes Assigned to CMS2 on Cancer Stem Cells
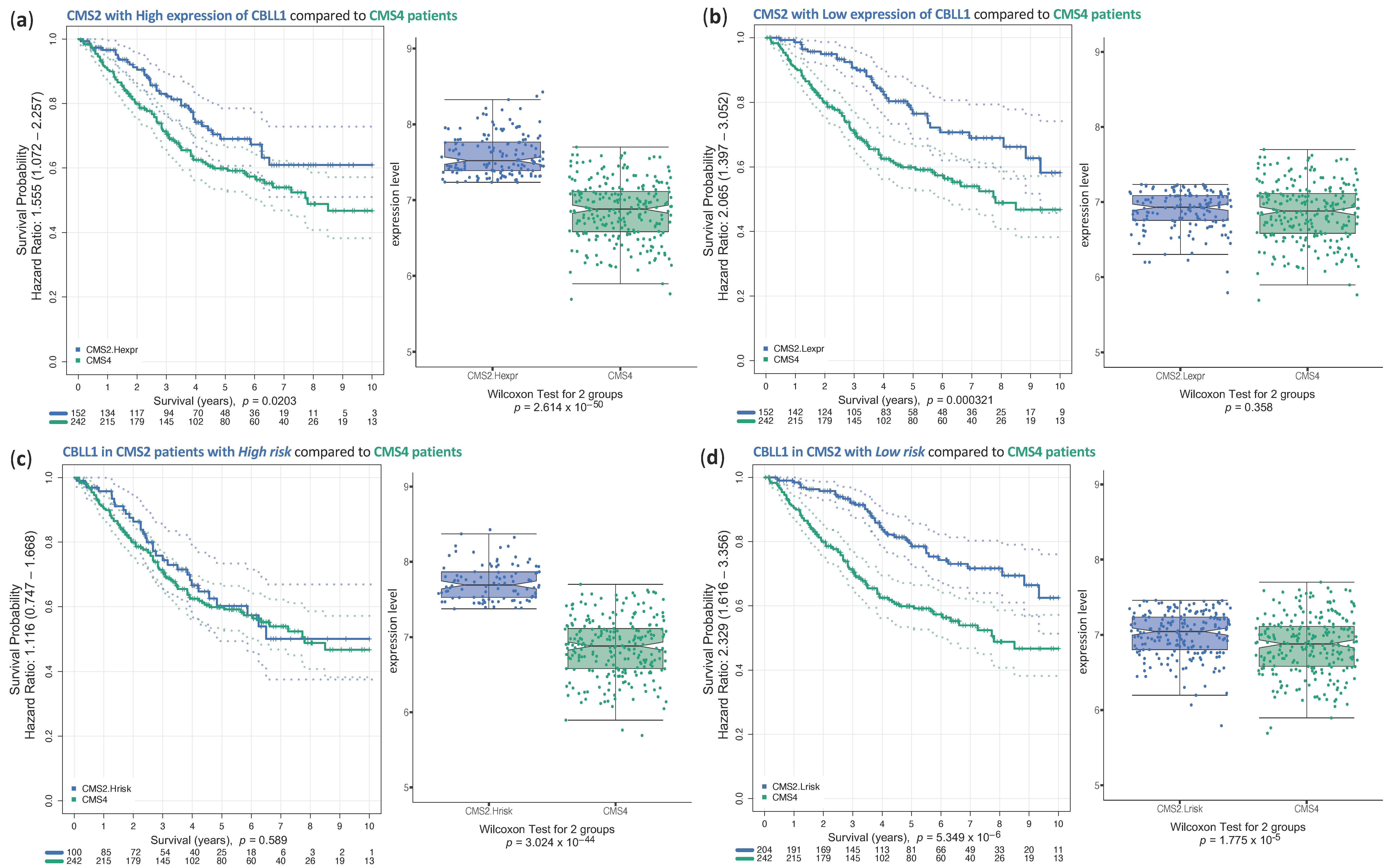
2.3. Analysis of Survival of CRC Samples Classified in CMS2
3. Discussion
4. Materials and Methods
4.1. HT29 and HCT116 Monolayer Cell Culture
4.2. Cancer Stem Cell Tumoursphere-Derived HCT116 and HT29 Cell Lines
4.3. Real-Time Quantitative PCR (RT-qPCR) and Western Blot Analysis
4.4. Data Integration and Analysis Using Bioinformatic
4.5. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fakih, M.G. Metastatic colorectal cancer: Current state and future directions. J. Clin. Oncol. 2015, 33, 1809–1824. [Google Scholar] [CrossRef]
- Milanez-Almeida, P.; Martins, A.J.; Germain, R.N.; Tsang, J.S. Cancer prognosis with shallow tumor RNA sequencing. Nat. Med. 2020, 26, 188–192. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, J.; Brozovic, A.; Izraely, S.; Casas-Pais, A.; Witz, I.P.; Figueroa, A. Cancer drug resistance induced by EMT: Novel therapeutic strategies. Arch. Toxicol. 2021, 95, 2279–2297. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Han, Y.; Xue, X.; Jiang, M.; Guo, X.; Li, P.; Liu, F.; Yuan, B.; Shen, Y.; Zhi, Q.; Zhao, H. LGR5, a relevant marker of cancer stem cells, indicates a poor prognosis in colorectal cancer patients: A meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, B.; Han, X.; Zheng, Z.; Huang, J.; Liu, J.; Huang, Y.; Wei, H. LGR5 is required for the maintenance of spheroid-derived colon cancer stem cells. Int. J. Mol. Med. 2014, 34, 35–42. [Google Scholar] [CrossRef]
- Li, F.; Song, X.; Li, X.; Zhang, X.; Feng, X.; Wang, L.; Xu, L.; Luo, J.; Zhu, B.; Ren, W.; et al. Lgr5 maintains stemness and regulates cell property in nasopharyngeal carcinoma through Wnt/β-catenin signaling pathway. Stem Cell Res. 2020, 47, 101916. [Google Scholar] [CrossRef]
- van Schaijik, B.; Davis, P.F.; Wickremesekera, A.C.; Tan, S.T.; Itinteang, T. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: A review. J. Clin. Pathol. 2018, 71, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Krause, G.; Scheffner, M.; Zechner, D.; Leddy, H.; Behrens, J.; Sommer, T.; Birchmeier, W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002, 4, 222–231. [Google Scholar] [CrossRef]
- Figueroa, A.; Kotani, H.; Toda, Y.; Mazan-Mamczarz, K.; Mueller, E.C.; Otto, A.; Disch, L.; Norman, M.; Ramdasi, R.M.; Keshtgar, M.; et al. Novel roles of hakai in cell proliferation and oncogenesis. Mol. Biol. Cell 2009, 20, 3533–3542. [Google Scholar] [CrossRef]
- Castosa, R.; Martinez-Iglesias, O.; Roca-Lema, D.; Casas-Pais, A.; Díaz-Díaz, A.; Iglesias, P.; Santamarina, I.; Graña, B.; Calvo, L.; Valladares-Ayerbes, M.; et al. Hakai overexpression effectively induces tumour progression and metastasis in vivo. Sci. Rep. 2018, 8, 3466. [Google Scholar] [CrossRef]
- Rodríguez-Alonso, A.; Casas-Pais, A.; Roca-Lema, D.; Graña, B.; Romay, G.; Figueroa, A. Regulation of Epithelial-Mesenchymal Plasticity by the E3 Ubiquitin-Ligases in Cancer. Cancers 2020, 12, 3093. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, M.; Rodríguez-Alonso, A.; Alfonsín, G.; Rodríguez, J.J.E.; Breijo, S.M.; Chantada, V.; Figueroa, A. Protein Degradation by E3 Ubiquitin Ligases in Cancer Stem Cells. Cancers 2022, 14, 990. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A.; Kaneko, T.; Li, S.S. Cell regulation by phosphotyrosine-targeted ubiquitin ligases. Mol. Cell Biol. 2015, 35, 1886–1897. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Jing-Song, F.; Ramachandran, S.; Guy, G.R.; Sivaraman, J. Dimeric switch of Hakai-truncated monomers during substrate recognition: Insights from solution studies and NMR structure. J. Biol. Chem. 2014, 289, 25611–25623. [Google Scholar] [CrossRef]
- Roca-Lema, D.; Quiroga, M.; Khare, V.; Díaz-Díaz, A.; Barreiro-Alonso, A.; Rodríguez-Alonso, A.; Concha, Á.; Romay, G.; Cerdán, M.E.; Gasche, C.; et al. Role of the E3 ubiquitin-ligase Hakai in intestinal inflammation and cancer bowel disease. Sci. Rep. 2022, 12, 17571. [Google Scholar] [CrossRef]
- Martinez-Romero, J.; Bueno-Fortes, S.; Martín-Merino, M.; Ramirez de Molina, A.; De Las Rivas, J. Survival marker genes of colorectal cancer derived from consistent transcriptomic profiling. BMC Genom. 2018, 19, 857. [Google Scholar] [CrossRef]
- Eide, P.W.; Bruun, J.; Lothe, R.A.; Sveen, A. CMScaller: An R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci. Rep. 2017, 7, 16618. [Google Scholar] [CrossRef]
- Baran, B.; Mert Ozupek, N.; Yerli Tetik, N.; Acar, E.; Bekcioglu, O.; Baskin, Y. Difference Between Left-Sided and Right-Sided Colorectal Cancer: A Focused Review of Literature. Gastroenterol. Res. 2018, 11, 264–273. [Google Scholar] [CrossRef]
- Vatandoust, S.; Price, T.J.; Karapetis, C.S. Colorectal cancer: Metastases to a single organ. World J. Gastroenterol. 2015, 21, 11767–11776. [Google Scholar] [CrossRef] [PubMed]
- Benedix, F.; Kube, R.; Meyer, F.; Schmidt, U.; Gastinger, I.; Lippert, H.; Group CRCPTS. Comparison of 17,641 patients with right- and left-sided colon cancer: Differences in epidemiology, perioperative course, histology, and survival. Dis. Colon Rectum 2010, 53, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef] [PubMed]
- Bueno-Fortes, S.; Muenzner, J.K.; Berral-Gonzalez, A.; Hampel, C.; Lindner, P.; Berninger, A.; Huebner, K.; Kunze, P.; Bäuerle, T.; Erlenbach-Wuensch, K.; et al. A gene signature derived from the loss of CDKN1A (p21) is associated with CMS4 Colorectal Cancer. Cancers 2021, 14, 136. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Berral-González, A.; López-Cade, I.; Galindo-Pumariño, C.; Bueno-Fortes, S.; Martín-Merino, M.; Carrato, A.; Ocaña, A.; De La Pinta, C.; López-Alfonso, A.; et al. Cancer-associated fibroblast-derived gene signatures determine prognosis in colon cancer patients. Mol. Cancer 2021, 20, 73. [Google Scholar] [CrossRef]
- Bueno-Fortes, S.; Berral-Gonzalez, A.; Sánchez-Santos, J.M.; Martin-Merino, M.; De Las Rivas, J. Identification of a gene expression signature associated with breast cancer survival and risk that improves clinical genomic platforms. Bioinform. Adv. 2023, 3, vbad037. [Google Scholar] [CrossRef]
- Ros, J.; Baraibar, I.; Martini, G.; Salvà, F.; Saoudi, N.; Cuadra-Urteaga, J.L.; Dienstmann, R.; Tabernero, J.; Élez, E. The Evolving Role of Consensus Molecular Subtypes: A Step Beyond Inpatient Selection for Treatment of Colorectal Cancer. Curr. Treat. Options Oncol. 2021, 22, 113. [Google Scholar] [CrossRef]
- Janda, E.; Nevolo, M.; Lehmann, K.; Downward, J.; Beug, H.; Grieco, M. Raf plus TGFbeta-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene 2006, 25, 7117–7130. [Google Scholar] [CrossRef]
- Xu, L.; Lin, W.; Wen, L.; Li, G. Lgr5 in cancer biology: Functional identification of Lgr5 in cancer progression and potential opportunities for novel therapy. Stem Cell Res. Ther. 2019, 10, 219. [Google Scholar] [CrossRef]
- Wen, J.; Lv, R.; Ma, H.; Shen, H.; He, C.; Wang, J.; Jiao, F.; Liu, H.; Yang, P.; Tan, L.; et al. Zc3h13 Regulates Nuclear RNA m. Mol. Cell 2018, 69, 1028–1038.e1026. [Google Scholar] [CrossRef]
- Horiuchi, K.; Kawamura, T.; Iwanari, H.; Ohashi, R.; Naito, M.; Kodama, T.; Hamakubo, T. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J. Biol. Chem. 2013, 288, 33292–33302. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m. Cell Discov. 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.; Xiong, X.; Kagan, O.; Kwan, J.; Bezginov, A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; Fang, J.M.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef]
- Vignot, S.; Lefebvre, C.; Frampton, G.M.; Meurice, G.; Yelensky, R.; Palmer, G.; Capron, F.; Lazar, V.; Hannoun, L.; Miller, V.A.; et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: Evaluation of concordance between genomic and transcriptional profiles. Eur. J. Cancer 2015, 51, 791–799. [Google Scholar] [CrossRef]
- Phipps, O.; Quraishi, M.N.; Dickson, E.A.; Steed, H.; Kumar, A.; Acheson, A.G.; Beggs, A.D.; Brookes, M.J.; Al-Hassi, H.O. Differences in the On- and Off-Tumor Microbiota between Right- and Left-Sided Colorectal Cancer. Microorganisms 2021, 9, 1108. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfonsín, G.; Berral-González, A.; Rodríguez-Alonso, A.; Quiroga, M.; De Las Rivas, J.; Figueroa, A. Stratification of Colorectal Patients Based on Survival Analysis Shows the Value of Consensus Molecular Subtypes and Reveals the CBLL1 Gene as a Biomarker of CMS2 Tumours. Int. J. Mol. Sci. 2024, 25, 1919. https://doi.org/10.3390/ijms25031919
Alfonsín G, Berral-González A, Rodríguez-Alonso A, Quiroga M, De Las Rivas J, Figueroa A. Stratification of Colorectal Patients Based on Survival Analysis Shows the Value of Consensus Molecular Subtypes and Reveals the CBLL1 Gene as a Biomarker of CMS2 Tumours. International Journal of Molecular Sciences. 2024; 25(3):1919. https://doi.org/10.3390/ijms25031919
Chicago/Turabian StyleAlfonsín, Gloria, Alberto Berral-González, Andrea Rodríguez-Alonso, Macarena Quiroga, Javier De Las Rivas, and Angélica Figueroa. 2024. "Stratification of Colorectal Patients Based on Survival Analysis Shows the Value of Consensus Molecular Subtypes and Reveals the CBLL1 Gene as a Biomarker of CMS2 Tumours" International Journal of Molecular Sciences 25, no. 3: 1919. https://doi.org/10.3390/ijms25031919
APA StyleAlfonsín, G., Berral-González, A., Rodríguez-Alonso, A., Quiroga, M., De Las Rivas, J., & Figueroa, A. (2024). Stratification of Colorectal Patients Based on Survival Analysis Shows the Value of Consensus Molecular Subtypes and Reveals the CBLL1 Gene as a Biomarker of CMS2 Tumours. International Journal of Molecular Sciences, 25(3), 1919. https://doi.org/10.3390/ijms25031919