
PP2A Affects Angiogenesis via Its Interaction with a Novel Phosphorylation Site of TSP1



Abstract
1. Introduction
2. Results
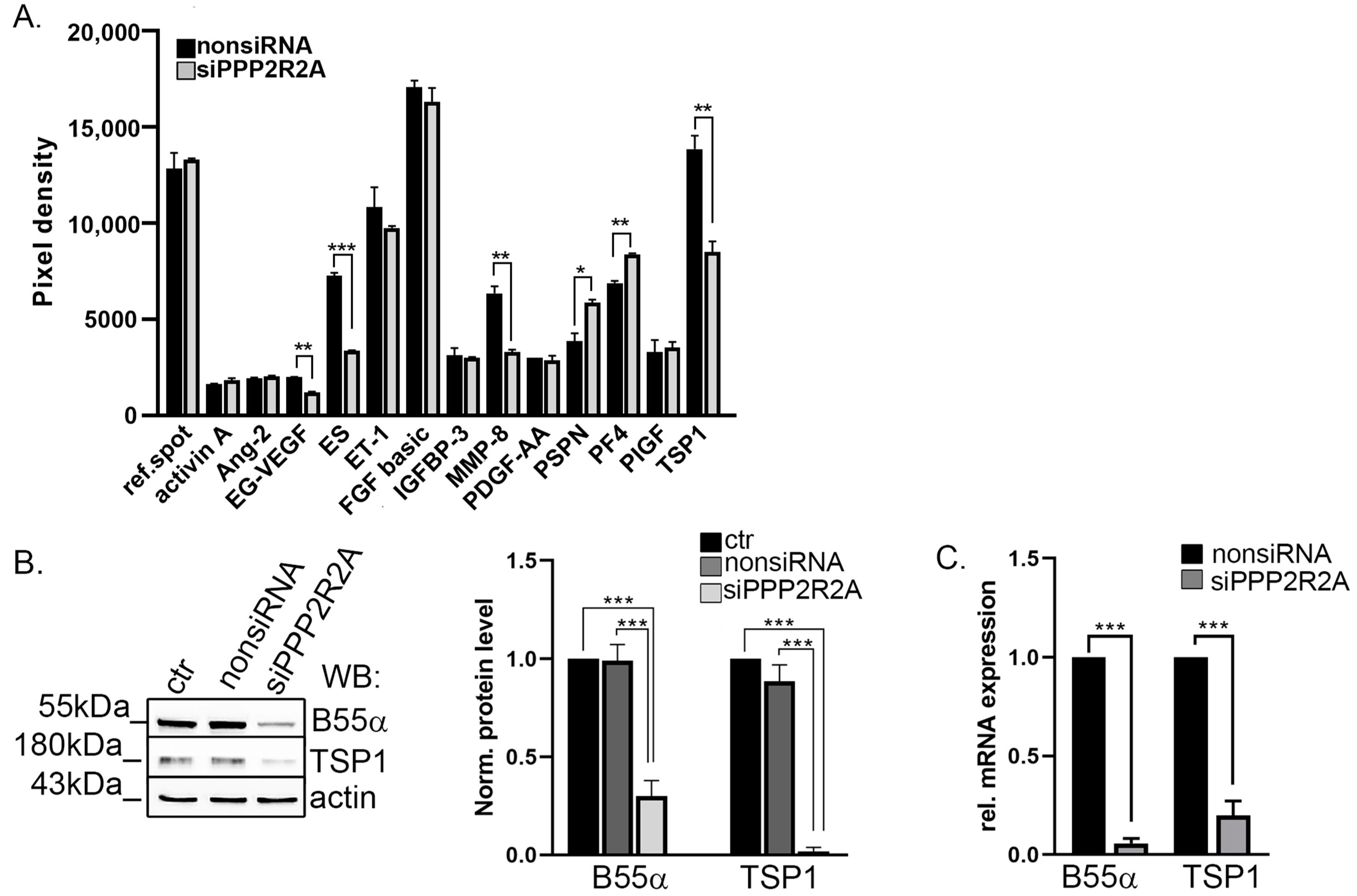
2.1. TSP1 Is Downregulated in B55α-Depleted Endothelial Cells
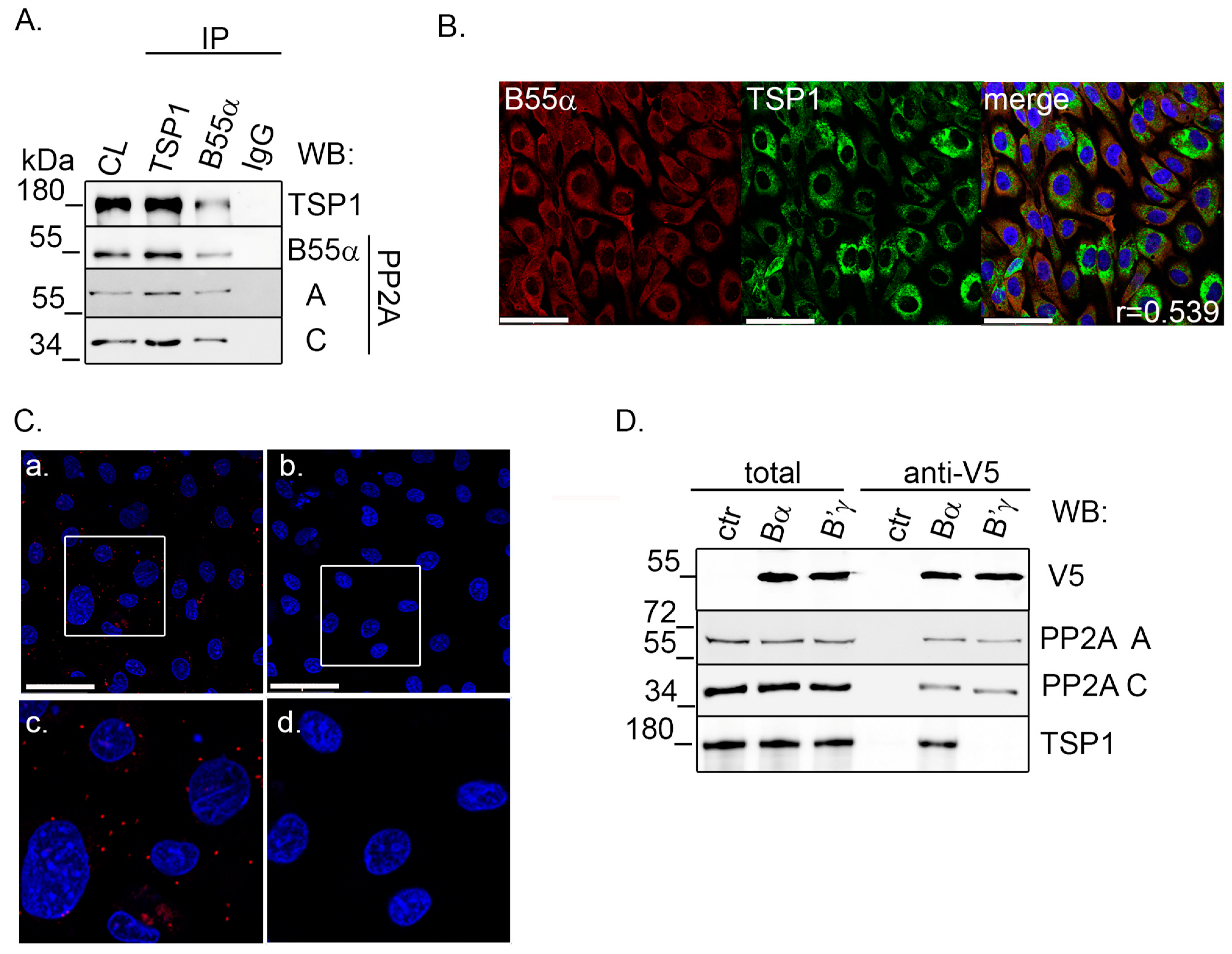
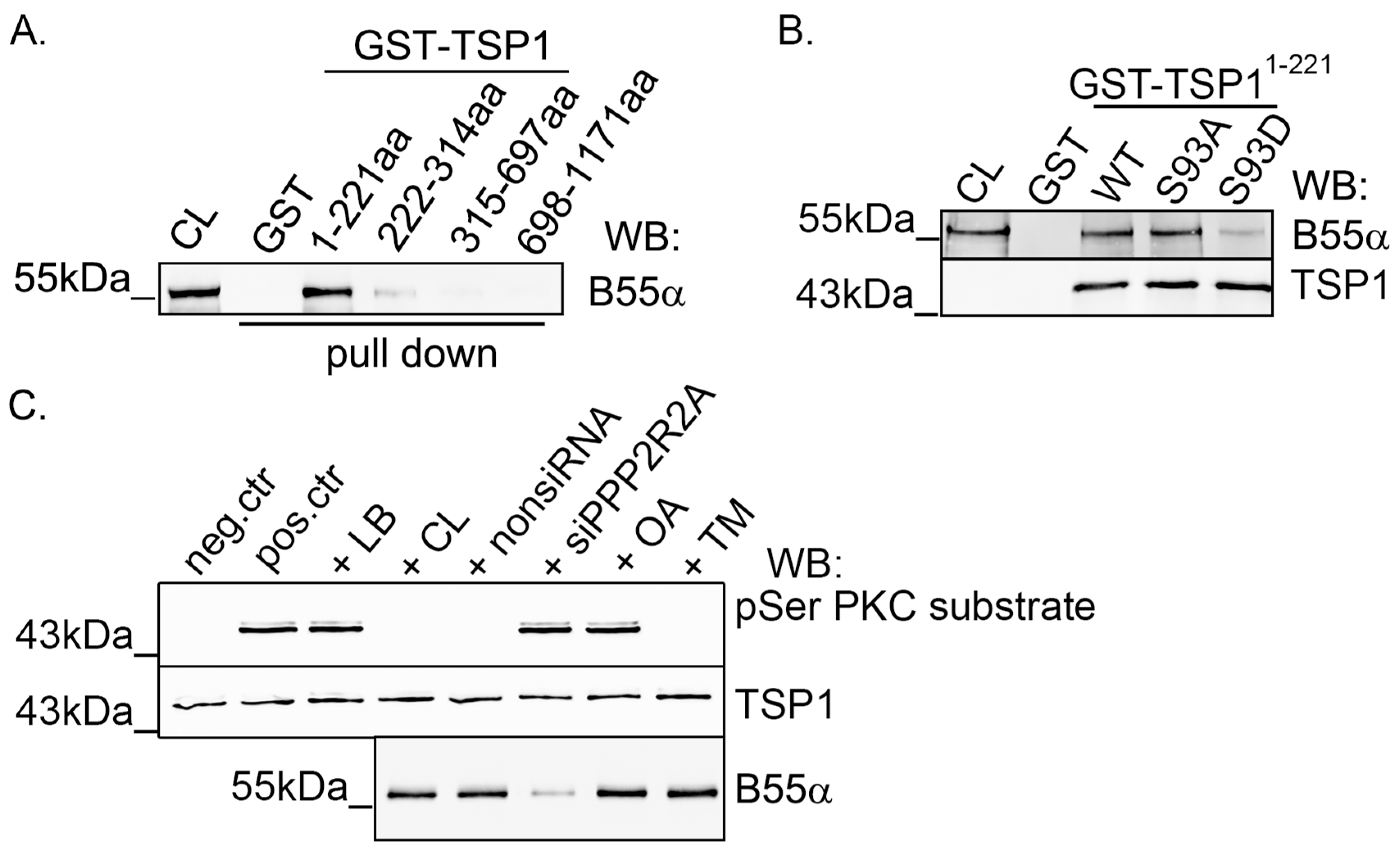
2.2. TSP1 Interacts with B55α Containing PP2A Holoenzyme in Endothelial Cells
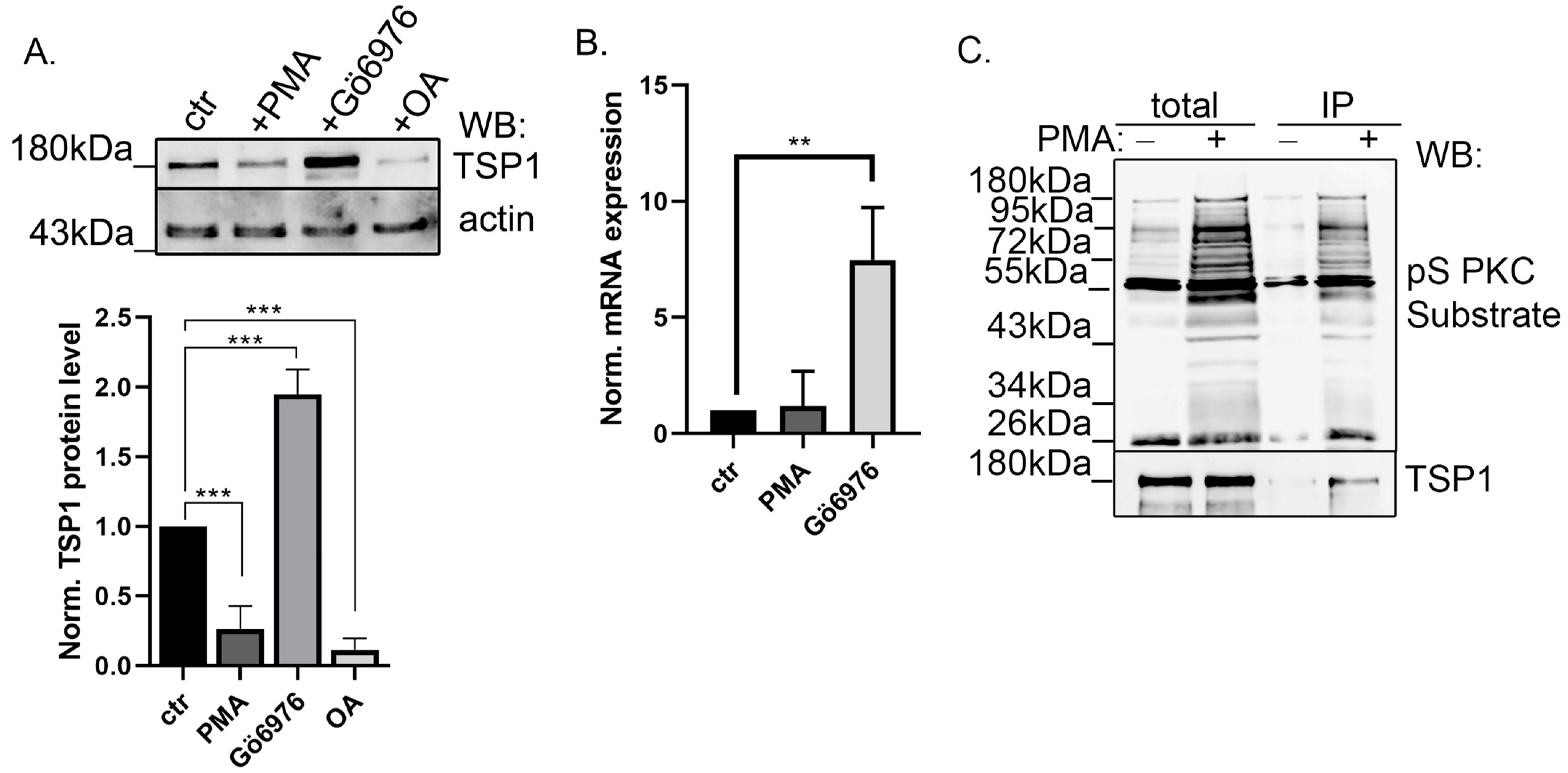
2.3. Modulation of PKC Activity Regulates TSP1
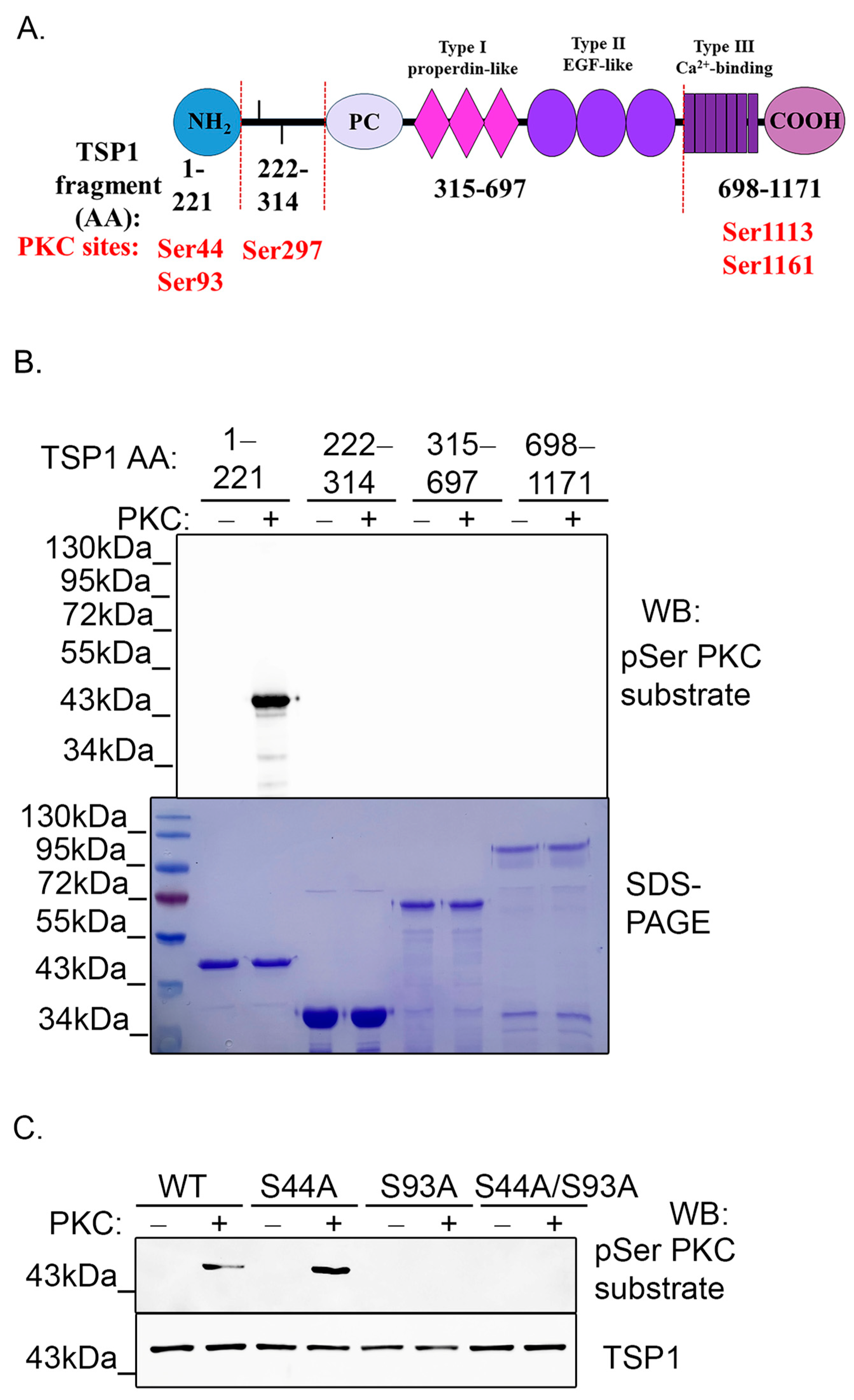
2.4. PKC Phosphorylates TSP1 on Ser93 Residue
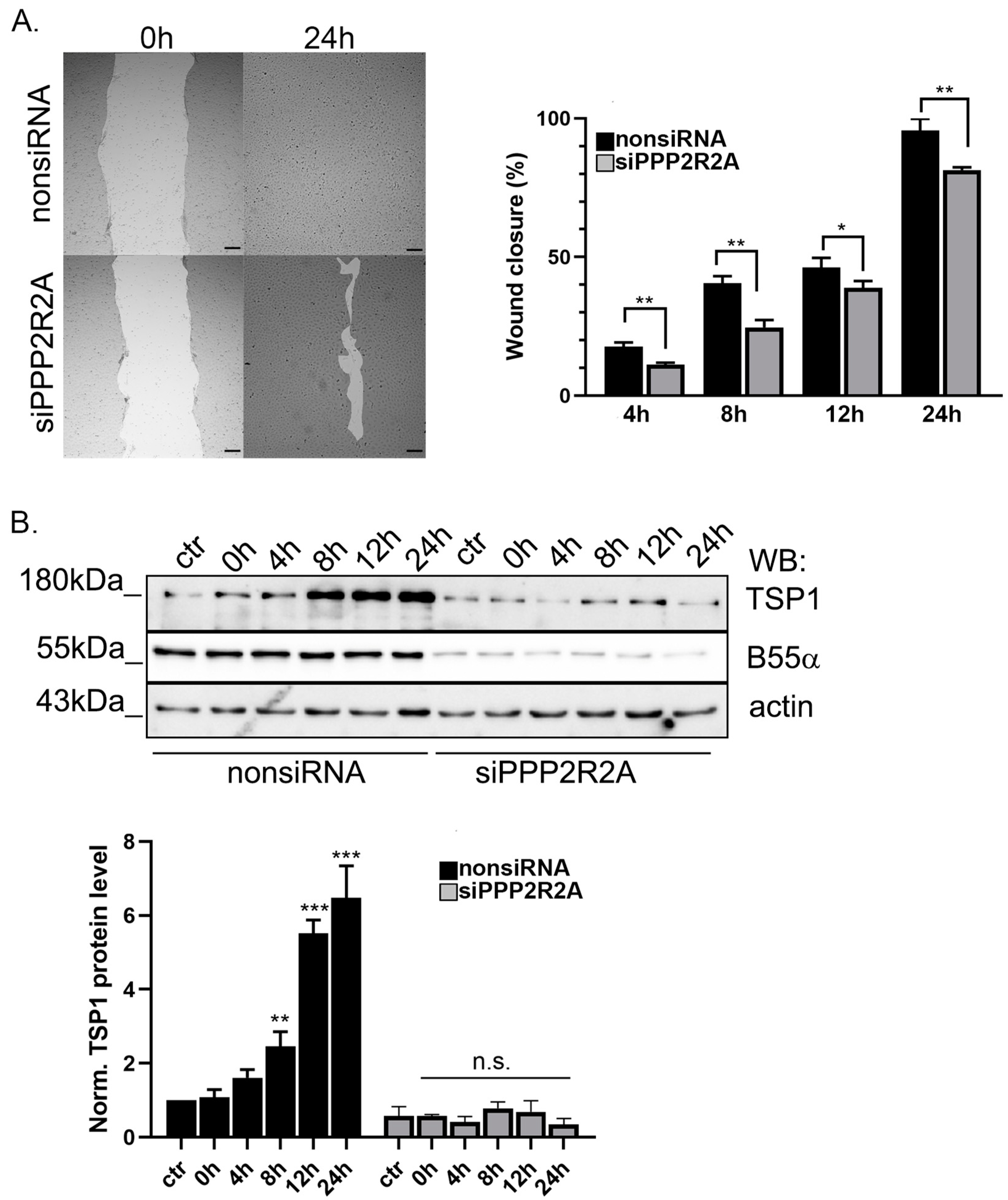
2.5. B55α Is Essential for TSP1 Upregulation in Wound Healing
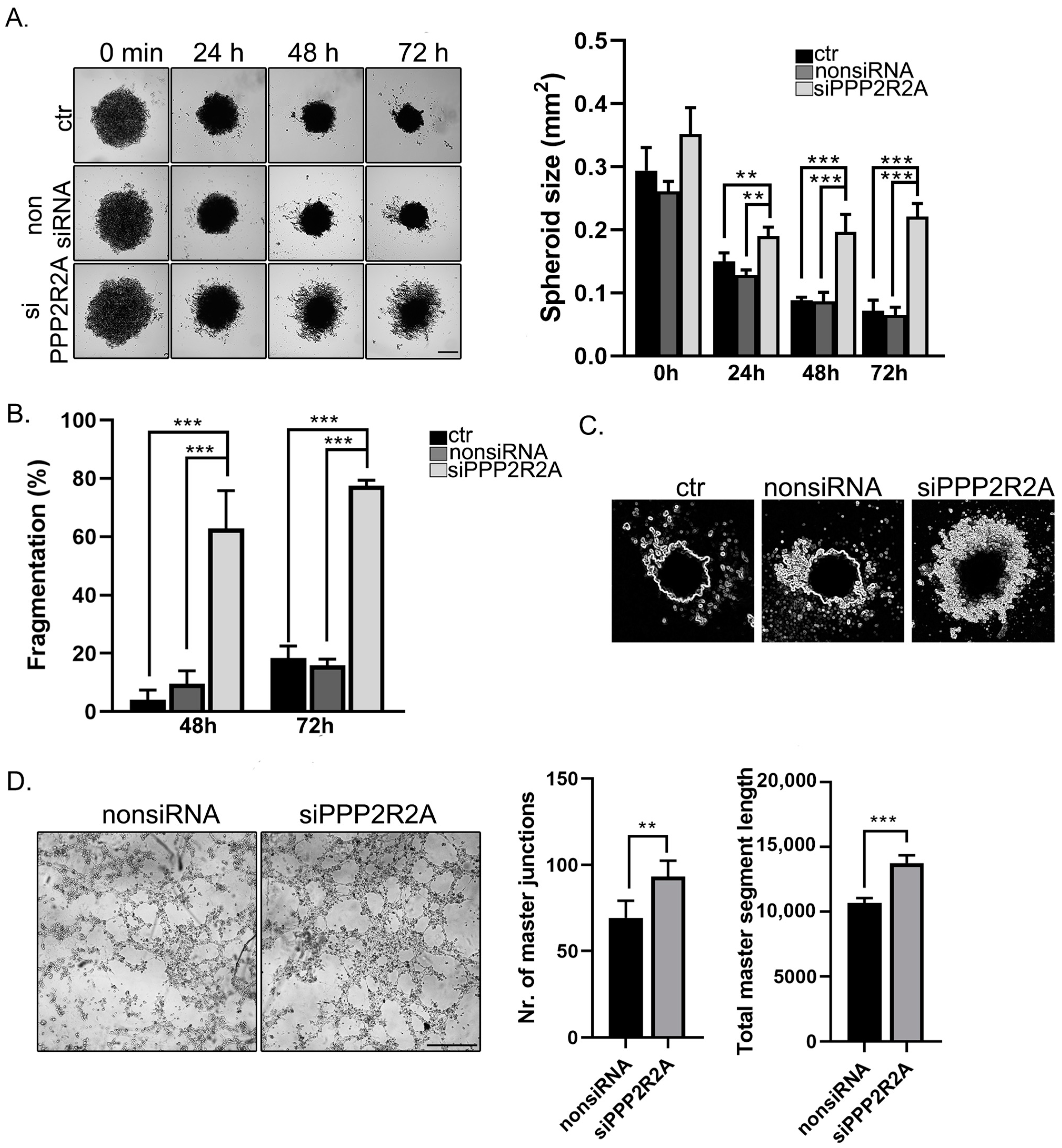
2.6. B55α Is Involved in Spheroid Stability and Tube Formation of EC
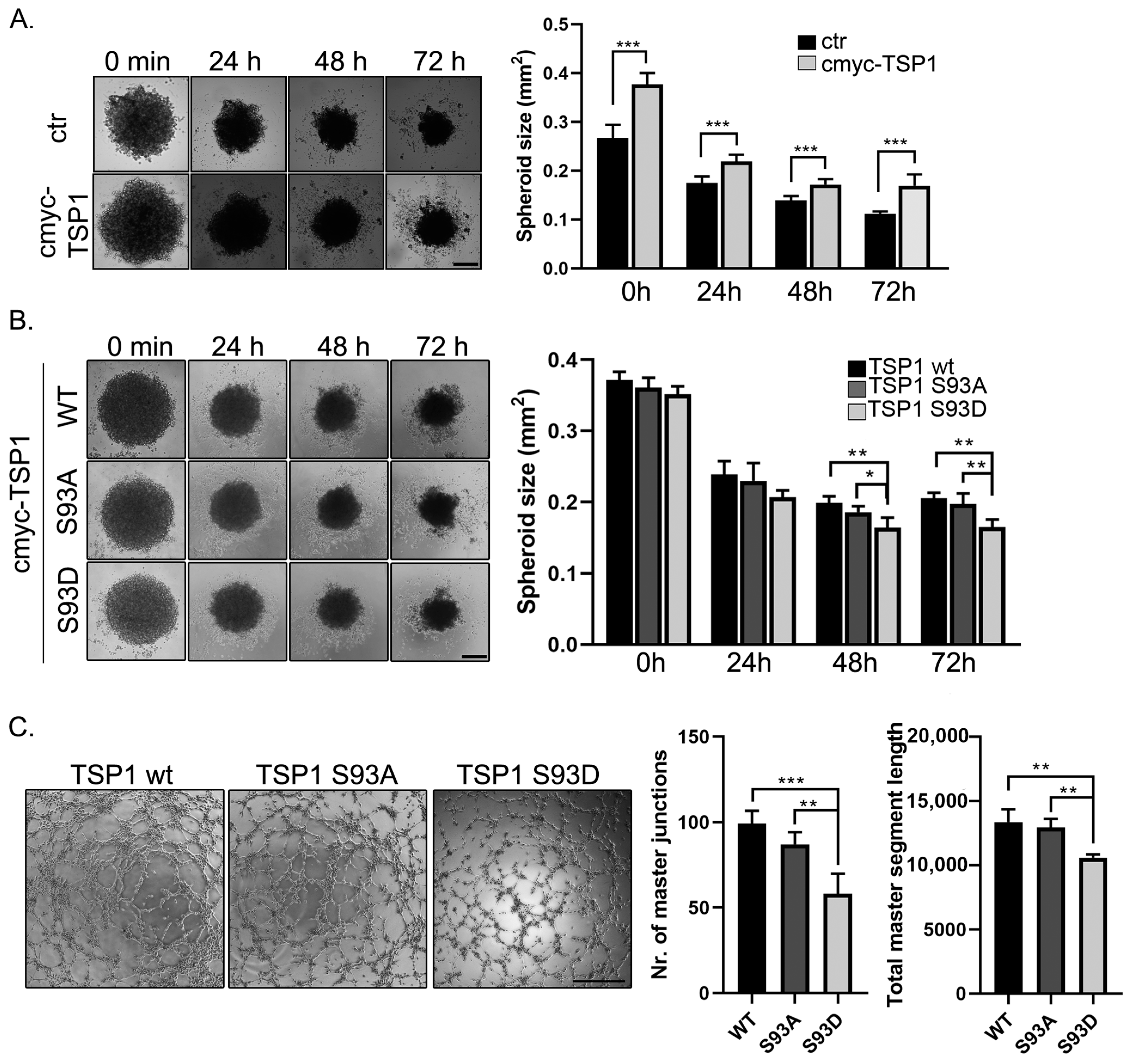
2.7. TSP1 S93D Mutants Inhibits In Vivo Angiogenesis
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatments
4.3. Gene Silencing and Transfection
4.4. RT-PCR and Quantitative Real-Time PCR (qPCR) Measurement
4.5. SDS-PAGE and Western Blotting
4.6. Immunoprecipitation, Immunofluorescence Staining, and Proximity Ligation Assay
4.7. Anti-V5 Agarose Affinity Gel
4.8. Generation of TSP1 Constructs
4.9. Bacterial Recombinant Protein Expression and GST Pull down Assay
4.10. In Vitro PKC Kinase Assay
4.11. Proteome Profiler™ Human Angiogenesis Antibody Array
4.12. Matrigel In Vitro Tube Formation Assay
4.13. Scratch Assay
4.14. Magnetic 3D Cell Culturing
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baenziger, N.L.; Brodie, G.N.; Majerus, P.W. A thrombin-sensitive protein of human platelet membranes. Proc. Natl. Acad. Sci. USA 1971, 68, 240–243. [Google Scholar] [CrossRef]
- Bornstein, P. Thrombospondins: Structure and regulation of expression. FASEB J. 1992, 6, 3290–3299. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, A.; Moura, R.; Hoylaerts, M.F. The evolving role of thrombospondin-1 in hemostasis and vascular biology. Cell Mol. Life Sci. 2008, 65, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.C. Thrombospondins: Multifunctional regulators of cell interactions. Annu. Rev. Cell Dev. Biol. 2001, 17, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.C.; Tucker, R.P. The thrombospondin type 1 repeat (TSR) superfamily: Diverse proteins with related roles in neuronal development. Dev. Dyn. 2000, 218, 280–299. [Google Scholar] [CrossRef]
- Lawler, J.W.; Slayter, H.S.; Coligan, J.E. Isolation and characterization of a high molecular weight glycoprotein from human blood platelets. J. Biol. Chem. 1978, 253, 8609–8616. [Google Scholar] [CrossRef] [PubMed]
- McPherson, J.; Sage, H.; Bornstein, P. Isolation and characterization of a glycoprotein secreted by aortic endothelial cells in culture. Apparent identity with platelet thrombospondin. J. Biol. Chem. 1981, 256, 11330–11336. [Google Scholar] [CrossRef] [PubMed]
- Mosher, D.F.; Doyle, M.J.; Jaffe, E.A. Synthesis and secretion of thrombospondin by cultured human endothelial cells. J. Cell Biol. 1982, 93, 343–348. [Google Scholar] [CrossRef]
- Raugi, G.J.; Mumby, S.M.; Abbott-Brown, D.; Bornstein, P. Thrombospondin: Synthesis and secretion by cells in culture. J. Cell Biol. 1982, 95, 351–354. [Google Scholar] [CrossRef]
- Jaffe, E.A.; Ruggiero, J.T.; Leung, L.K.; Doyle, M.J.; McKeown-Longo, P.J.; Mosher, D.F. Cultured human fibroblasts synthesize and secrete thrombospondin and incorporate it into extracellular matrix. Proc. Natl. Acad. Sci. USA 1983, 80, 998–1002. [Google Scholar] [CrossRef]
- Sage, H.; Farin, F.M.; Striker, G.E.; Fisher, A.B. Granular pneumocytes in primary culture secrete several major components of the extracellular matrix. Biochemistry 1983, 22, 2148–2155. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.A.; Ruggiero, J.T.; Falcone, D.J. Monocytes and macrophages synthesize and secrete thrombospondin. Blood 1985, 65, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Mosher, D.F. Physiology of thrombospondin. Annu. Rev. Med. 1990, 41, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Varani, J.; O'Rourke, K.; Dixit, V.M. Thrombospondin binding by human squamous carcinoma and melanoma cells: Relationship to biological activity. Exp. Cell Res. 1988, 174, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Riser, B.L.; Hughes, L.A.; Carey, T.E.; Fligiel, S.E.; Dixit, V.M. Characterization of thrombospondin synthesis, secretion and cell surface expression by human tumor cells. Clin. Exp. Metastasis 1989, 7, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Resovi, A.; Pinessi, D.; Chiorino, G.; Taraboletti, G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014, 37, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Hellewell, A.L.; Adams, J.C. Insider trading: Extracellular matrix proteins and their non-canonical intracellular roles. Bioessays 2016, 38, 77–88. [Google Scholar] [CrossRef]
- Baek, K.H.; Bhang, D.; Zaslavsky, A.; Wang, L.C.; Vachani, A.; Kim, C.F.; Albelda, S.M.; Evan, G.I.; Ryeom, S. Thrombospondin-1 mediates oncogenic Ras-induced senescence in premalignant lung tumors. J. Clin. Investig. 2013, 123, 4375–4389. [Google Scholar] [CrossRef]
- Roberts, D.D. Regulation of tumor growth and metastasis by thrombospondin-1. FASEB J. 1996, 10, 1183–1191. [Google Scholar] [CrossRef]
- Yabkowitz, R.; Dixit, V.M.; Guo, N.; Roberts, D.D.; Shimizu, Y. Activated T-cell adhesion to thrombospondin is mediated by the alpha 4 beta 1 (VLA-4) and alpha 5 beta 1 (VLA-5) integrins. J. Immunol. 1993, 151, 149–158. [Google Scholar] [CrossRef]
- Barazi, H.O.; Li, Z.; Cashel, J.A.; Krutzsch, H.C.; Annis, D.S.; Mosher, D.F.; Roberts, D.D. Regulation of integrin function by CD47 ligands. Differential effects on alpha vbeta 3 and alpha 4beta1 integrin-mediated adhesion. J. Biol. Chem. 2002, 277, 42859–42866. [Google Scholar] [CrossRef]
- Tan, K.; Duquette, M.; Liu, J.H.; Dong, Y.; Zhang, R.; Joachimiak, A.; Lawler, J.; Wang, J.H. Crystal structure of the TSP-1 type 1 repeats: A novel layered fold and its biological implication. J. Cell Biol. 2002, 159, 373–382. [Google Scholar] [CrossRef]
- Bull, H.A.; Brickell, P.M.; Dowd, P.M. Src-related protein tyrosine kinases are physically associated with the surface antigen CD36 in human dermal microvascular endothelial cells. FEBS Lett. 1994, 351, 41–44. [Google Scholar] [CrossRef]
- Gao, A.G.; Lindberg, F.P.; Dimitry, J.M.; Brown, E.J.; Frazier, W.A. Thrombospondin modulates alpha v beta 3 function through integrin-associated protein. J. Cell Biol. 1996, 135, 533–544. [Google Scholar] [CrossRef]
- Good, D.J.; Polverini, P.J.; Rastinejad, F.; Le Beau, M.M.; Lemons, R.S.; Frazier, W.A.; Bouck, N.P. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc. Natl. Acad. Sci. USA 1990, 87, 6624–6628. [Google Scholar] [CrossRef]
- Lawler, P.R.; Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Herndon, M.E.; Lawler, J. The cell biology of thrombospondin-1. Matrix Biol. 2000, 19, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Dieude, M.; Bell, C.; Turgeon, J.; Beillevaire, D.; Pomerleau, L.; Yang, B.; Hamelin, K.; Qi, S.; Pallet, N.; Beland, C.; et al. The 20S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci. Transl. Med. 2015, 7, 318ra200. [Google Scholar] [CrossRef] [PubMed]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef]
- Iruela-Arispe, M.L. Regulation of thrombospondin1 by extracellular proteases. Curr. Drug Targets 2008, 9, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Csortos, C.; Kolosova, I.; Verin, A.D. Regulation of vascular endothelial cell barrier function and cytoskeleton structure by protein phosphatases of the PPP family. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L843–L854. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.M.; Garcia, J.G. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Hahn, W.C. Involvement of PP2A in viral and cellular transformation. Oncogene 2005, 24, 7746–7755. [Google Scholar] [CrossRef]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 2009, 1795, 1–15. [Google Scholar] [CrossRef]
- Westermarck, J.; Hahn, W.C. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 2008, 14, 152–160. [Google Scholar] [CrossRef]
- Tung, H.Y.; Alemany, S.; Cohen, P. The protein phosphatases involved in cellular regulation. 2. Purification, subunit structure and properties of protein phosphatases-2A0, 2A1, and 2A2 from rabbit skeletal muscle. Eur. J. Biochem. 1985, 148, 253–263. [Google Scholar] [CrossRef]
- Alberts, A.S.; Thorburn, A.M.; Shenolikar, S.; Mumby, M.C.; Feramisco, J.R. Regulation of cell cycle progression and nuclear affinity of the retinoblastoma protein by protein phosphatases. Proc. Natl. Acad. Sci. USA 1993, 90, 388–392. [Google Scholar] [CrossRef]
- Glenn, G.M.; Eckhart, W. Mutation of a cysteine residue in polyomavirus middle T antigen abolishes interactions with protein phosphatase 2A, pp60c-src, and phosphatidylinositol-3 kinase, activation of c-fos expression, and cellular transformation. J. Virol. 1993, 67, 1945–1952. [Google Scholar] [CrossRef]
- Ronne, H.; Carlberg, M.; Hu, G.Z.; Nehlin, J.O. Protein phosphatase 2A in Saccharomyces cerevisiae: Effects on cell growth and bud morphogenesis. Mol. Cell Biol. 1991, 11, 4876–4884. [Google Scholar] [PubMed]
- Schonthal, A.H. Role of serine/threonine protein phosphatase 2A in cancer. Cancer Lett. 2001, 170, 1–13. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J.; Van Hoof, C. PP2A: The expected tumor suppressor. Curr. Opin. Genet. Dev. 2005, 15, 34–41. [Google Scholar] [CrossRef]
- Kasa, A.; Czikora, I.; Verin, A.D.; Gergely, P.; Csortos, C. Protein phosphatase 2A activity is required for functional adherent junctions in endothelial cells. Microvasc. Res. 2013, 89, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Tar, K.; Birukova, A.A.; Csortos, C.; Bako, E.; Garcia, J.G.; Verin, A.D. Phosphatase 2A is involved in endothelial cell microtubule remodeling and barrier regulation. J. Cell. Biochem. 2004, 92, 534–546. [Google Scholar] [CrossRef]
- Tar, K.; Csortos, C.; Czikora, I.; Olah, G.; Ma, S.F.; Wadgaonkar, R.; Gergely, P.; Garcia, J.G.; Verin, A.D. Role of protein phosphatase 2A in the regulation of endothelial cell cytoskeleton structure. J. Cell. Biochem. 2006, 98, 931–953. [Google Scholar] [CrossRef]
- Thalwieser, Z.; Kiraly, N.; Fonodi, M.; Csortos, C.; Boratko, A. Protein phosphatase 2A-mediated flotillin-1 dephosphorylation up-regulates endothelial cell migration and angiogenesis regulation. J. Biol. Chem. 2019, 294, 20196–20206. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Zhou, J.; Zhang, Y.; Shi, Y.; Peng, D.; Xue, Y. GPS 5.0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genom. Proteom. Bioinform. 2020, 18, 72–80. [Google Scholar] [CrossRef]
- Agah, A.; Kyriakides, T.R.; Lawler, J.; Bornstein, P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am. J. Pathol. 2002, 161, 831–839. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Huang, K.Y.; Yang, C.H.; Yang, Y.S.; Lee, W.Y.; Chiang, C.W. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J. Biol. Chem. 2008, 283, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhang, Q.; Ding, J.; Yu, M.; Jiang, J.; Yang, F.; Wang, S.; Wang, A.; Wang, L.; Wu, S.; et al. Roles of I(2)(PP2A) in the downregulation of eNOS Ser1177 phosphorylation by angiotensin II-activated PP2A. Biochem. Biophys. Res. Commun. 2019, 516, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Greif, D.M.; Kou, R.; Michel, T. Site-specific dephosphorylation of endothelial nitric oxide synthase by protein phosphatase 2A: Evidence for crosstalk between phosphorylation sites. Biochemistry 2002, 41, 15845–15853. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Hu, J.; Wu, Z.; Cafarello, S.T.; Di Matteo, M.; Shen, Y.; Dong, X.; Adler, H.; Mazzone, M.; Ruiz de Almodovar, C.; et al. Protein Phosphatase 2A Mediates YAP Activation in Endothelial Cells Upon VEGF Stimulation and Matrix Stiffness. Front. Cell Dev. Biol. 2021, 9, 675562. [Google Scholar] [CrossRef]
- Mazhar, S.; Taylor, S.E.; Sangodkar, J.; Narla, G. Targeting PP2A in cancer: Combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 51–63. [Google Scholar] [CrossRef]
- Liu, B.; Yang, H.; Song, Y.S.; Sorenson, C.M.; Sheibani, N. Thrombospondin-1 in vascular development, vascular function, and vascular disease. Semin. Cell Dev. Biol. 2024, 155 Pt B, 32–44. [Google Scholar] [CrossRef]
- Dawson, D.W.; Pearce, S.F.; Zhong, R.; Silverstein, R.L.; Frazier, W.A.; Bouck, N.P. CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. J. Cell Biol. 1997, 138, 707–717. [Google Scholar] [CrossRef]
- Tolsma, S.S.; Volpert, O.V.; Good, D.J.; Frazier, W.A.; Polverini, P.J.; Bouck, N. Peptides derived from two separate domains of the matrix protein thrombospondin-1 have anti-angiogenic activity. J. Cell Biol. 1993, 122, 497–511. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Ridnour, L.A.; Dimitry, J.; Frazier, W.A.; Wink, D.A.; Roberts, D.D. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J. Biol. Chem. 2006, 281, 26069–26080. [Google Scholar] [CrossRef]
- Nicosia, R.F.; Tuszynski, G.P. Matrix-bound thrombospondin promotes angiogenesis in vitro. J. Cell Biol. 1994, 124, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Wang, T.N.; Rothman, V.L.; Nicosia, R.F.; Tuszynski, G.P. Thrombospondin-1 modulates angiogenesis in vitro by up-regulation of matrix metalloproteinase-9 in endothelial cells. Exp. Cell Res. 1997, 235, 403–412. [Google Scholar] [CrossRef]
- Taraboletti, G.; Morbidelli, L.; Donnini, S.; Parenti, A.; Granger, H.J.; Giavazzi, R.; Ziche, M. The heparin binding 25 kDa fragment of thrombospondin-1 promotes angiogenesis and modulates gelatinase and TIMP-2 production in endothelial cells. FASEB J. 2000, 14, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Forbes, T.; Pauza, A.G.; Adams, J.C. In the balance: How do thrombospondins contribute to the cellular pathophysiology of cardiovascular disease? Am. J. Physiol. Cell Physiol. 2021, 321, C826–C845. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.R.; Liu, Y.; Mosher, D.F. Modification of EGF-like module 1 of thrombospondin-1, an animal extracellular protein, by O-linked N-acetylglucosamine. PLoS ONE 2012, 7, e32762. [Google Scholar] [CrossRef] [PubMed]
- Hofsteenge, J.; Huwiler, K.G.; Macek, B.; Hess, D.; Lawler, J.; Mosher, D.F.; Peter-Katalinic, J. C-mannosylation and O-fucosylation of the thrombospondin type 1 module. J. Biol. Chem. 2001, 276, 6485–6498. [Google Scholar] [CrossRef] [PubMed]
- Ivovic, D.; Kabelikova, P.; Jurkovicova, D. Unraveling the complexity: A comprehensive analysis of the PP2A in cancer and its potential for novel targeted therapies. Neoplasma 2023, 70, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2022, 118, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Rege, T.A.; Stewart, J., Jr.; Dranka, B.; Benveniste, E.N.; Silverstein, R.L.; Gladson, C.L. Thrombospondin-1-induced apoptosis of brain microvascular endothelial cells can be mediated by TNF-R1. J. Cell Physiol. 2009, 218, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Martin-Manso, G.; Maxhimer, J.B.; Roberts, D.D. Regulation of nitric oxide signalling by thrombospondin 1: Implications for anti-angiogenic therapies. Nat. Rev. Cancer 2009, 9, 182–194. [Google Scholar] [CrossRef]
- Swerlick, R.A.; Lee, K.H.; Wick, T.M.; Lawley, T.J. Human dermal microvascular endothelial but not human umbilical vein endothelial cells express CD36 in vivo and in vitro. J. Immunol. 1992, 148, 78–83. [Google Scholar] [CrossRef]
- Lee, N.V.; Sato, M.; Annis, D.S.; Loo, J.A.; Wu, L.; Mosher, D.F.; Iruela-Arispe, M.L. ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. EMBO J. 2006, 25, 5270–5283. [Google Scholar] [CrossRef] [PubMed]
- Schultz-Cherry, S.; Murphy-Ullrich, J.E. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J. Cell Biol. 1993, 122, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.E.; Ingram, J.A.; Norton, H.J.; Hanley, E.N., Jr. Senescence in cells of the aging and degenerating intervertebral disc: Immunolocalization of senescence-associated beta-galactosidase in human and sand rat discs. Spine 2007, 32, 321–327. [Google Scholar] [CrossRef] [PubMed]
- DiPietro, L.A.; Nissen, N.N.; Gamelli, R.L.; Koch, A.E.; Pyle, J.M.; Polverini, P.J. Thrombospondin 1 synthesis and function in wound repair. Am. J. Pathol. 1996, 148, 1851–1860. [Google Scholar]
- Passaniti, A.; Kleinman, H.K.; Martin, G.R. Matrigel: History/background, uses, and future applications. J. Cell Commun. Signal. 2022, 16, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Bao, X.; Yu, J.; Chen, W.; Wang, L.; Zhang, Z.; Xu, Q. Disruption and inactivation of the PP2A complex promotes the proliferation and angiogenesis of hemangioma endothelial cells through activating AKT and ERK. Oncotarget 2015, 6, 25660–25676. [Google Scholar] [CrossRef]
- Martin, M.; Geudens, I.; Bruyr, J.; Potente, M.; Bleuart, A.; Lebrun, M.; Simonis, N.; Deroanne, C.; Twizere, J.C.; Soubeyran, P.; et al. PP2A regulatory subunit Balpha controls endothelial contractility and vessel lumen integrity via regulation of HDAC7. EMBO J. 2013, 32, 2491–2503. [Google Scholar] [CrossRef]
- Vailhe, B.; Feige, J.J. Thrombospondins as anti-angiogenic therapeutic agents. Curr. Pharm. Des. 2003, 9, 583–588. [Google Scholar] [CrossRef]
- Anderson, J.C.; Grammer, J.R.; Wang, W.; Nabors, L.B.; Henkin, J.; Stewart, J.E., Jr.; Gladson, C.L. ABT-510, a modified type 1 repeat peptide of thrombospondin, inhibits malignant glioma growth in vivo by inhibiting angiogenesis. Cancer Biol. Ther. 2007, 6, 454–462. [Google Scholar] [CrossRef]
- Luo, P.; Zhou, K.; Li, G.; Tao, T.; Tao, J.; Han, R.P.S.; Tu, Y. Preclinical Evaluation of CD36-Targeting Antiangiogenic Peptide ABT-510 for Near-Infrared Fluorescence Molecular Imaging of Colorectal Cancer. Anal. Chem. 2023, 95, 7344–7353. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lv, L.; Zhao, Y.; Yang, N. Okadaic acid inhibits cell multiplication and induces apoptosis in a549 cells, a human lung adenocarcinoma cell line. Int. J. Clin. Exp. Med. 2014, 7, 2025–2030. [Google Scholar]
- Ferron, P.J.; Hogeveen, K.; Fessard, V.; Le Hegarat, L. Comparative analysis of the cytotoxic effects of okadaic acid-group toxins on human intestinal cell lines. Mar. Drugs 2014, 12, 4616–4634. [Google Scholar] [CrossRef]
- Li, W.; Xie, L.; Chen, Z.; Zhu, Y.; Sun, Y.; Miao, Y.; Xu, Z.; Han, X. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci. 2010, 101, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Kok, S.H.; Chui, C.H.; Lam, W.S.; Chen, J.; Lau, F.Y.; Cheng, G.Y.; Wong, R.S.; Lai, P.P.; Leung, T.W.; Tang, J.C.; et al. Apoptotic activity of a novel synthetic cantharidin analogue on hepatoma cell lines. Int. J. Mol. Med. 2006, 17, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.S.; Ho, W.; Zhang, C.; Yang, C.; Elder, J.B.; Zhuang, Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 2015, 16, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Boratko, A.; Gergely, P.; Csortos, C. RACK1 is involved in endothelial barrier regulation via its two novel interacting partners. Cell Commun. Signal. 2013, 11, 2. [Google Scholar] [CrossRef]
- Wu, T.C.; Chang, C.C.; Leu, H.B.; Huang, P.H.; Lin, S.J.; Chen, J.W. Phorbol ester-induced angiogenesis of endothelial progenitor cells: The role of NADPH oxidase-mediated, redox-related matrix metalloproteinase pathways. PLoS ONE 2019, 14, e0209426. [Google Scholar] [CrossRef] [PubMed]
- Hanana, H.; Talarmin, H.; Pennec, J.P.; Droguet, M.; Morel, J.; Dorange, G. Effect of okadaic acid on cultured clam heart cells: Involvement of MAPkinase pathways. Biol. Open 2012, 1, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Kohn, E.A.; Yoo, C.J.; Eastman, A. The protein kinase C inhibitor Go6976 is a potent inhibitor of DNA damage-induced S and G2 cell cycle checkpoints. Cancer Res. 2003, 63, 31–35. [Google Scholar]
- Fonodi, M.; Thalwieser, Z.; Csortos, C.; Boratko, A. TIMAP, a Regulatory Subunit of Protein Phosphatase 1, Inhibits In Vitro Neuronal Differentiation. Int. J. Mol. Sci. 2023, 24, 17360. [Google Scholar] [CrossRef]
- Boratko, A.; Peter, M.; Thalwieser, Z.; Kovacs, E.; Csortos, C. Elongation factor-1A1 is a novel substrate of the protein phosphatase 1-TIMAP complex. Int. J. Biochem. Cell Biol. 2015, 69, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Boratko, A.; Gergely, P.; Csortos, C. Cell cycle dependent association of EBP50 with protein phosphatase 2A in endothelial cells. PLoS ONE 2012, 7, e35595. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Dilution | Vendor (cat#) |
|---|---|---|
| PP2A Bα subunit (100C1) | 1:1000 | Cell Signaling Technologies (Danvers, MA, USA) (#2290) |
| PPP2R2A (2G9) | 1:1000 | Cell Signaling Technologies (Danvers, MA, USA) (#5689) |
| PP2A A subunit | 1:1000 | Cell Signaling Technologies (Danvers, MA, USA) (#2039) |
| PP2A C subunit | 1:1000 | Cell Signaling Technologies (Danvers, MA, USA) (#2038) |
| phospho-(Ser) PKC substrate | 1:1000 | Cell Signaling Technologies (Danvers, MA, USA) (#2261) |
| thrombospondin-1 | 1:1000 | Cell Signaling Technologies (Danvers, MA, USA)(#14778) |
| thrombospondin-1 (C-8) | 1:1000 | Santa Cruz Biotechnology (Dallas, TX, USA) (sc-393504) |
| actin | 1:1000 | Sigma (St. Louis, MO, USA) (A5060) |
| V5-tag (D3H8Q) | 1:1000 | Cell Signaling Technologies (Danvers, MA, USA) (#13202) |
| V5-tag | 1:1000 | Thermo Scientific (Vantaa, Finland) (R960-25) |
| c-myc | 1:1000 | Invitrogen (Carlsbad, CA, USA) (13-2500) |
| anti-rabbit IgG HRP-linked | 1:5000 | Cell Signaling Technologies (Danvers, MA, USA) (#7074) |
| anti-mouse IgG HRP-linked | 1:5000 | Cell Signaling Technologies (Danvers, MA, USA) (#7076) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thalwieser, Z.; Fonódi, M.; Király, N.; Csortos, C.; Boratkó, A. PP2A Affects Angiogenesis via Its Interaction with a Novel Phosphorylation Site of TSP1. Int. J. Mol. Sci. 2024, 25, 1844. https://doi.org/10.3390/ijms25031844
Thalwieser Z, Fonódi M, Király N, Csortos C, Boratkó A. PP2A Affects Angiogenesis via Its Interaction with a Novel Phosphorylation Site of TSP1. International Journal of Molecular Sciences. 2024; 25(3):1844. https://doi.org/10.3390/ijms25031844
Chicago/Turabian StyleThalwieser, Zsófia, Márton Fonódi, Nikolett Király, Csilla Csortos, and Anita Boratkó. 2024. "PP2A Affects Angiogenesis via Its Interaction with a Novel Phosphorylation Site of TSP1" International Journal of Molecular Sciences 25, no. 3: 1844. https://doi.org/10.3390/ijms25031844
APA StyleThalwieser, Z., Fonódi, M., Király, N., Csortos, C., & Boratkó, A. (2024). PP2A Affects Angiogenesis via Its Interaction with a Novel Phosphorylation Site of TSP1. International Journal of Molecular Sciences, 25(3), 1844. https://doi.org/10.3390/ijms25031844