


A Multi-Omics Approach Reveals Enrichment in Metabolites Involved in the Regulation of the Glutathione Pathway in LIN28B-Dependent Cancer Cells
 ,
,  , ,
, ,  ,
, 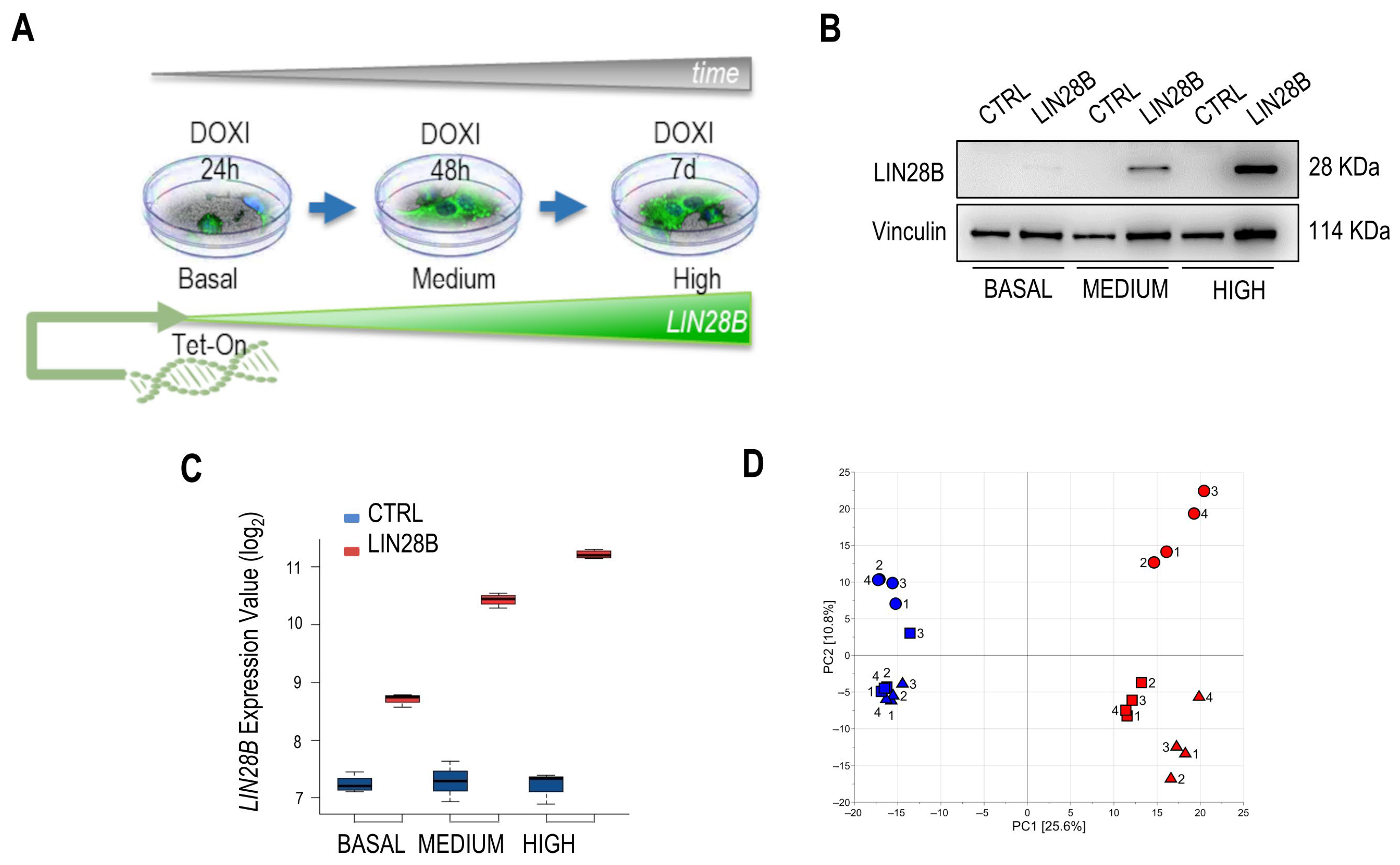{kind=link}
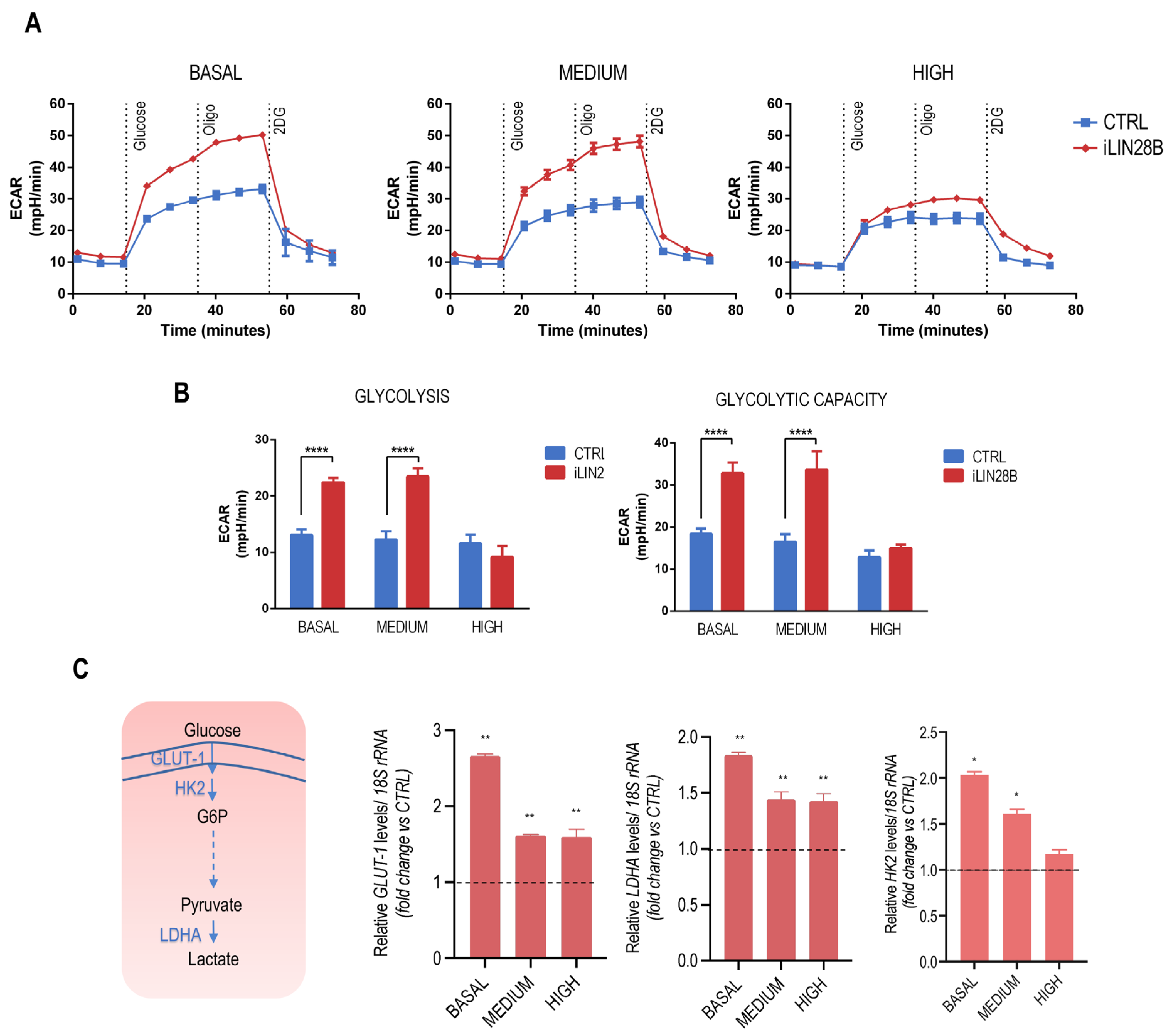{kind=link}
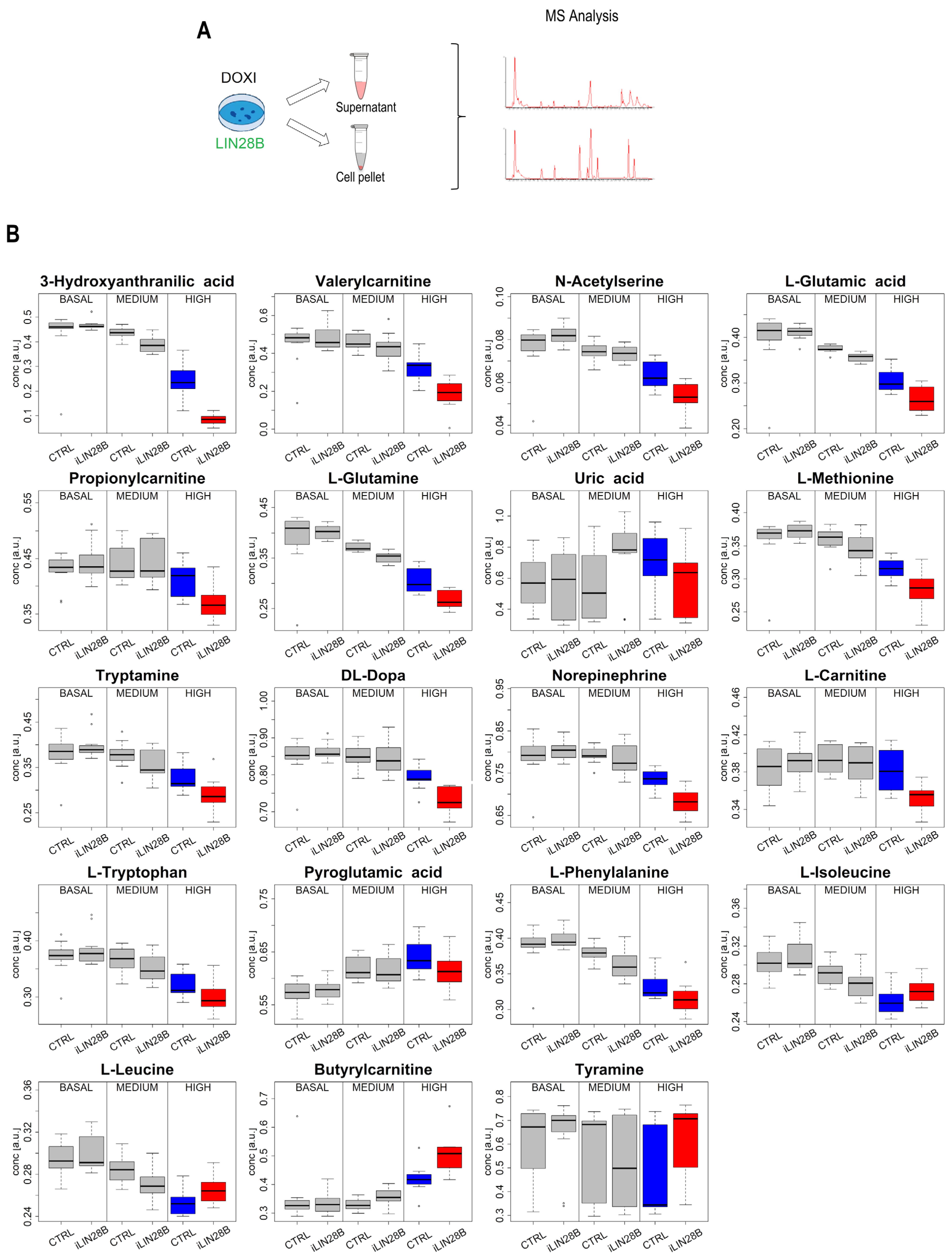{kind=link}
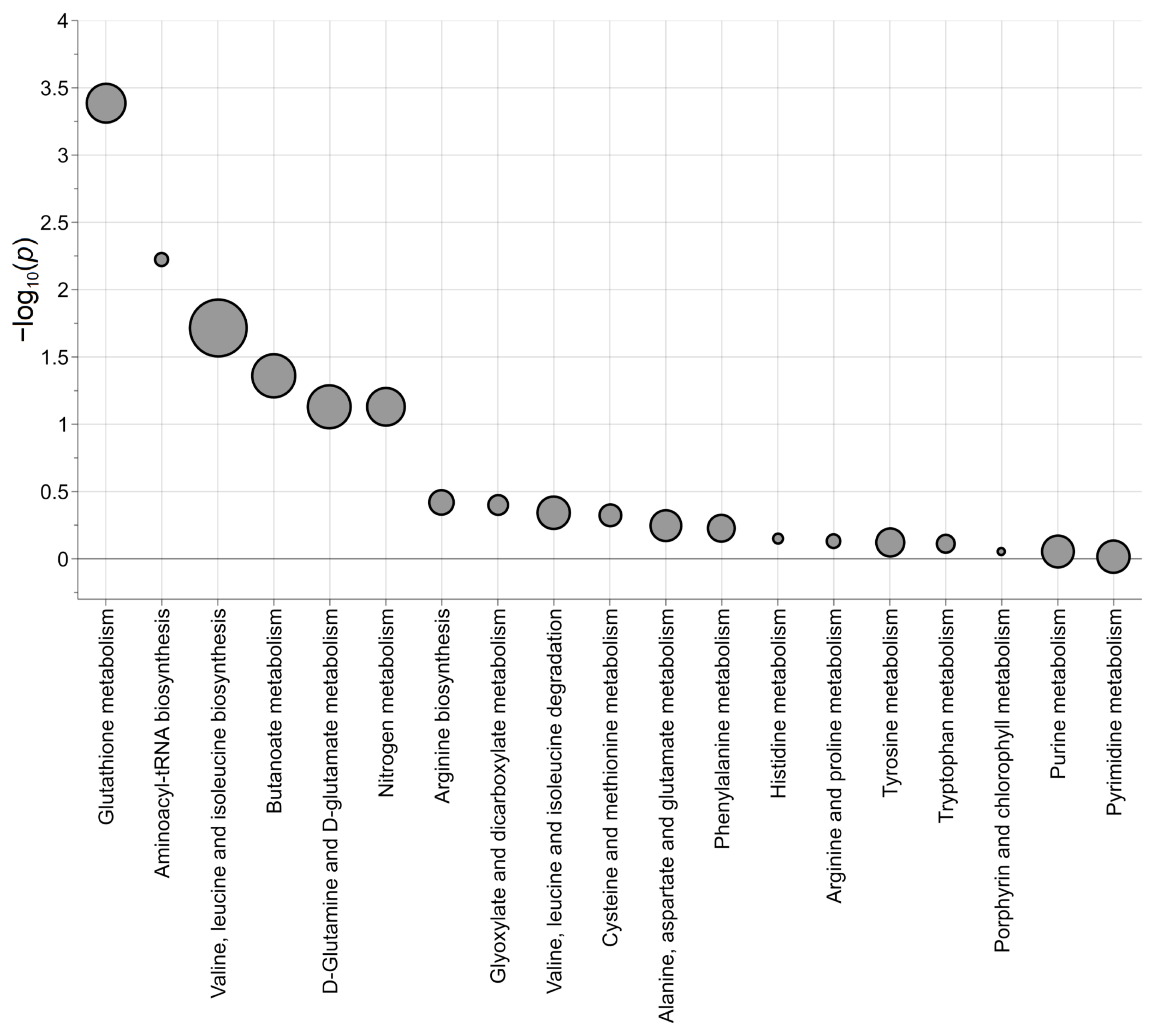{kind=link}
Abstract
1. Introduction
2. Results
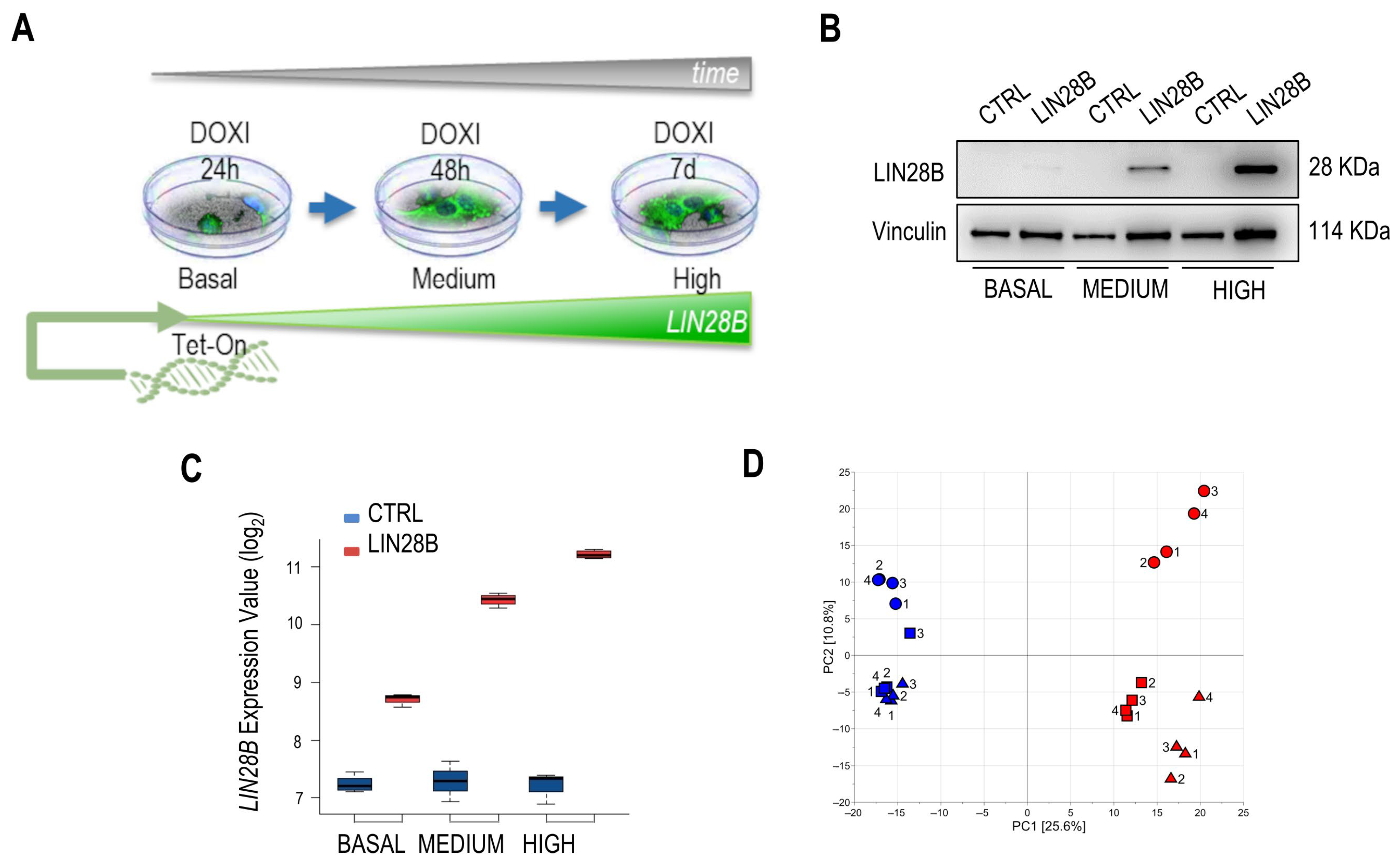
2.1. Exploratory Transcriptomics Data Analysis Highlights Potentially Relevant Transcripts for the Metabolic Diversity of iLIN28B Cells
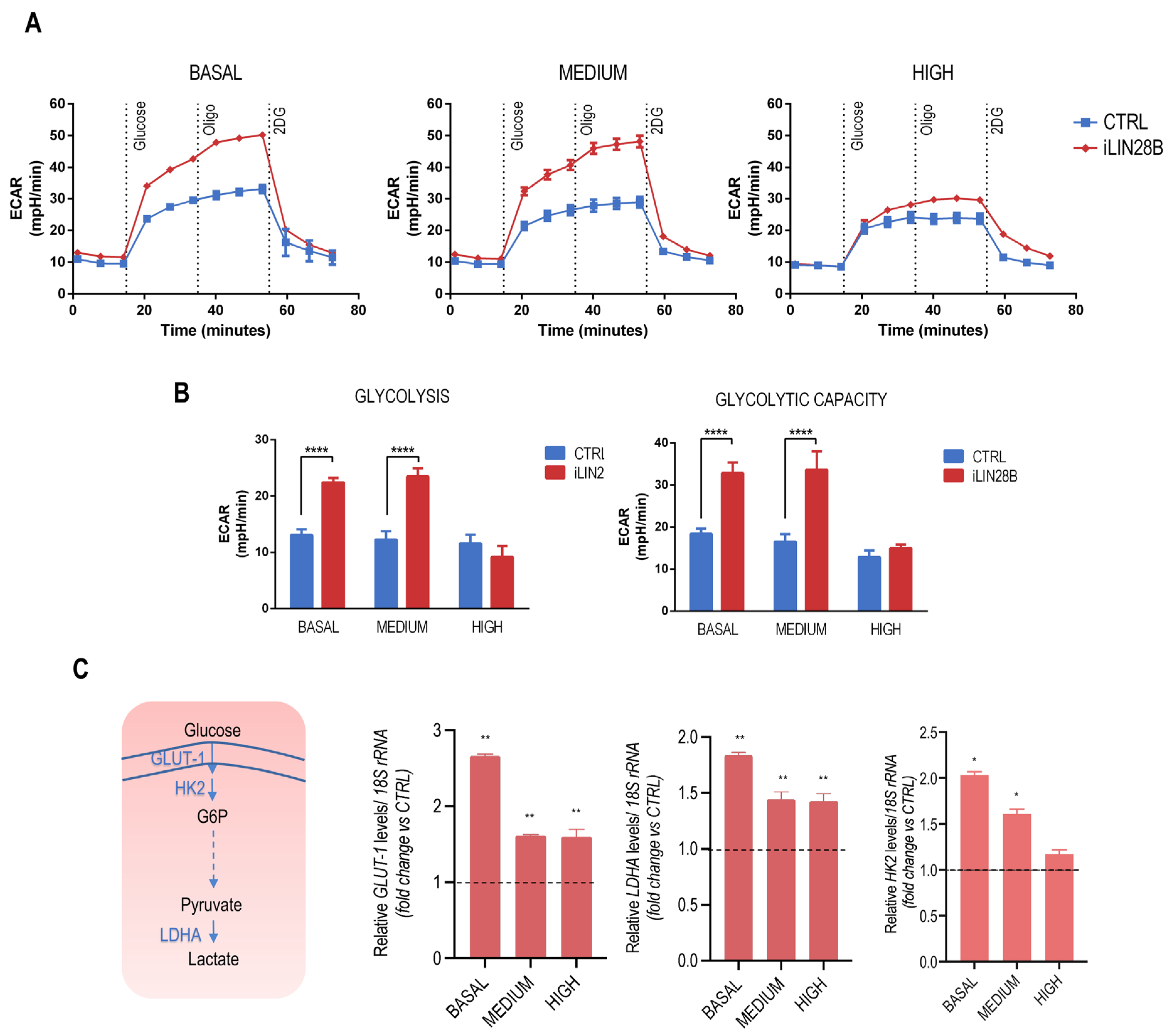
2.2. iLIN28B Triggers Glycolysis in Neuroblastoma Cells
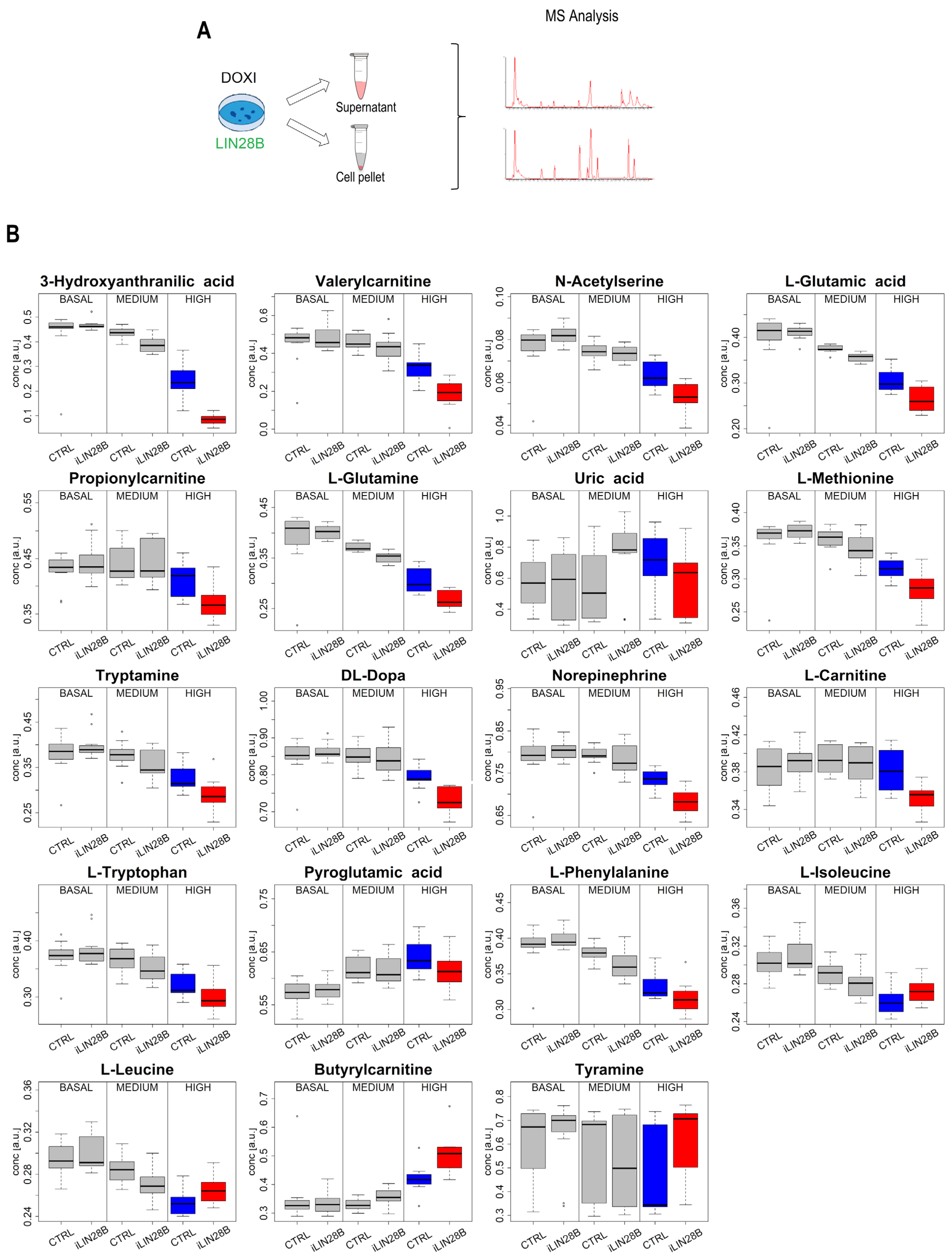
2.3. LIN28B Defines a Metabolic Rewiring in Neuroblastoma Cells
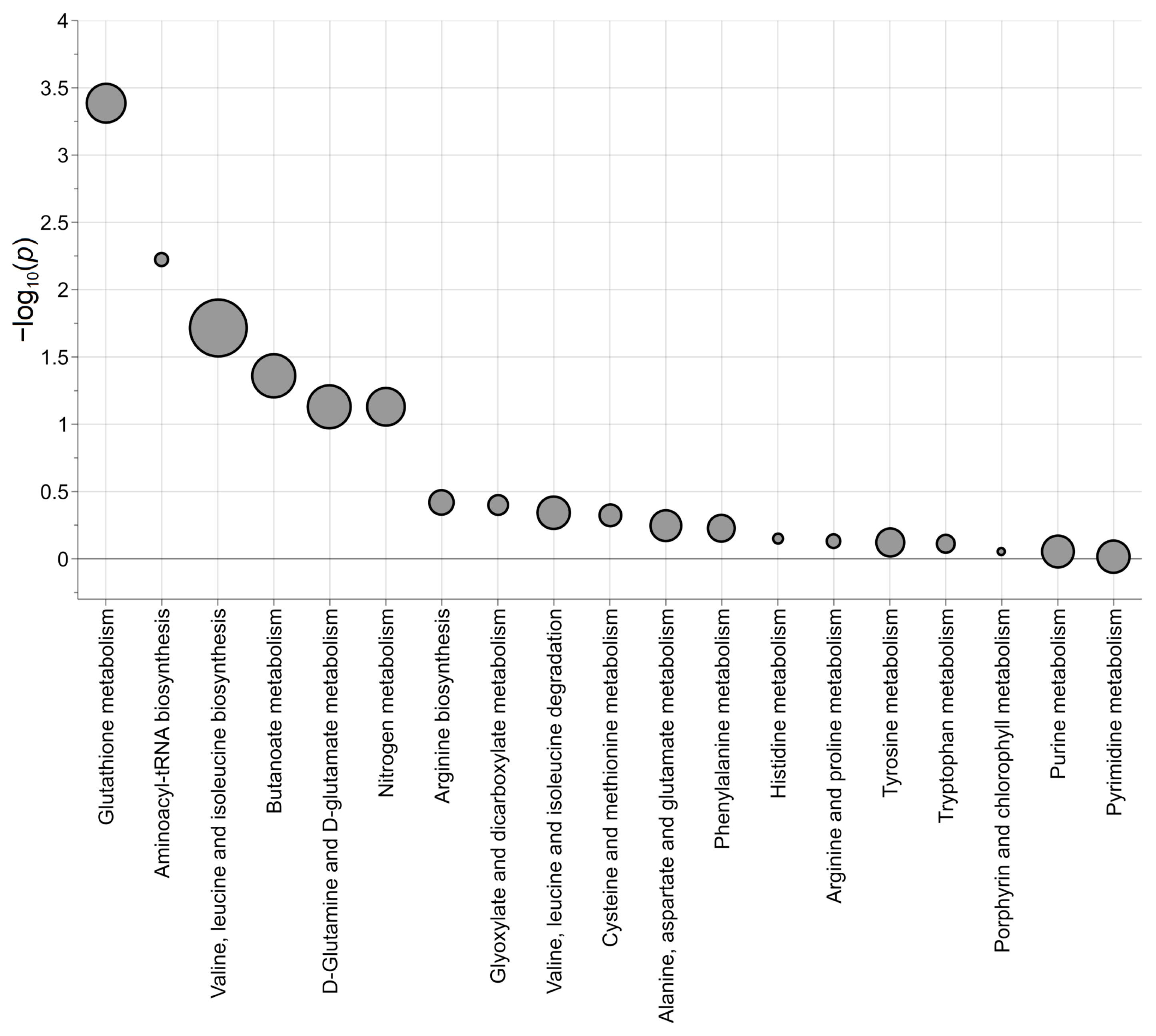
2.4. Omics Data Integration Potentiates the Discovery of LIN28B-Dependent Regulatory Pathways
3. Discussion
4. Material and Methods
4.1. Experimental Design
4.2. Glycolysis Function Assays
4.3. RNA Isolation, cDNA Synthesis, and a Real-Time Quantitative PCR (qPCR) Assay
4.4. Immunoblot Assay
4.5. Gene Expression Analysis and Data Interpretation
4.6. Metabolomics Investigation
4.6.1. Sample Preparation
4.6.2. Untargeted Metabolomics Analysis
4.6.3. Data Pre-Processing
4.6.4. Variable Annotation
4.7. Statistical Analysis of Metabolomics and Transcriptomics Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desai, A.V.; Applebaum, M.A.; Karrison, T.G.; Oppong, A.; Yuan, C.; Berg, K.R.; MacQuarrie, K.; Sokol, E.; Hall, A.G.; Pinto, N.; et al. Efficacy of post-induction therapy for high-risk neuroblastoma patients with end-induction residual disease. Cancer 2022, 128, 2967–2977. [Google Scholar] [CrossRef]
- DuBois, S.G.; Macy, M.E.; Henderson, T.O. High-Risk and Relapsed Neuroblastoma: Toward More Cures and Better Outcomes. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 768–780. [Google Scholar] [CrossRef]
- Meany, H.J. Non-high-risk neuroblastoma: Classification and achievements in therapy. Children 2019, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, B.; Beckers, A.; Lindner, S.; Kristina, A.; De Preter, K.; Depuydt, P.; Mestdagh, P.; Sante, T.; Lefever, S.; Hertwig, F.; et al. The mutational landscape of MYCN, Lin28b and ALK F1174L driven murine neuroblastoma mimics human disease. Oncotarget 2018, 9, 8334–8349. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Bao, R.; Lin, M.; Han, X.R.; Ai, Y.J.; Gao, Y.; Guan, K.L.; Xiong, Y.; Yuan, H.X. ALK fusion promotes metabolic reprogramming of cancer cells by transcriptionally upregulating PFKFB3. Oncogene 2022, 41, 4547–4559. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Bai, L.; Cao, F.; Wang, S.; He, H.; Song, M.; Chen, H.; Liu, Y.; Guo, J.; Si, Q.; et al. Targeting LIN28B reprograms tumor glucose metabolism and acidic microenvironment to suppress cancer stemness and metastasis. Oncogene 2019, 38, 4527–4539. [Google Scholar] [CrossRef]
- Tjaden, B.; Baum, K.; Marquardt, V.; Simon, M.; Trajkovic-Arsic, M.; Kouril, T.; Siebers, B.; Lisec, J.; Siveke, J.T.; Schulte, J.H.; et al. N-Myc-induced metabolic rewiring creates novel therapeutic vulnerabilities in neuroblastoma. Sci. Rep. 2020, 10, 7157. [Google Scholar] [CrossRef]
- Ruiz-Pérez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN protein in health and disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef]
- Oliynyk, G.; Ruiz-Pérez, M.V.; Sainero-Alcolado, L.; Dzieran, J.; Zirath, H.; Gallart-Ayala, H.; Wheelock, C.; Johansson, H.J.; Nilsson, R.; Lehtiö, J.; et al. MYCN-enhanced Oxidative and Glycolytic Metabolism Reveals Vulnerabilities for Targeting Neuroblastoma. iScience 2019, 21, 188–204. [Google Scholar] [CrossRef]
- Xia, Y.; Ye, B.; Ding, J.; Yu, Y.; Alptekin, A.; Thangaraju, M.; Prasad, P.D.; Ding, Z.C.; Park, E.J.; Choi, J.H.; et al. Metabolic reprogramming by MYCN confers dependence on the serine-glycine-one-carbon biosynthetic pathway. Cancer Res. 2019, 79, 3837–3850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ratanasirintrawoot, S.; Chandrasekaran, S.; Wu, Z.; Ficarro, S.B.; Yu, C.; Ross, C.A.; Cacchiarelli, D.; Xia, Q.; Seligson, M.; et al. LIN28 Regulates Stem Cell Metabolism and Conversion to Primed Pluripotency. Cell Stem Cell 2016, 19, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Gewalt, T.; Noh, K.W.; Meder, L. The role of LIN28B in tumor progression and metastasis in solid tumor entities. Oncol. Res. 2023, 31, 101–115. [Google Scholar] [CrossRef]
- Zhu, H.; Ng, S.C.; Segr, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Tsialikas, J.; Romer-Seibert, J. LIN28: Roles and regulation in development and beyond. Development 2015, 142, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, C.; Sun, L.; Huang, D.; Li, T.; He, X.; Wu, G.; Yang, Z.; Zhong, X.; Song, L.; et al. Lin28/let-7 axis regulates aerobic glycolysis and cancer progression via PDK1. Nat. Commun. 2014, 5, 6212. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Robinton, D.A.; Seligson, M.T.; Wu, L.; Li, L.; Rakheja, D.; Comerford, S.A.; Ramezani, S.; Sun, X.; Parikh, M.S.; et al. Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer Cell 2014, 26, 248–261. [Google Scholar] [CrossRef]
- Zhou, J.; Bi, C.; Ching, Y.Q.; Chooi, J.Y.; Lu, X.; Quah, J.Y.; Toh, S.H.M.; Chan, Z.L.; Tan, T.Z.; Chong, P.S.; et al. Inhibition of LIN28B impairs leukemia cell growth and metabolism in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 138. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Domingo-Fernández, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; Van Sluis, P.; Valentijn, L.J.; Van Nes, J.; et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012, 44, 1199–1206. [Google Scholar] [CrossRef]
- Corallo, D.; Donadon, M.; Pantile, M.; Sidarovich, V.; Cocchi, S.; Ori, M.; De Sarlo, M.; Candiani, S.; Frasson, C.; Distel, M.; et al. LIN28B increases neural crest cell migration and leads to transformation of trunk sympathoadrenal precursors. Cell Death Differ. 2020, 27, 1225–1242. [Google Scholar] [CrossRef]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Celeste Simon, M. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sun, B.; Nie, A.; Yu, D.; Bian, M. Roles of Aminoacyl-tRNA Synthetases in Cancer. Front. Cell Dev. Biol. 2020, 8, 599765. [Google Scholar] [CrossRef] [PubMed]
- Mandela, P.; Ordway, G.A. The norepinephrine transporter and its regulation. J. Neurochem. 2006, 97, 310–333. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.; Bak, L.; Schousboe, A.; Iversen, P.; Sorensen, M.; Keiding, S.; Vilstrup, H.; Gjedde, A.; Ott, P.; Waagepetersen, H. The metabolic role of isoleucine in detoxification of ammonia in cultured mouse neurons and astrocytes. Neurochem. Int. 2007, 50, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Guerrouahen, B.S.; Pasquier, J.; Diboun, I.; Karoly, E.D.; Suhre, K.; Rafii, A. Metabolic signatures differentiate ovarian from colon cancer cell lines. J. Transl. Med. 2015, 13, 223. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, G.; Zhu, H.; Yang, F.; Yang, S.; Vuong, A.M.; Li, J.; Zhu, D.; Sun, Y.; Tao, W. Circulating Carnitine Levels and Breast Cancer: A Matched Retrospective Case-Control Study. Front. Oncol. 2022, 12, 891619. [Google Scholar] [CrossRef]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.B.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef]
- Čuperlović-Culf, M.; Barnett, D.A.; Culf, A.S.; Chute, I. Cell culture metabolomics: Applications and future directions. Drug Discov. Today 2010, 15, 610–621. [Google Scholar] [CrossRef]
- Liu, Y.; Hyde, A.S.; Simpson, M.A.; Barycki, J.J. Emerging Regulatory Paradigms in Glutathione Metabolism. Adv. Cancer Res. 2014, 122, 69–101. [Google Scholar]
- Floros, K.V.; Cai, J.; Jacob, S.; Kurupi, R.; Fairchild, C.K.; Shende, M.; Coon, C.M.; Powell, K.M.; Belvin, B.R.; Hu, B.; et al. MYCN -amplified neuroblastoma is addicted to iron and vulnerable to inhibition of the system xc-/glutathione axis. Cancer Res. 2021, 81, 1896–1908. [Google Scholar] [CrossRef]
- Tan, F.E.; Sathe, S.; Wheeler, E.C.; Nussbacher, J.K.; Peter, S.; Yeo, G.W. A Transcriptome-wide Translational Program Defined by LIN28B Expression Level. Mol. Cell 2019, 73, 304–313.e3. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Jeet, V.; Clements, J.A.; Gunter, J.H.; Batra, J. Emergence of MicroRNAs as Key Players in Cancer Cell Metabolism. Clin. Chem. 2019, 65, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Marengo, B.; Pulliero, A.; Izzotti, A.; Domenicotti, C. miRNA Regulation of Glutathione Homeostasis in Cancer Initiation, Progression and Therapy Resistance. MicroRNA 2019, 9, 187–197. [Google Scholar] [CrossRef]
- Aveic, S.; Pantile, M.; Seydel, A.; Esposito, M.R.; Zanon, C.; Li, G.; Tonini, G.P. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget 2016, 7, 5646–5663. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, F.; Montella, S.; Stocchero, M.; Pirillo, P.; Bozzetto, S.; Giordano, G.; Poeta, M.; Baraldi, E. Effects of pidotimod and bifidobacteria mixture on clinical symptoms and urinary metabolomic profile of children with recurrent respiratory infections: A randomized placebo-controlled trial. Pulm. Pharmacol. Ther. 2019, 58, 101818. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Jolliffe, I. Principal Component Analysis, 2nd ed.; Springer: New York, NY, USA, 2002. [Google Scholar] [CrossRef]
- Stocchero, M.; Locci, E.; D’Aloja, E.; Nioi, M.; Baraldi, E.; Giordano, G. PLS2 in metabolomics. Metabolites 2019, 9, 51. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Stocchero, M. Relevant and irrelevant predictors in PLS2. J. Chemom. 2020, 34, e3237. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; De Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stocchero, M.; Corallo, D.; Bresolin, S.; Pantile, M.; Pirillo, P.; Bortolozzi, R.; Menegazzo, S.; Boso, D.; Viola, G.; Baraldi, E.; et al. A Multi-Omics Approach Reveals Enrichment in Metabolites Involved in the Regulation of the Glutathione Pathway in LIN28B-Dependent Cancer Cells. Int. J. Mol. Sci. 2024, 25, 1602. https://doi.org/10.3390/ijms25031602
Stocchero M, Corallo D, Bresolin S, Pantile M, Pirillo P, Bortolozzi R, Menegazzo S, Boso D, Viola G, Baraldi E, et al. A Multi-Omics Approach Reveals Enrichment in Metabolites Involved in the Regulation of the Glutathione Pathway in LIN28B-Dependent Cancer Cells. International Journal of Molecular Sciences. 2024; 25(3):1602. https://doi.org/10.3390/ijms25031602
Chicago/Turabian StyleStocchero, Matteo, Diana Corallo, Silvia Bresolin, Marcella Pantile, Paola Pirillo, Roberta Bortolozzi, Sara Menegazzo, Daniele Boso, Giampietro Viola, Eugenio Baraldi, and et al. 2024. "A Multi-Omics Approach Reveals Enrichment in Metabolites Involved in the Regulation of the Glutathione Pathway in LIN28B-Dependent Cancer Cells" International Journal of Molecular Sciences 25, no. 3: 1602. https://doi.org/10.3390/ijms25031602
APA StyleStocchero, M., Corallo, D., Bresolin, S., Pantile, M., Pirillo, P., Bortolozzi, R., Menegazzo, S., Boso, D., Viola, G., Baraldi, E., Biffi, A., Giordano, G., & Aveic, S. (2024). A Multi-Omics Approach Reveals Enrichment in Metabolites Involved in the Regulation of the Glutathione Pathway in LIN28B-Dependent Cancer Cells. International Journal of Molecular Sciences, 25(3), 1602. https://doi.org/10.3390/ijms25031602