
Atrazine Desorption Mechanism from an Hydrated Calcium Montmorillonite—A DFT Molecular Dynamics Study




Abstract
1. Introduction
2. Results and Discussion
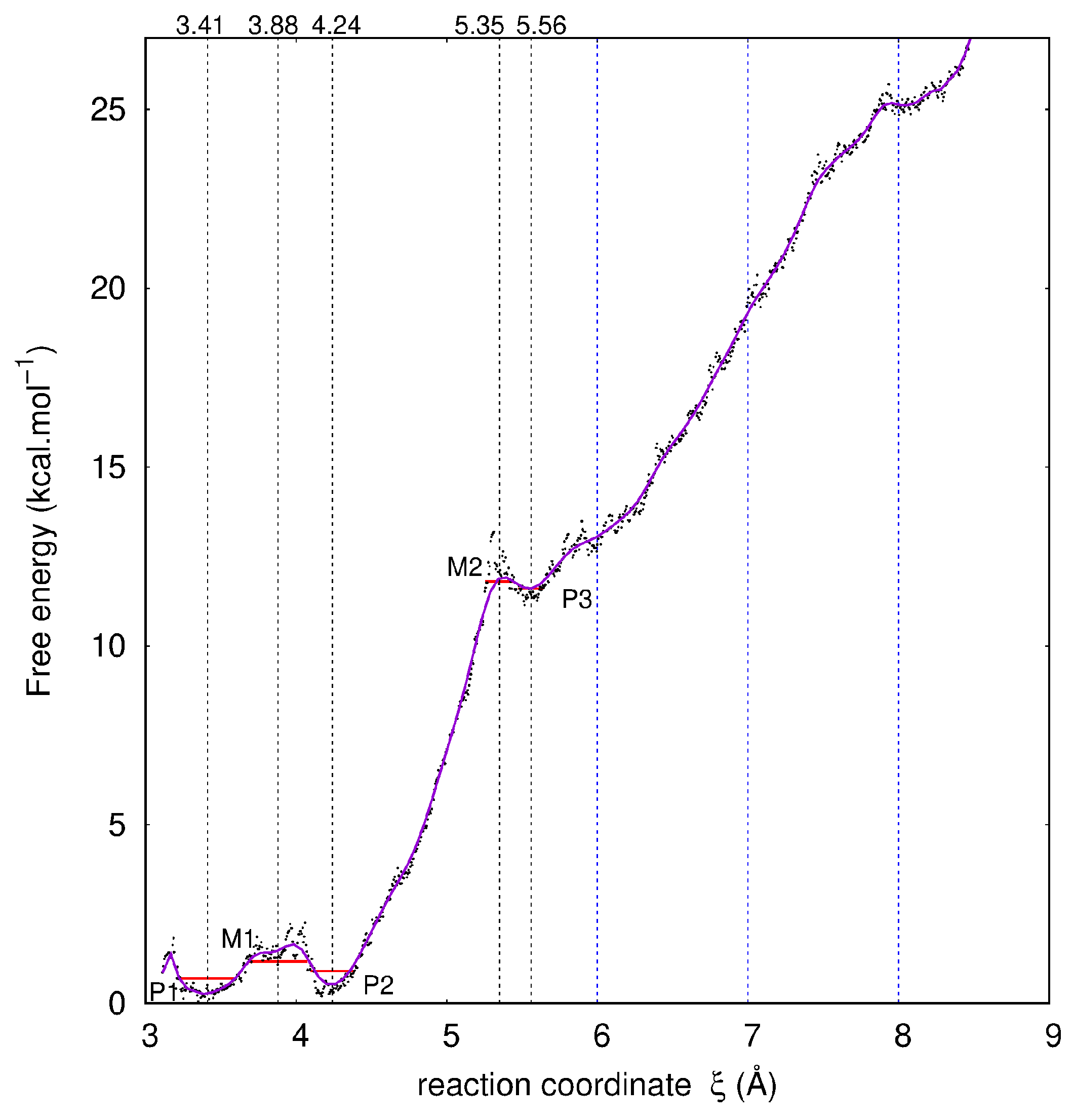
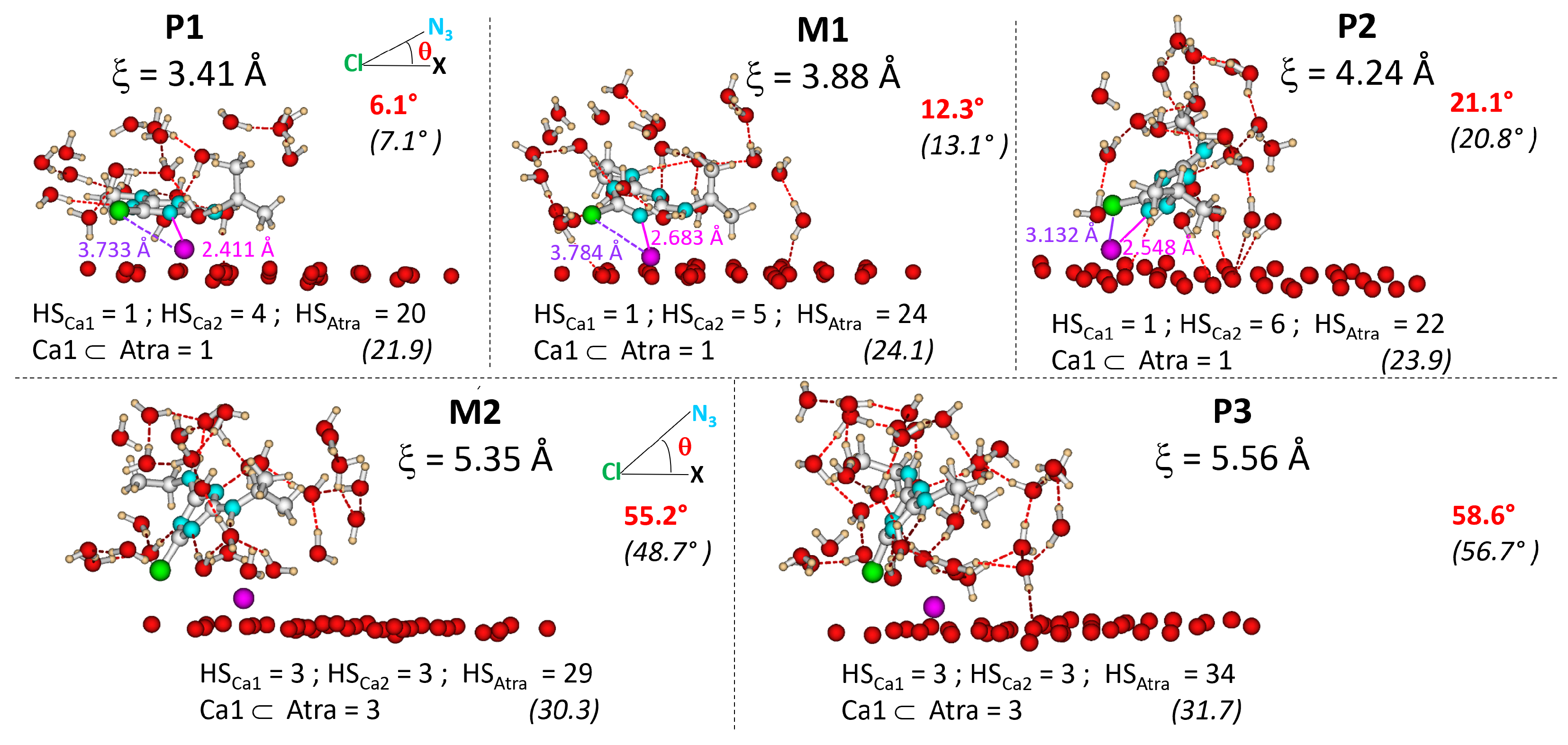
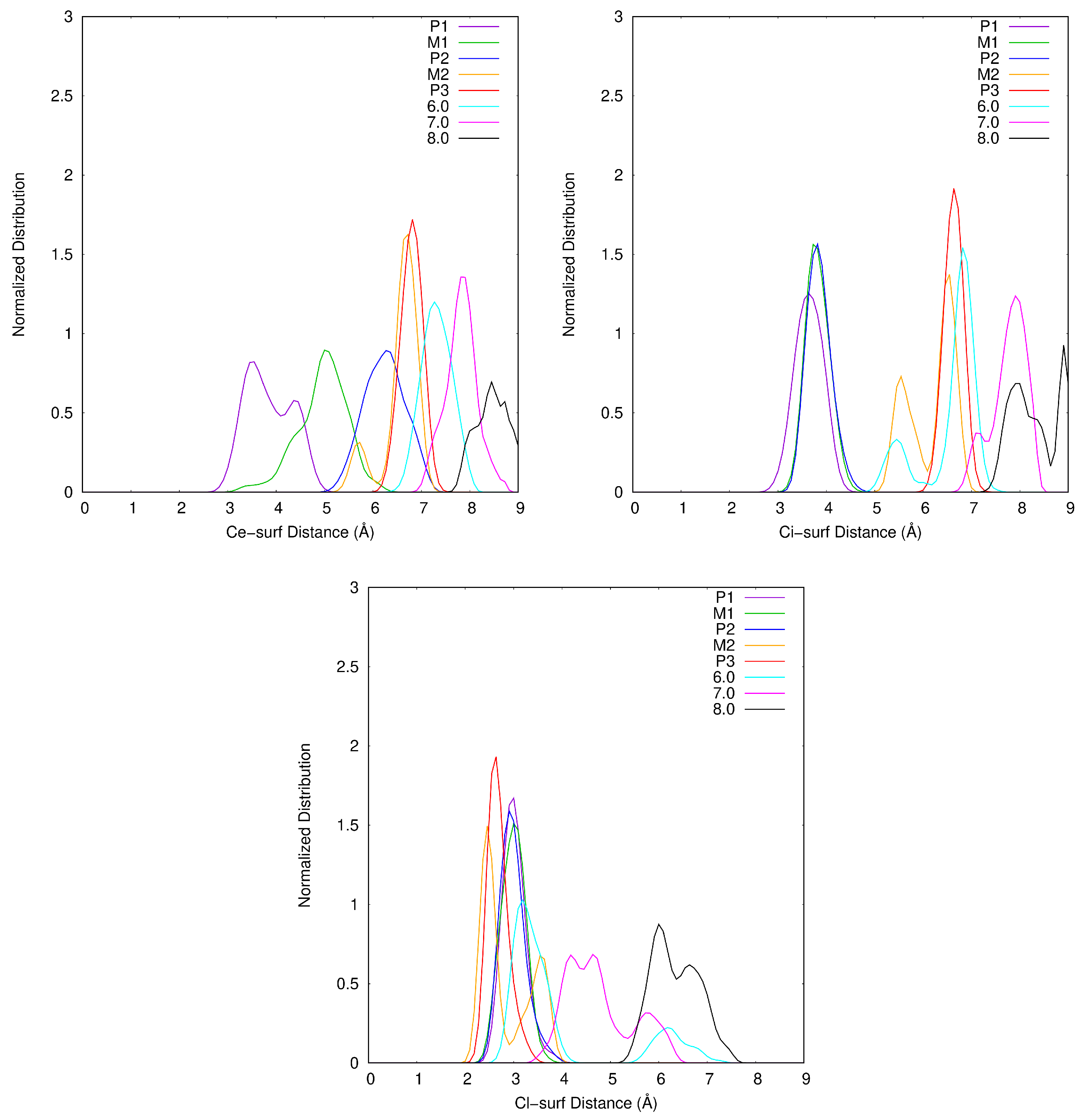
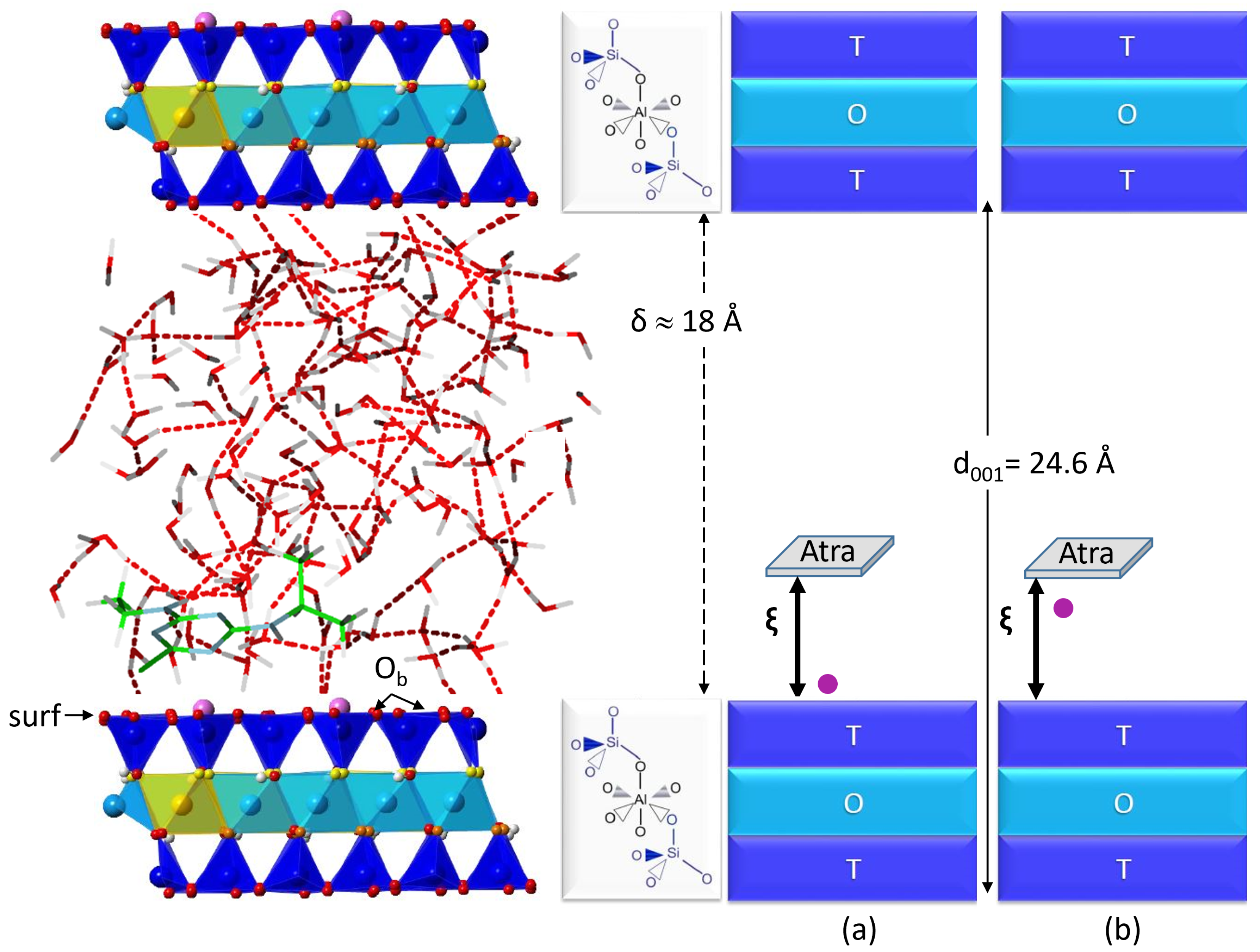
2.1. Description of the Desorption Profile
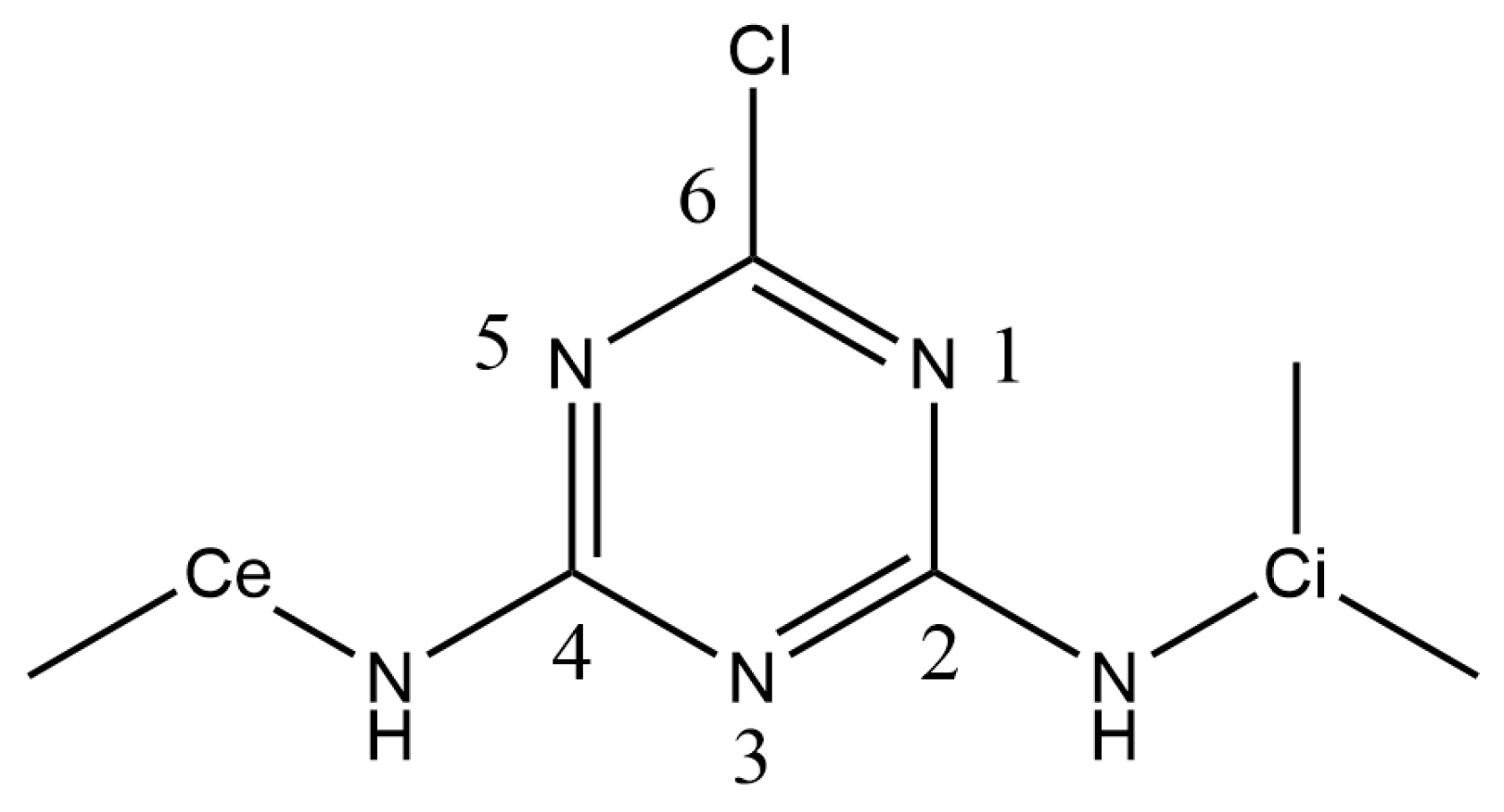
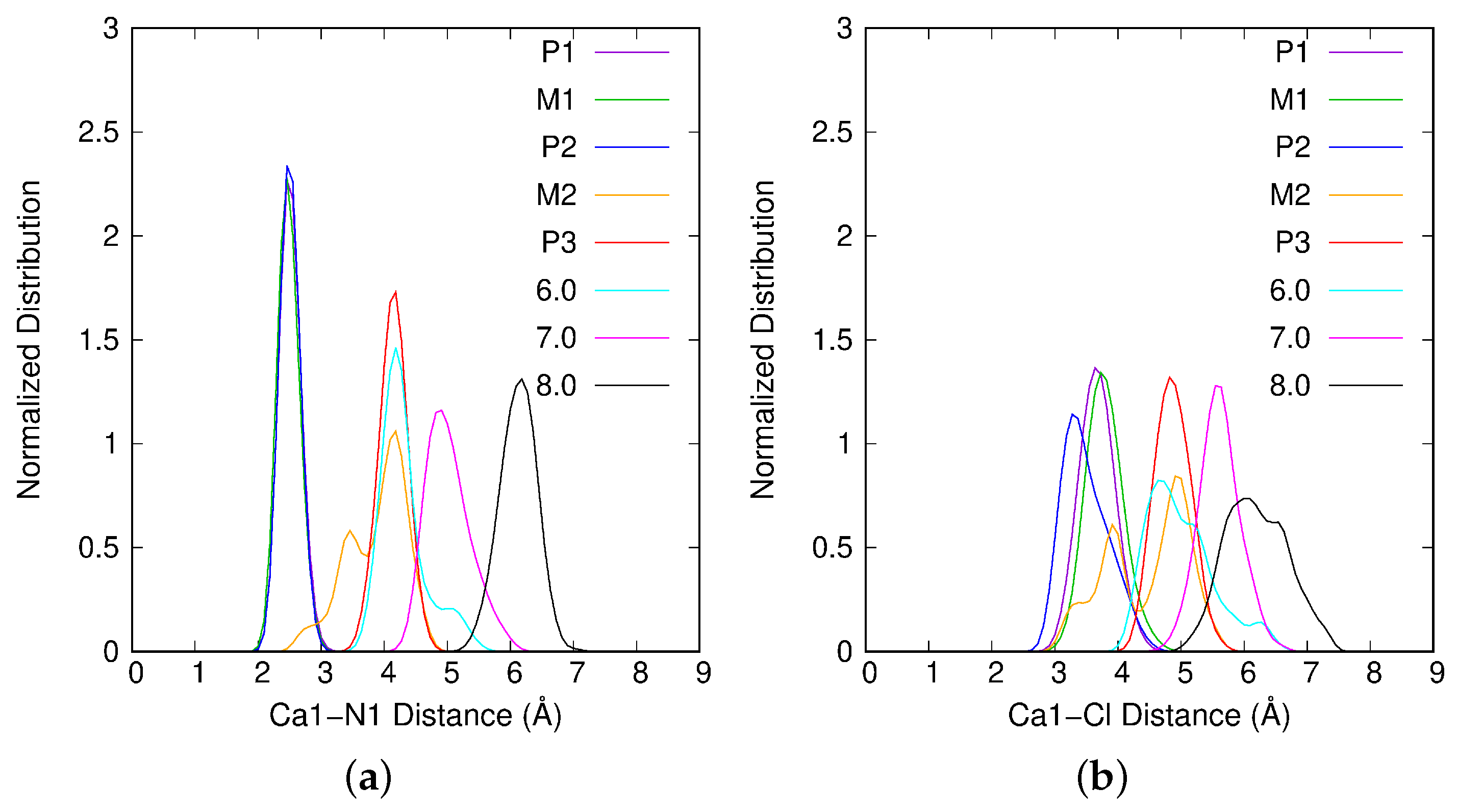
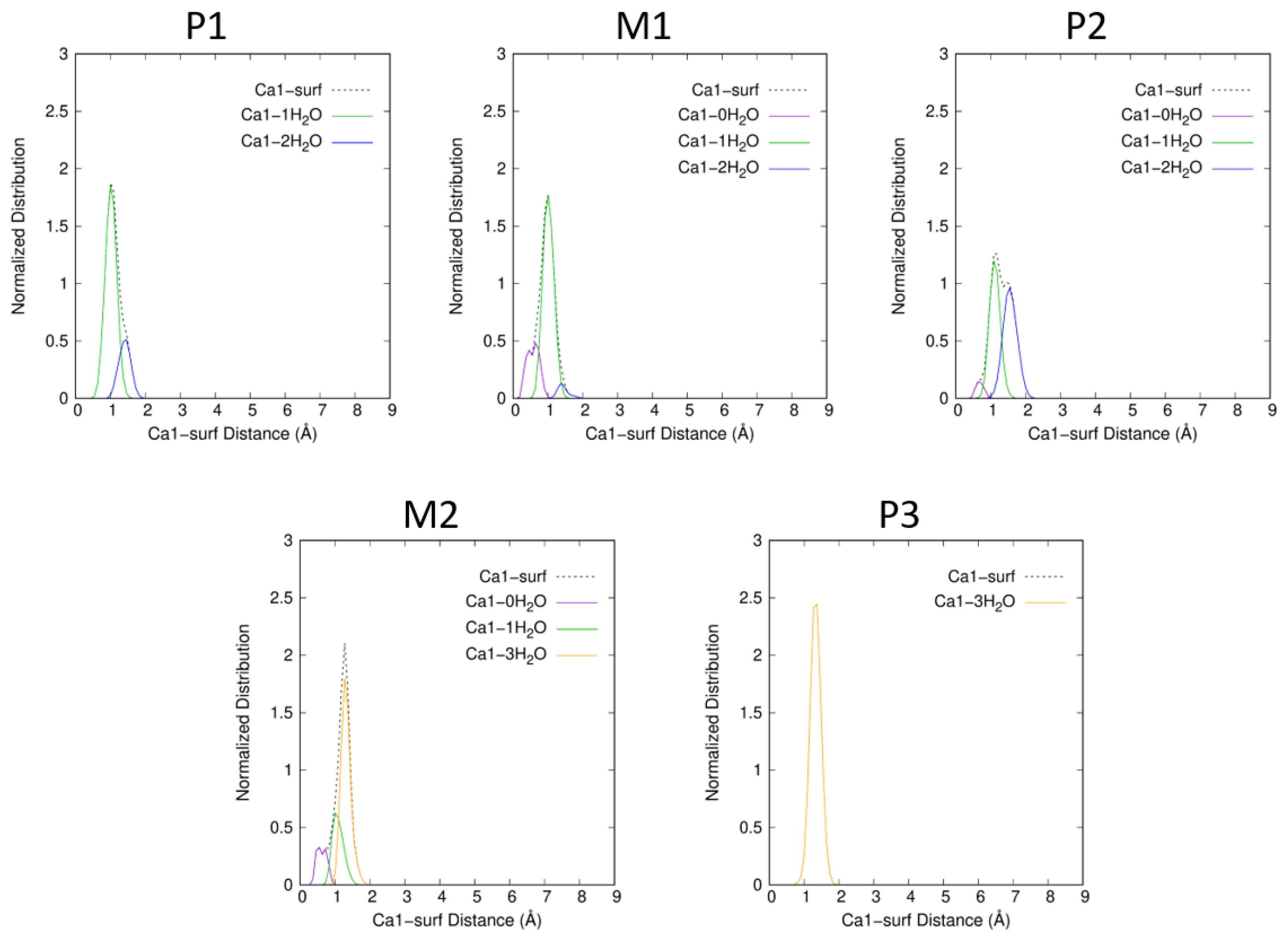
2.2. Interaction of Atrazine with the Ca1 Cation
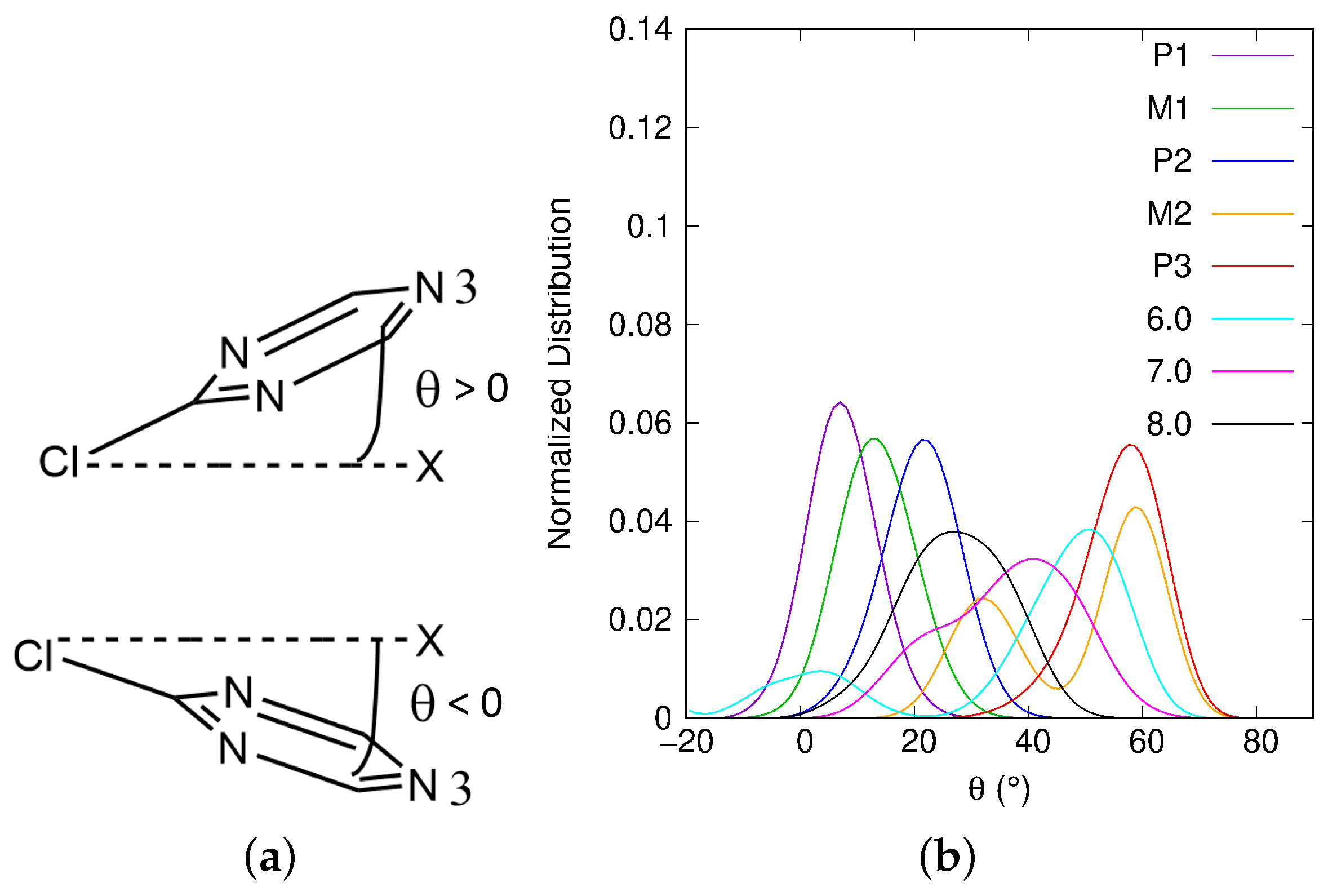
2.3. Atrazine Inclination with Respect to the Surface
2.4. Dispersion Effects and Atrazine and Cation Hydration States
3. Materials and Methods
3.1. Montmorillonite Model
3.2. Molecular Dynamics Simulations
3.3. Potential of Mean Force
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vieira, Y.; Silveira, J.P.; Dotto, G.L.; Knani, S.; Vieillard, J.; Georgin, J.; Franco, D.S.P.; Lima, E.C. Mechanistic insights and steric interpretations through statistical physics modelling and density functional theory calculations for the adsorption of the pesticides atrazine and diuron by Hovenia dulcis biochar. J. Mol. Liq. 2022, 367, 120418. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, B.; Shen, J.; Yan, P.; Kang, J.; Wang, W.; Bi, L.; Zhu, X.; Li, Y.; Wang, S.; et al. Preparation of novel N-doped biochar and its high adsorption capacity for atrazine based on Pi–Pi electron donor-acceptor interaction. J. Hazard. Mater. 2022, 432, 128757. [Google Scholar] [CrossRef] [PubMed]
- Vieira, Y.; Schnorr, C.; Piazzi, A.C.; Netto, M.S.; Piccini, W.M.; Franco, D.S.P.; Mallmann, E.S.; Georgin, J.; Silva, L.F.O.; Dotto, G.L. An advanced combination of density functional theory simulations and statistical physics modeling in the unveiling and prediction of adsorption mechanisms of 2,4-D pesticide to activated carbon. J. Mol. Liq. 2022, 361, 119639. [Google Scholar] [CrossRef]
- Jablonowski, N.D.; Schäffer, A.; Burauel, P. Still present after all these years: Persistence plus potential toxicity raise questions about the use of atrazine. Environ. Sci. Pollut. Res. 2011, 18, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Council Regulation (EEC) No. 793/93 on the Evaluation and Control of the Risks of Existing Substances. Official Journal L 84. 5 April 1993, pp. 1–75. Available online: https://www.fao.org/faolex/results/details/en/c/LEX-FAOC037952 (accessed on 21 January 2024).
- Rostami, S.; Jafari, S.; Moeini, Z.; Jaskulak, M.; Keshtgar, L.; Badeenezhad, A.; Azhdarpoor, A.; Rostami, M.; Zorena, K.; Dehghani, M. Current methods and technologies for degradation of atrazine in contaminated soil and water: A review. Environ. Technol. Innov. 2021, 24, 102019. [Google Scholar] [CrossRef]
- Abate, G.; Masini, J.C. Adsorption of Atrazine, Hydroxyatrazine, Deethylatrazine, and Deisopropylatrazine onto Fe(III) Polyhydroxy Cations Intercalated Vermiculite and Montmorillonite. J. Agric. Food Chem. 2005, 53, 1612–1619. [Google Scholar] [CrossRef]
- Han, Y.; Gu, L.; Zhang, M.; Li, Z.; Yang, W.; Tang, X.; Xie, G. Computer-aided design of molecularly imprinted polymers for recognition of atrazine. Comput. Theor. Chem. 2017, 1121, 29–34. [Google Scholar] [CrossRef]
- França, V.L.B.; Amaral, J.L.; Martins, Y.A.; Caetano, E.W.S.; Brunaldi, K.; Freire, V.N. Characterization of the binding interaction between atrazine and human serum albumin: Fluorescence spectroscopy, molecular dynamics and quantum biochemistry. Chem.-Biol. Interact. 2022, 366, 110130. [Google Scholar] [CrossRef]
- Moreno-Rodriguez, D.; Jankovic, L.; Scholtzova, E.; Tunega, D. Stability of Atrazine–Smectite Intercalates: Density Functional Theory and Experimental Study. Minerals 2021, 11, 554. [Google Scholar] [CrossRef]
- Belzunces, B.; Hoyau, S.; Cuny, J.; Bessac, F. Desorption Mechanism of a Pesticide from a Hydrated Calcium Montmorillonite Unraveled by Molecular Dynamics Simulations. J. Phys. Chem. C 2023, 127, 12953–12966. [Google Scholar] [CrossRef]
- Bessac, F.; Hoyau, S. Pesticide interaction with environmentally important cations: A theoretical study of atrazine. Comput. Theor. Chem. 2011, 966, 284–298. [Google Scholar] [CrossRef]
- Bessac, F.; Hoyau, S. Pesticide interaction with environmentally important cations: A theoretical study of atrazine in interaction with two Ca2+ cations. Comput. Theor. Chem. 2013, 1022, 6–13. [Google Scholar] [CrossRef]
- Belzunces, B.; Hoyau, S.; Benoit, M.; Tarrat, N.; Bessac, F. Theoretical study of the atrazine pesticide interaction with pyrophyllite and Ca2+-montmorillonite clay surfaces. J. Comput. Chem. 2016, 38, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Belzunces, B.; Hoyau, S.; Bessac, F. Interaction of Metamitron and Fenhexamid with Ca2+-Montmorillonite Clay Surfaces: A Density Functional Theory Molecular Dynamics Study. J. Comput. Chem. 2019, 40, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Wardle, R.; Brindley, G.W. The crystal structures of pyrophyllite, 1Tc, and of its dehydroxylate. Am. Mineral. 1972, 57, 732–750. [Google Scholar]
- Belzunces, B.; Hoyau, S.; Cuny, J.; Bessac, F. Pesticide interaction with environmentally important cations: A molecular dynamics and DFT study of metamitron and fenhexamid. Comput. Theor. Chem. 2017, 1117, 220–234. [Google Scholar] [CrossRef]
- CPMD. Copyright IBM Corp 1990–2015, Copyright MPI für Festkörperforschung Stuttgart 1997–2001. Available online: https://github.com/CPMD-code (accessed on 21 January 2024).
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Voora, V.K.; Al-Saidi, W.A.; Jordan, K.D. Density Functional Theory Study of Pyrophyllite and M-Montmorillonites (M = Li, Na, K, Mg, and Ca): Role of Dispersion Interactions. J. Phys. Chem. A 2011, 115, 9695–9703. [Google Scholar] [CrossRef]
- Tunega, D.; Bučko, T.; Zaoui, A. Assessment of ten DFT methods in predicting structures of sheet silicates: Importance of dispersion corrections. J. Chem. Phys. 2012, 137, 114105. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Hamann, D.R.; Schlüter, M.; Chiang, C. Norm-Conserving Pseudopotentials. Phys. Rev. Lett. 1979, 43, 1494–1497. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Ceriotti, M.; Bussi, G.; Parrinello, M. Langevin Equation with Colored Noise for Constant-Temperature Molecular Dynamics Simulations. Phys. Rev. Lett. 2009, 102, 020601. [Google Scholar] [CrossRef] [PubMed]
- Soper, A.K. The radial distribution functions of water and ice from 220 to 673 K and at pressures up to 400 MPa. Chem. Phys. 2000, 258, 121–137. [Google Scholar] [CrossRef]
- Skinner, L.B.; Huang, C.; Schlesinger, D.; Pettersson, L.G.M.; Nilsson, A.; Benmore, C.J. Benchmark oxygen-oxygen pair-distribution function of ambient water from x-ray diffraction measurements with a wide Q-range. J. Chem. Phys. 2013, 138, 074506. [Google Scholar] [CrossRef] [PubMed]
- Bankura, A.; Karmakar, A.; Carnevale, V.; Chandra, A.; Klein, M.L. Structure, Dynamics, and Spectral Diffusion of Water from First-Principles Molecular Dynamics. J. Phys. Chem. C 2014, 118, 29401–29411. [Google Scholar] [CrossRef]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Monte Carlo free energy estimates using non-Boltzmann sampling: Application to the sub-critical Lennard-Jones fluid. Chem. Phys. Lett. 1974, 28, 578–581. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- The PLUMED Consortium. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670. [Google Scholar] [CrossRef] [PubMed]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED2: New feathers for an old bird. Comp. Phys. Comm. 2014, 185, 604. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free energy calculations with molecular dynamics. Comp. Phys. Comm. 2009, 180, 1961. [Google Scholar] [CrossRef]
- Kollman, P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 1993, 93, 2395–2417. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. Multidimensional free-energy calculations using the weighted histogram analysis method. J. Comput. Chem. 1995, 16, 1339–1350. [Google Scholar] [CrossRef]
- Roux, B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 1995, 91, 275–282. [Google Scholar] [CrossRef]
- Trzesniak, D.; Kunz, A.P.E.; van Gunsteren, W.F. A Comparison of Methods to Compute the Potential of Mean Force. ChemPhysChem 2007, 8, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Grossfield, A. WHAM: The Weighted Histogram Analysis Method, Version 2.0.9.1. Available online: http://membrane.urmc.rochester.edu/wordpress/?page_id=126 (accessed on 21 January 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ca1 ⊂ Atra a | Ca1–Ca2 (Å) | (∘) | ||||||
|---|---|---|---|---|---|---|---|---|
| P1 | 26.4 | 21.9 | 1.2 | 21.9 | 1.2 | 4.5 | 9.10 | 7.1 |
| M1 | 28.6 | 24.1 | 0.8 | 24.1 | 0.8 | 4.5 | 8.57 | 13.1 |
| P2 | 29.2 | 23.9 | 1.4 | 23.9 | 1.4 | 5.2 | 9.74 | 20.8 |
| M2 | 33.1 | 30.4 | 2.1 | 30.3 | 2.1 | 2.7 | 10.34 | 48.7 |
| P3 | 34.9 | 31.9 | 2.8 | 31.7 | 3.0 | 3.0 | 10.29 | 56.7 |
| 6.0 Å | 37.8 | 33.6 | 2.7 | 33.3 | 3.0 | 4.2 | 10.33 | 40.3 |
| 7.0 Å | 41.6 | 37.7 | 1.9 | 36.6 | 3.0 | 3.9 | 9.70 | 36.5 |
| 8.0 Å | 41.6 | 38.1 | 1.1 | 35.8 | 3.3 | 3.5 | 10.00 | 27.1 |
| MIN | MAX | MEAN | |
|---|---|---|---|
| P1 | 14 | 29 | 21.9 |
| M1 | 15 | 32 | 24.1 |
| P2 | 16 | 30 | 23.9 |
| M2 | 24 | 35 | 30.3 |
| P3 | 24 | 38 | 31.7 |
| 6.0 Å | 25 | 41 | 33.3 |
| 7.0 Å | 30 | 44 | 36.6 |
| 8.0 Å | 26 | 43 | 35.8 |
| i | (Å) | (Å−2) | () |
|---|---|---|---|
| 1 | 3.583 | 28.6 | 13 |
| 2 | 3.628 | 14.3 | 16 |
| 3 | 3.805 | 57.1 | 4 |
| 4 | 3.805 | 28.6 | 9 |
| 5 | 4.157 | 14.3 | 12 |
| 6 | 4.486 | 14.3 | 14 |
| 7 | 5.216 | 14.3 | 12 |
| 8 | 5.421 | 57.1 | 5 |
| 9 | 5.421 | 28.6 | 7 |
| 10 | 5.615 | 28.6 | 13 |
| 11 | 5.745 | 14.3 | 14 |
| 12 | 6.274 | 14.3 | 12 |
| 13 | 6.803 | 14.3 | 14 |
| 14 | 7.332 | 14.3 | 14 |
| 15 | 7.832 | 57.1 | 4 |
| 16 | 7.832 | 28.6 | 10 |
| 17 | 7.861 | 14.3 | 12 |
| 18 | 8.391 | 14.3 | 12 |
| 19 | 8.920 | 14.3 | 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desdion, Q.; Bessac, F.; Hoyau, S. Atrazine Desorption Mechanism from an Hydrated Calcium Montmorillonite—A DFT Molecular Dynamics Study. Int. J. Mol. Sci. 2024, 25, 1604. https://doi.org/10.3390/ijms25031604
Desdion Q, Bessac F, Hoyau S. Atrazine Desorption Mechanism from an Hydrated Calcium Montmorillonite—A DFT Molecular Dynamics Study. International Journal of Molecular Sciences. 2024; 25(3):1604. https://doi.org/10.3390/ijms25031604
Chicago/Turabian StyleDesdion, Quentin, Fabienne Bessac, and Sophie Hoyau. 2024. "Atrazine Desorption Mechanism from an Hydrated Calcium Montmorillonite—A DFT Molecular Dynamics Study" International Journal of Molecular Sciences 25, no. 3: 1604. https://doi.org/10.3390/ijms25031604
APA StyleDesdion, Q., Bessac, F., & Hoyau, S. (2024). Atrazine Desorption Mechanism from an Hydrated Calcium Montmorillonite—A DFT Molecular Dynamics Study. International Journal of Molecular Sciences, 25(3), 1604. https://doi.org/10.3390/ijms25031604