
Antifibrotic Drugs against Idiopathic Pulmonary Fibrosis and Pulmonary Fibrosis Induced by COVID-19: Therapeutic Approaches and Potential Diagnostic Biomarkers

 ,
,  and
and 
Abstract
1. Introduction
1.1. Viral Transmission and Clinical Features of COVID-19 Disease
1.2. Post-COVID-19 Syndrome
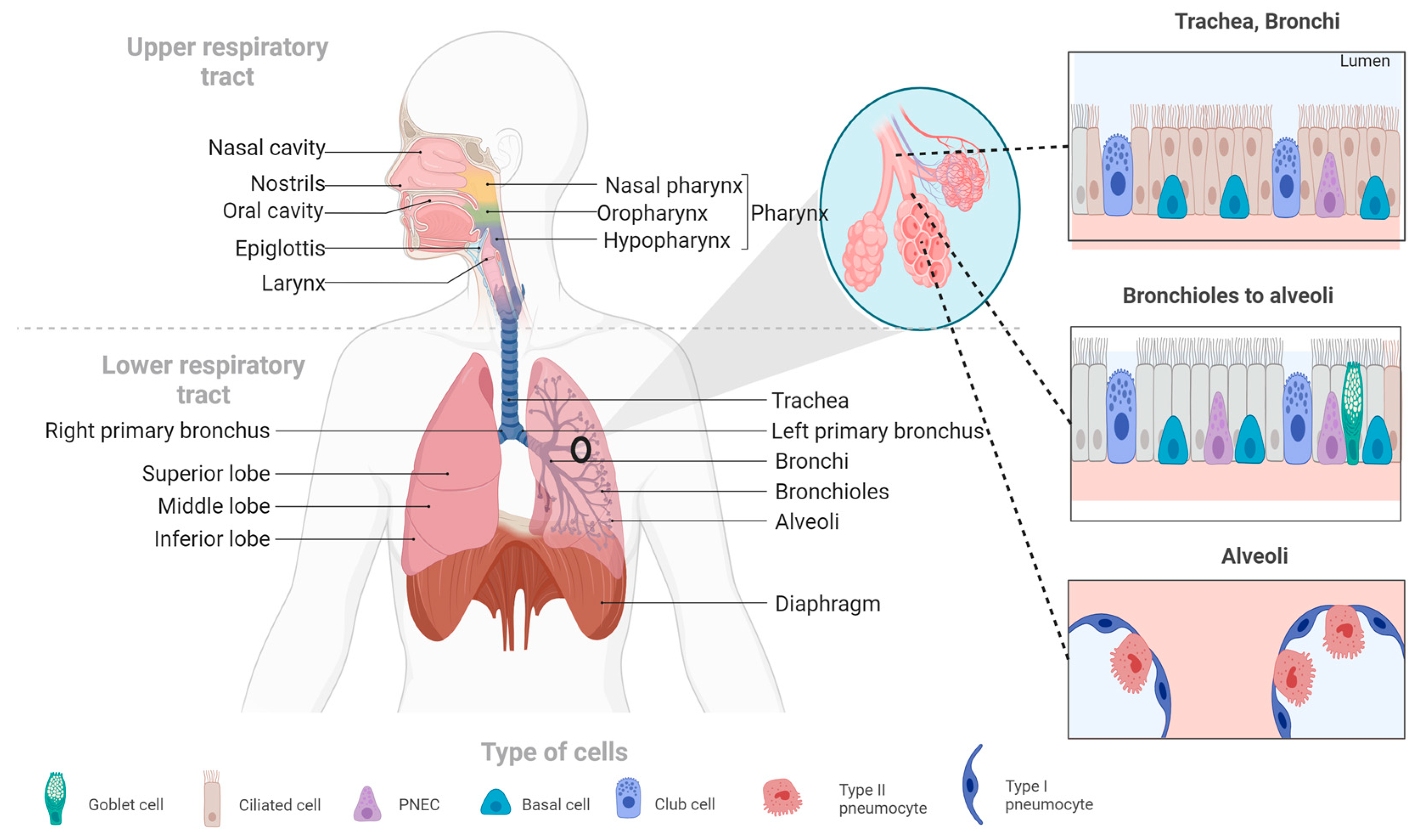
2. Overview of the Respiratory System
3. Pulmonary Fibrosis (PF)
3.1. Risk Factors for PF Associated with COVID-19
3.1.1. Age and Sex
3.1.2. Smoking and Alcoholism
3.1.3. Comorbidities
3.1.4. Mechanical Ventilation and ICU Length of Stay
3.1.5. Acute Respiratory Distress Syndrome
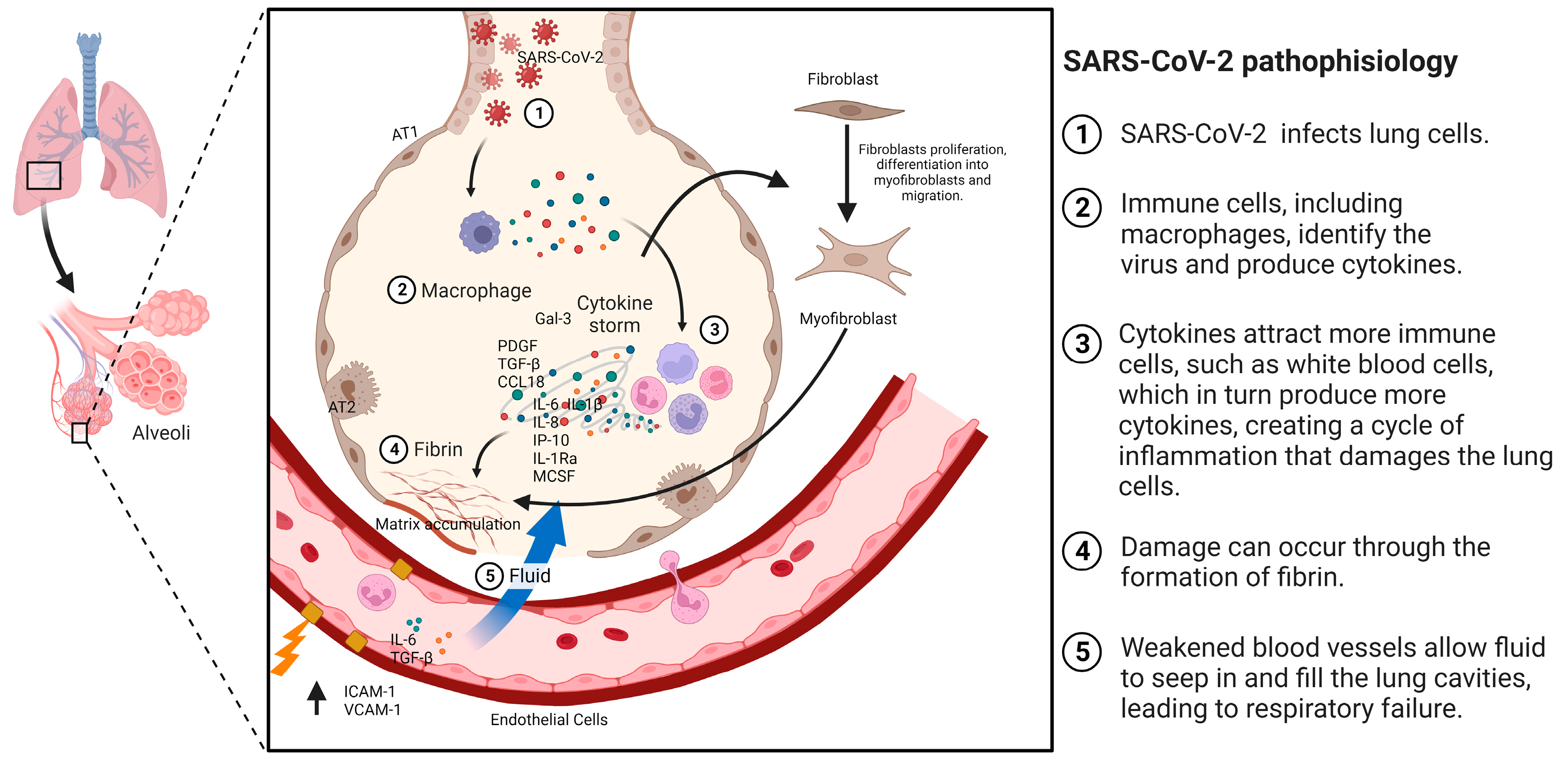
4. Mechanisms Involved in the Development of IPF
4.1. Cells Involved in the Development of IPF
4.1.1. Macrophage Activation
4.1.2. Activation of Fibroblasts and its Differentiation to Myofibroblasts
4.2. Signaling Pathways Involved in the Development of IPF
4.2.1. TGF-β Signaling
4.2.2. Activation of Integrins
4.3. Growth Factors Involved in Idiopathic Pulmonary Fibrosis
- -
- PDGF is a mitogenic molecule that affects various connective tissue cells and other cell types [99]. This factor is composed of polypeptide chains A and B linked by a disulfide bond, which can form homo- and heterodimers [100]. PDGF is produced by several cell types, including macrophages, platelets, endothelial cells, and fibroblasts [101]. It has four isoforms that bind to two receptor tyrosine kinases (PDGFR α and β). These receptors are expressed in higher quantities in cells such as fibroblasts and myofibroblasts, where they participate in survival, proliferation, and migration [102]. PDGF works together with TGF-β to promote the release of activated alveolar macrophages and epithelial cells, which are directly involved in the self-activation cycle of fibrosis [103]. High levels of PDGF have been associated with FP, both in lung tissue and in bronchoalveolar lavage fluid, as demonstrated in various animal studies with models of PF [104]. PDGF-A and PDGF-C have also been found to be overexpressed in various cell types in a mouse model of injured lungs [105]. During pulmonary fibrosis, alveolar macrophages promote the release of PDGF, which contributes to the proliferation of alveolar myofibroblasts and fibrogenesis [106].
- -
- Fibroblast growth factors (FGFs) belong to a family of 22 members [107,108]. These can be divided into hormone-like FGFs (FGF 15/19, 21 and 23), canonical FGFs (FGF 1–10, 16–18, 20 and 22), and intracellular FGFs (FGF 11–14) [109]. FGFs are involved in several biological responses, interacting with heparan sulfate glycosaminoglycans (HPSGs). Released FGFs interact with cell surface receptor (FGFR) domains [110]; a complex of FGF, FGFR, and HPSG is formed, and thus, signaling is carried out [111]. Abnormal FGF signaling is implicated in various pathologies and is known to participate in the pathogenesis of PF [112]. The inhibition of FGF signaling by using FGFR2c decreased lung fibroblast proliferation and differentiation in vitro and in vivo in a murine model [113]. The alteration in FGFR1 and FGF1 signaling is critical for fibroblast migration in PF [114]. In SARS-CoV infection, the EGFR signaling pathway remains active even after the virus is eliminated. This pathway is believed to be responsible for the effect of FGF in the conduction to PF [115].
- -
- Connective tissue growth factor (CTGF) belongs to the IGFBP family, which is known as insulin-like growth factor-binding protein 8 (IGFBP-8) [116,117]. This 38 kDa mitogenic peptide is involved in fibrotic processes and stimulates migration, fibroblast proliferation, and ECM production [118]. CTGF is a cytokine that participates in the activation of fibroblasts; it was observed that in alveolar epithelial cells, the expression levels of CTGF were drastically reduced by inhibiting the Rho pathway. Additionally, the epithelial cells of the lung in the mice indicators of CTGF increased the activity of the promoter of CTGF when the authors treated them with bleomycin, an inducer of PF [119]. CTGF can interact with other molecules to develop its fibrotic effects, thus contributing to the generation of fibrosis; among the molecules with which it interacts, there are cytokines and growth factors such as IGF1, BMP4, BMP7, TGF-β and VEGF. This interaction with other molecules may positively or negatively alter the signaling pathways of which they are participants, resulting in changes in cell adhesion, migration, differentiation, angiogenesis, myofibroblast activation, deposition, and remodeling of ECM, which causes changes in structure and alters tissue remodeling [120].
- -
- Metalloproteinases (MMPs) belong to the family of extracellular endopeptidases, whose primary function is the degradation of ECM components, while tissue inhibitors of metalloproteinases (TIMPs) block the activity of MMPs [121]. The participation of proinflammatory cytokines affects the overexpression of MMPs, which increases its activity and thus participates in the remodeling of the airways [122]. The level of TIMPs is increased in the presence of a fibrotic process such as PF [123]. MMP-2 and MMP-9 are downregulated in severe COVID-19 patients [124]. MMP-2 has shown a correlation with mortality in patients with COVID-19, so it could be a potential prognostic predictor for patients with COVID-19 [125]. One study reported that MMP-7 correlates with clinical and functional predictors of disease severity and mortality, so it can be used to distinguish IPF from other chronic diseases [126].
5. Antifibrotic Treatments and Evidence Level
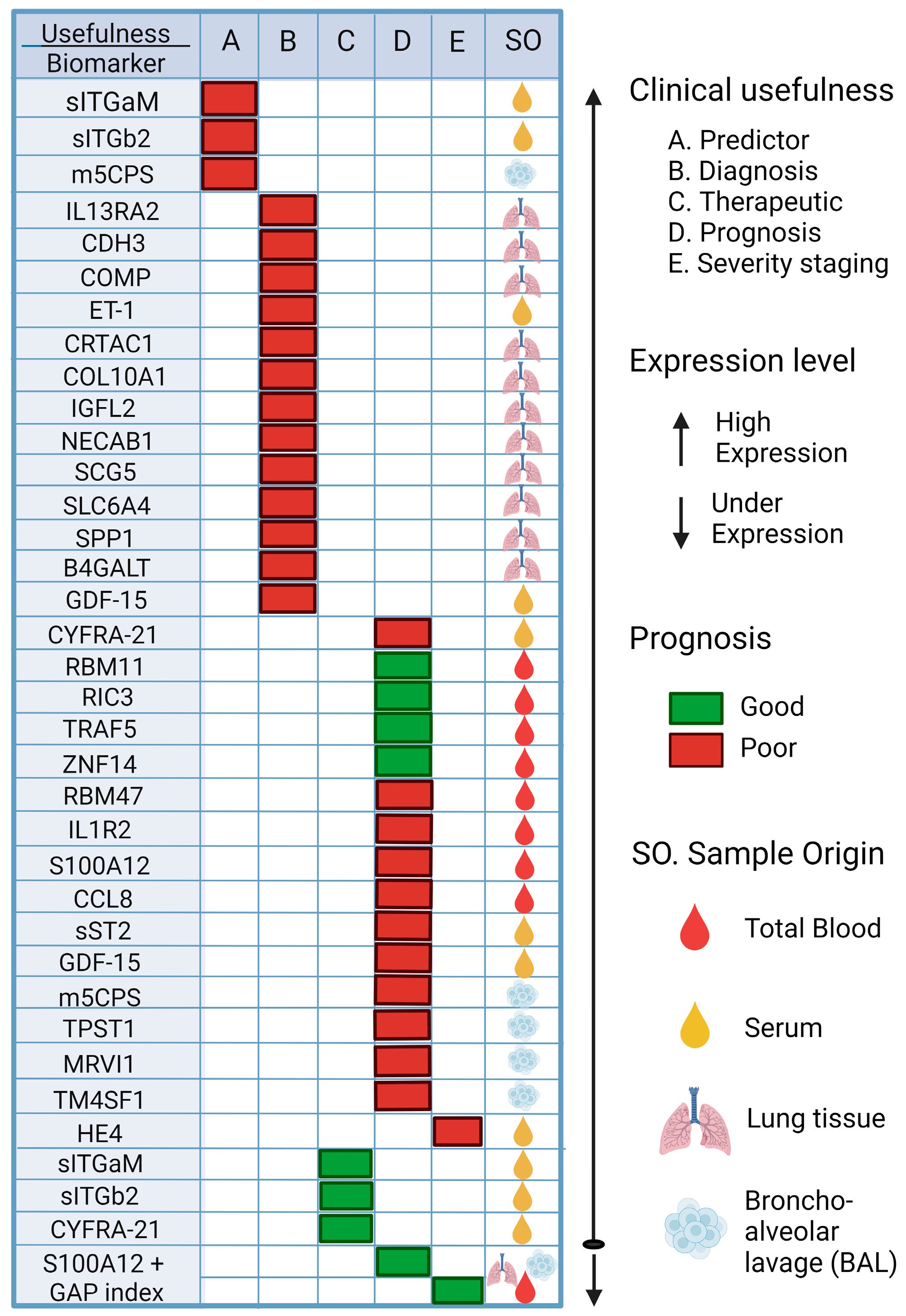
6. Biomarkers Associated with IPF, Its Progression, and Its Possible Application in PF Associated with COVID-19
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhao, S.; Lin, Q.; Ran, J.; Musa, S.S.; Yang, G.; Wang, W.; Lou, Y.; Gao, D.; Yang, L.; He, D.; et al. Preliminary estimation of the basic reproduction number of novel coronavirus (2019-nCoV) in China, from 2019 to 2020: A data-driven analysis in the early phase of the outbreak. Int. J. Infect. Dis. 2020, 92, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Boger, B.; Fachi, M.M.; Vilhena, R.O.; Cobre, A.F.; Tonin, F.S.; Pontarolo, R. Systematic review with meta-analysis of the accuracy of diagnostic tests for COVID-19. Am. J. Infect. Control 2021, 49, 21–29. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Coronavirus (COVID-19) Dashboard; WHO: Geneva, Switzerland, 2023. [Google Scholar]
- DGE. COVID-19 Tablero México—CONAHCYT. Available online: https://datos.covid-19.conacyt.mx/ (accessed on 7 November 2023).
- Gralton, J.; Tovey, E.; McLaws, M.L.; Rawlinson, W.D. The role of particle size in aerosolised pathogen transmission: A review. J. Infect. 2011, 62, 1–13. [Google Scholar] [CrossRef]
- Fernstrom, A.; Goldblatt, M. Aerobiology and its role in the transmission of infectious diseases. J. Pathog. 2013, 2013, 493960. [Google Scholar] [CrossRef]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Singhal, T. A Review of Coronavirus Disease-2019 (COVID-19). Indian J. Pediatr. 2020, 87, 281–286. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Emrani, J.; Ahmed, M.; Jeffers-Francis, L.; Teleha, J.C.; Mowa, N.; Newman, R.H.; Thomas, M.D. SARS-CoV-2, infection, transmission, transcription, translation, proteins, and treatment: A review. Int. J. Biol. Macromol. 2021, 193, 1249–1273. [Google Scholar] [CrossRef]
- Stokes, E.K.; Zambrano, L.D.; Anderson, K.N.; Marder, E.P.; Raz, K.M.; El Burai Felix, S.; Tie, Y.; Fullerton, K.E. Coronavirus Disease 2019 Case Surveillance—United States, January 22–May 30, 2020. MMWR Morb. Mortal Wkly. Rep. 2020, 69, 759–765. [Google Scholar] [CrossRef]
- Tian, S.; Hu, N.; Lou, J.; Chen, K.; Kang, X.; Xiang, Z.; Chen, H.; Wang, D.; Liu, N.; Liu, D.; et al. Characteristics of COVID-19 infection in Beijing. J. Infect. 2020, 80, 401–406. [Google Scholar] [CrossRef]
- Guarner, J. Three Emerging Coronaviruses in Two Decades. Am. J. Clin. Pathol. 2020, 153, 420–421. [Google Scholar] [CrossRef]
- Fernandez-de-Las-Penas, C.; Palacios-Cena, D.; Gomez-Mayordomo, V.; Cuadrado, M.L.; Florencio, L.L. Defining Post-COVID Symptoms (Post-Acute COVID, Long COVID, Persistent Post-COVID): An Integrative Classification. Int. J. Env. Res. Public Health 2021, 18, 2621. [Google Scholar] [CrossRef]
- van Kessel, S.A.M.; Olde Hartman, T.C.; Lucassen, P.; van Jaarsveld, C.H.M. Post-acute and long-COVID-19 symptoms in patients with mild diseases: A systematic review. Fam. Pr. 2022, 39, 159–167. [Google Scholar] [CrossRef]
- Cares-Marambio, K.; Montenegro-Jimenez, Y.; Torres-Castro, R.; Vera-Uribe, R.; Torralba, Y.; Alsina-Restoy, X.; Vasconcello-Castillo, L.; Vilaro, J. Prevalence of potential respiratory symptoms in survivors of hospital admission after coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. Chron Respir. Dis. 2021, 18, 14799731211002240. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 Long-term effects of COVID-19: A systematic review and meta-analysis. medRxiv 2021, 11, 16144. [Google Scholar] [CrossRef]
- Kamal, M.; Abo Omirah, M.; Hussein, A.; Saeed, H. Assessment and characterisation of post-COVID-19 manifestations. Int. J. Clin. Pr. 2021, 75, e13746. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-de-Las-Penas, C.; Palacios-Cena, D.; Gomez-Mayordomo, V.; Rodriuez-Jimenez, J.; Palacios-Cena, M.; Velasco-Arribas, M.; Guijarro, C.; de-la-Llave-Rincon, A.I.; Fuensalida-Novo, S.; Elvira-Martinez, C.M.; et al. Long-term post-COVID symptoms and associated risk factors in previously hospitalized patients: A multicenter study. J. Infect. 2021, 83, 237–279. [Google Scholar] [CrossRef] [PubMed]
- Ceban, F.; Ling, S.; Lui, L.M.W.; Lee, Y.; Gill, H.; Teopiz, K.M.; Rodrigues, N.B.; Subramaniapillai, M.; Di Vincenzo, J.D.; Cao, B.; et al. Fatigue and cognitive impairment in Post-COVID-19 Syndrome: A systematic review and meta-analysis. Brain Behav. Immun. 2022, 101, 93–135. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Haddad, M.; Sharma, S. Physiology, Lung; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chaudhry, R.; Bordoni, B. Anatomy, Thorax, Lungs; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Brinkman, J.E.; Sharma, S. Physiology, Pulmonary; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Lofrese, J.J.; Tupper, C.; Denault, D.; Lappin, S.L. Physiology, Residual Volume; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Burney, P.; Perez-Padilla, R.; Marks, G.; Wong, G.; Bateman, E.; Jarvis, D. Chronic Lower Respiratory Tract Diseases. In Cardiovascular, Respiratory, and Related Disorders, 3rd ed.; Prabhakaran, D., Anand, S., Gaziano, T.A., Mbanya, J.C., Wu, Y., Nugent, R., Eds.; The International Bank for Reconstruction and Development/The World Bank: Washington, DC, USA, 2017. [Google Scholar] [CrossRef]
- Martinez-Pitre, P.J.; Sabbula, B.R.; Cascella, M. Restrictive Lung Disease; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Kalchiem-Dekel, O.; Galvin, J.R.; Burke, A.P.; Atamas, S.P.; Todd, N.W. Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History. J. Clin. Med. 2018, 7, 476. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, F.; Solidoro, P.; Gavelli, F.; Apostolo, D.; Bellan, M. Idiopathic Pulmonary Fibrosis and Post-COVID-19 Lung Fibrosis: Links and Risks. Microorganisms 2023, 11, 895. [Google Scholar] [CrossRef]
- Bazdyrev, E.; Rusina, P.; Panova, M.; Novikov, F.; Grishagin, I.; Nebolsin, V. Lung Fibrosis after COVID-19: Treatment Prospects. Pharmaceuticals 2021, 14, 807. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.K.; Sharma, P.; Kumar, R. Post COVID 19 pulmonary fibrosis. Is it real threat? Indian J. Tuberc. 2021, 68, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 6175964. [Google Scholar] [CrossRef] [PubMed]
- Vaz de Paula, C.B.; Nagashima, S.; Liberalesso, V.; Collete, M.; da Silva, F.P.G.; Oricil, A.G.G.; Barbosa, G.S.; da Silva, G.V.C.; Wiedmer, D.B.; da Silva Deziderio, F.; et al. COVID-19: Immunohistochemical Analysis of TGF-beta Signaling Pathways in Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 23, 168. [Google Scholar] [CrossRef]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef]
- Qiao, J.; Zhang, M.; Bi, J.; Wang, X.; Deng, G.; He, G.; Luan, Z.; Lv, N.; Xu, T.; Zhao, L. Pulmonary fibrosis induced by H5N1 viral infection in mice. Respir. Res. 2009, 10, 107. [Google Scholar] [CrossRef]
- Huang, W.J.; Tang, X.X. Virus infection induced pulmonary fibrosis. J. Transl. Med. 2021, 19, 496. [Google Scholar] [CrossRef]
- Shi, X.; Young, C.D.; Zhou, H.; Wang, X. Transforming Growth Factor-beta Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules 2020, 10, 1666. [Google Scholar] [CrossRef]
- Duong-Quy, S.; Vo-Pham-Minh, T.; Tran-Xuan, Q.; Huynh-Anh, T.; Vo-Van, T.; Vu-Tran-Thien, Q.; Nguyen-Nhu, V. Post-COVID-19 Pulmonary Fibrosis: Facts-Challenges and Futures: A Narrative Review. Pulm. Ther. 2023, 9, 295–307. [Google Scholar] [CrossRef]
- Alrajhi, N.N. Post-COVID-19 pulmonary fibrosis: An ongoing concern. Ann. Thorac. Med. 2023, 18, 173–181. [Google Scholar] [CrossRef]
- Peckham, H.; de Gruijter, N.M.; Raine, C.; Radziszewska, A.; Ciurtin, C.; Wedderburn, L.R.; Rosser, E.C.; Webb, K.; Deakin, C.T. Male sex identified by global COVID-19 meta-analysis as a risk factor for death and ITU admission. Nat. Commun. 2020, 11, 6317. [Google Scholar] [CrossRef]
- McGroder, C.F.; Zhang, D.; Choudhury, M.A.; Salvatore, M.M.; D’Souza, B.M.; Hoffman, E.A.; Wei, Y.; Baldwin, M.R.; Garcia, C.K. Pulmonary fibrosis 4 months after COVID-19 is associated with severity of illness and blood leucocyte telomere length. Thorax 2021, 76, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Umnuaypornlert, A.; Kanchanasurakit, S.; Lucero-Prisno, D.E.I.; Saokaew, S. Smoking and risk of negative outcomes among COVID-19 patients: A systematic review and meta-analysis. Tob. Induc. Dis. 2021, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tao, Z.W.; Wang, L.; Yuan, M.L.; Liu, K.; Zhou, L.; Wei, S.; Deng, Y.; Liu, J.; Liu, H.G.; et al. Analysis of factors associated with disease outcomes in hospitalized patients with 2019 novel coronavirus disease. Chin. Med. J. 2020, 133, 1032–1038. [Google Scholar] [CrossRef]
- Simou, E.; Leonardi-Bee, J.; Britton, J. The Effect of Alcohol Consumption on the Risk of ARDS: A Systematic Review and Meta-Analysis. Chest 2018, 154, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Balan, I.; Yadav, S.; Matos, W.F.; Kharawala, A.; Gaddam, M.; Sarabia, N.; Koneru, S.C.; Suddapalli, S.K.; Marzban, S. Post-COVID-19 Pulmonary Fibrosis. Cureus 2022, 14, e22770. [Google Scholar] [CrossRef]
- Enomoto, T.; Usuki, J.; Azuma, A.; Nakagawa, T.; Kudoh, S. Diabetes mellitus may increase risk for idiopathic pulmonary fibrosis. Chest 2003, 123, 2007–2011. [Google Scholar] [CrossRef]
- Aul, D.R.; Gates, D.J.; Draper, D.A.; Dunleavy, D.A.; Ruickbie, D.S.; Meredith, D.H.; Walters, D.N.; van Zeller, D.C.; Taylor, D.V.; Bridgett, D.M.; et al. Complications after discharge with COVID-19 infection and risk factors associated with development of post-COVID pulmonary fibrosis. Respir. Med. 2021, 188, 106602. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Duffy, H.S. Fibroblasts and myofibroblasts: What are we talking about? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, L.; Wang, M.; Zhou, S.; Lu, Y.; Cui, H.; Racanelli, A.C.; Zhang, L.; Ye, T.; Ding, B.; et al. Targeting fibrosis, mechanisms and cilinical trials. Signal Transduct. Target Ther. 2022, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Rumende, C.M.; Susanto, E.C.; Sitorus, T.P. The Management of Pulmonary Fibrosis in COVID-19. Acta Med. Indones. 2021, 53, 233–241. [Google Scholar] [PubMed]
- Wen, J.H.; Li, D.Y.; Liang, S.; Yang, C.; Tang, J.X.; Liu, H.F. Macrophage autophagy in macrophage polarization, chronic inflammation and organ fibrosis. Front. Immunol. 2022, 13, 946832. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Perciani, C.T.; MacParland, S.A. Lifting the veil on macrophage diversity in tissue regeneration and fibrosis. Sci. Immunol. 2019, 4, eaaz0749. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef]
- Lang, R.; Patel, D.; Morris, J.J.; Rutschman, R.L.; Murray, P.J. Shaping gene expression in activated and resting primary macrophages by IL-10. J. Immunol. 2002, 169, 2253–2263. [Google Scholar] [CrossRef]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Fu, X.; Chen, X.; Han, X.; Dong, P. M2 macrophages induce EMT through the TGF-beta/Smad2 signaling pathway. Cell Biol. Int. 2017, 41, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wang, C.; Chen, X.; Hou, J.; Xiang, Z.; Shen, Y.; Han, X. Inhibition of Wnt/beta-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci. Rep. 2018, 8, 13644. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, T.; Xu, Y.; Xu, X.; Zhu, Z.; Zhang, Y.; Xu, J.; Xu, K.; Cheng, H.; Zhang, X.; et al. Increased levels of Gab1 and Gab2 adaptor proteins skew interleukin-4 (IL-4) signaling toward M2 macrophage-driven pulmonary fibrosis in mice. J. Biol. Chem. 2017, 292, 14003–14015. [Google Scholar] [CrossRef]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell 2021, 184, 6243–6261.e6227. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- White, E.S. Lung extracellular matrix and fibroblast function. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S1), S30–S33. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.; O’Connor, R. Idiopathic pulmonary fibrosis: Current understanding of the pathogenesis and the status of treatment. CMAJ 2004, 171, 153–160. [Google Scholar] [CrossRef]
- Li, X.; Bi, Z.; Liu, S.; Gao, S.; Cui, Y.; Huang, K.; Huang, M.; Mao, J.; Li, L.; Gao, J.; et al. Antifibrotic Mechanism of Cinobufagin in Bleomycin-Induced Pulmonary Fibrosis in Mice. Front. Pharmacol. 2019, 10, 1021. [Google Scholar] [CrossRef]
- Kwon, O.S.; Kim, K.T.; Lee, E.; Kim, M.; Choi, S.H.; Li, H.; Fornace, A.J., Jr.; Cho, J.H.; Lee, Y.S.; Lee, J.S.; et al. Induction of MiR-21 by Stereotactic Body Radiotherapy Contributes to the Pulmonary Fibrotic Response. PLoS ONE 2016, 11, e0154942. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018, 71–72, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Striker, L.J.; Hudson, L.D.; Striker, G.E. Extracellular matrix in normal and fibrotic human lungs. Am. Rev. Respir. Dis. 1985, 131, 281–289. [Google Scholar] [PubMed]
- Bellaye, P.S.; Shimbori, C.; Upagupta, C.; Sato, S.; Shi, W.; Gauldie, J.; Ask, K.; Kolb, M. Lysyl Oxidase-Like 1 Protein Deficiency Protects Mice from Adenoviral Transforming Growth Factor-beta1-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 461–470. [Google Scholar] [CrossRef]
- Philp, C.J.; Siebeke, I.; Clements, D.; Miller, S.; Habgood, A.; John, A.E.; Navaratnam, V.; Hubbard, R.B.; Jenkins, G.; Johnson, S.R. Extracellular Matrix Cross-Linking Enhances Fibroblast Growth and Protects against Matrix Proteolysis in Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Abdo Cuza, A.A.; Avila, J.P.; Martinez, R.M.; Gonzalez, J.J.; Aspuro, G.P.; Gutierrez Martinez, J.A.; Suzarte, M.R.; Hernandez, D.S.; Ane-Kouri, A.L.; Ramos, T.C. Nimotuzumab for COVID-19: Case series. Immunotherapy 2021, 14, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Saiphoklang, N.; Patanayindee, P.; Ruchiwit, P. The Effect of Nintedanib in Post-COVID-19 Lung Fibrosis: An Observational Study. Crit. Care Res. Pr. 2022, 2022, 9972846. [Google Scholar] [CrossRef]
- Londres, H.D.; Armada, J.J.; Martinez, A.H.; Abdo Cuza, A.A.; Sanchez, Y.H.; Rodriguez, A.G.; Figueroa, S.S.; Llanez Gregorich, E.M.; Torres Lahera, M.L.; Peire, F.G.; et al. Blocking EGFR with nimotuzumab: A novel strategy for COVID-19 treatment. Immunotherapy 2022, 14, 521–530. [Google Scholar] [CrossRef]
- Acat, M.; Yildiz Gulhan, P.; Oner, S.; Turan, M.K. Comparison of pirfenidone and corticosteroid treatments at the COVID-19 pneumonia with the guide of artificial intelligence supported thoracic computed tomography. Int. J. Clin. Pr. 2021, 75, e14961. [Google Scholar] [CrossRef]
- Gaughan, E.E.; Quinn, T.M.; Mills, A.; Bruce, A.M.; Antonelli, J.; MacKinnon, A.C.; Aslanis, V.; Li, F.; O’Connor, R.; Boz, C.; et al. An Inhaled Galectin-3 Inhibitor in COVID-19 Pneumonitis: A Phase Ib/IIa Randomized Controlled Clinical Trial (DEFINE). Am. J. Respir. Crit. Care Med. 2023, 207, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Flores, S.; Priego-Ranero, A.; Azamar-Llamas, D.; Olvera-Prado, H.; Rivas-Redonda, K.I.; Ochoa-Hein, E.; Perez-Ortiz, A.; Rendon-Macias, M.E.; Rojas-Castaneda, E.; Urbina-Teran, S.; et al. Effect of polymerised type I collagen on hyperinflammation of adult outpatients with symptomatic COVID-19. Clin. Transl. Med. 2022, 12, e763. [Google Scholar] [CrossRef] [PubMed]
- Oshitani, N.; Watanabe, K.; Sakuma, T.; Matsuda, M.; Oyama, Y. Tranilast, an antifibrotic agent and COVID-19-induced pulmonary fibrosis. QJM 2022, 115, 249–250. [Google Scholar] [CrossRef]
- Nan, D.; Abraira-Meriel, C.; de la Roz-Fernandez, S.; Maestre-Orozco, T.; Hernandez, J.L.; Fernandez-Ayala, M. Delayed Use of the Recombinant Human IL-1 Receptor Antagonist Anakinra in Five COVID-19 Patients with Pulmonary Fibrosis and Persistent Hypoxaemia: A Preliminary Report. Eur. J. Case Rep. Intern. Med. 2021, 8, 002821. [Google Scholar] [CrossRef]
- Hyytiainen, M.; Penttinen, C.; Keski-Oja, J. Latent TGF-beta binding proteins: Extracellular matrix association and roles in TGF-beta activation. Crit. Rev. Clin. Lab. Sci. 2004, 41, 233–264. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, J.; Taipale, J.; Keski-Oja, J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 1996, 15, 245–253. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-beta: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef]
- Sheppard, D. Transforming growth factor beta: A central modulator of pulmonary and airway inflammation and fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 413–417. [Google Scholar] [CrossRef]
- Xu, Y.D.; Hua, J.; Mui, A.; O’Connor, R.; Grotendorst, G.; Khalil, N. Release of biologically active TGF-beta1 by alveolar epithelial cells results in pulmonary fibrosis. Am. J. Physiol. Lung. Cell Mol. Physiol. 2003, 285, L527–L539. [Google Scholar] [CrossRef]
- Bartram, U.; Speer, C.P. The role of transforming growth factor beta in lung development and disease. Chest 2004, 125, 754–765. [Google Scholar] [CrossRef]
- Gentile, F.; Aimo, A.; Forfori, F.; Catapano, G.; Clemente, A.; Cademartiri, F.; Emdin, M.; Giannoni, A. COVID-19 and risk of pulmonary fibrosis: The importance of planning ahead. Eur. J. Prev. Cardiol. 2020, 27, 1442–1446. [Google Scholar] [CrossRef]
- Ye, Z.; Hu, Y. TGFbeta1: Gentlemanly orchestrator in idiopathic pulmonary fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Giacomelli, C.; Piccarducci, R.; Marchetti, L.; Romei, C.; Martini, C. Pulmonary fibrosis from molecular mechanisms to therapeutic interventions: Lessons from post-COVID-19 patients. Biochem. Pharmacol. 2021, 193, 114812. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Teoh, C.M.; Tan, S.S.; Tran, T. Integrins as Therapeutic Targets for Respiratory Diseases. Curr. Mol. Med. 2015, 15, 714–734. [Google Scholar] [CrossRef]
- Robles, J.P.; Zamora, M.; Adan-Castro, E.; Siqueiros-Marquez, L.; Martinez de la Escalera, G.; Clapp, C. The spike protein of SARS-CoV-2 induces endothelial inflammation through integrin alpha5beta1 and NF-kappaB signaling. J. Biol. Chem. 2022, 298, 101695. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, K. alphaV beta6 Integrin: An Intriguing Target for COVID-19 and Related Diseases. Chembiochem 2021, 22, 2516–2520. [Google Scholar] [CrossRef]
- Saini, G.; Porte, J.; Weinreb, P.H.; Violette, S.M.; Wallace, W.A.; McKeever, T.M.; Jenkins, G. alphavbeta6 integrin may be a potential prognostic biomarker in interstitial lung disease. Eur. Respir. J. 2015, 46, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef]
- Heldin, C.H.; Eriksson, U.; Ostman, A. New members of the platelet-derived growth factor family of mitogens. Arch. Biochem. Biophys. 2002, 398, 284–290. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef] [PubMed]
- Betsholtz, C. Biology of platelet-derived growth factors in development. Birth Defects Res. C Embryo Today 2003, 69, 272–285. [Google Scholar] [CrossRef]
- Abdollahi, A.; Li, M.; Ping, G.; Plathow, C.; Domhan, S.; Kiessling, F.; Lee, L.B.; McMahon, G.; Grone, H.J.; Lipson, K.E.; et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J. Exp. Med. 2005, 201, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004, 15, 255–273. [Google Scholar] [CrossRef]
- Zhuo, Y.; Zhang, J.; Laboy, M.; Lasky, J.A. Modulation of PDGF-C and PDGF-D expression during bleomycin-induced lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L182–L188. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef]
- Prudovsky, I. Cellular Mechanisms of FGF-Stimulated Tissue Repair. Cells 2021, 10, 1830. [Google Scholar] [CrossRef]
- Jones, S.A. Physiology of FGF15/19. Adv. Exp. Med. Biol. 2012, 728, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.N.; Kilgour, E.; Smith, P.D. Molecular pathways: Fibroblast growth factor signaling: A new therapeutic opportunity in cancer. Clin. Cancer Res. 2012, 18, 1855–1862. [Google Scholar] [CrossRef]
- Olsen, S.K.; Garbi, M.; Zampieri, N.; Eliseenkova, A.V.; Ornitz, D.M.; Goldfarb, M.; Mohammadi, M. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 2003, 278, 34226–34236. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef]
- Guzy, R.D.; Li, L.; Smith, C.; Dorry, S.J.; Koo, H.Y.; Chen, L.; Ornitz, D.M. Pulmonary fibrosis requires cell-autonomous mesenchymal fibroblast growth factor (FGF) signaling. J. Biol. Chem. 2017, 292, 10364–10378. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.H.; Wang, D.D.; Zhou, Z.Y.; He, S.L.; Chen, A.A.; Wang, J. Mutant soluble ectodomain of fibroblast growth factor receptor-2 IIIc attenuates bleomycin-induced pulmonary fibrosis in mice. Biol. Pharm. Bull. 2012, 35, 731–736. [Google Scholar] [CrossRef][Green Version]
- MacKenzie, B.; Korfei, M.; Henneke, I.; Sibinska, Z.; Tian, X.; Hezel, S.; Dilai, S.; Wasnick, R.; Schneider, B.; Wilhelm, J.; et al. Increased FGF1-FGFRc expression in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, T.; Coleman, C.M.; Frieman, M.B. Overactive Epidermal Growth Factor Receptor Signaling Leads to Increased Fibrosis after Severe Acute Respiratory Syndrome Coronavirus Infection. J. Virol. 2017, 91, e00182-17. [Google Scholar] [CrossRef]
- Kim, H.S.; Nagalla, S.R.; Oh, Y.; Wilson, E.; Roberts, C.T., Jr.; Rosenfeld, R.G. Identification of a family of low-affinity insulin-like growth factor binding proteins (IGFBPs): Characterization of connective tissue growth factor as a member of the IGFBP superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 12981–12986. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.T.; Knight, R.A.; Bloor, C.A.; Spiteri, M.A. Enhanced insulin-like growth factor binding protein-related protein 2 (Connective tissue growth factor) expression in patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am. J. Respir. Cell Mol. Biol. 1999, 21, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Alkhatib, A.; Kolls, J.K.; Kondoh, Y.; Lasky, J.A. Pharmacotherapy and adjunctive treatment for idiopathic pulmonary fibrosis (IPF). J. Thorac. Dis. 2019, 11, S1740–S1754. [Google Scholar] [CrossRef]
- Yang, J.; Velikoff, M.; Canalis, E.; Horowitz, J.C.; Kim, K.K. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L786–L796. [Google Scholar] [CrossRef]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012, 5, S24. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Amalinei, C.; Caruntu, I.D.; Balan, R.A. Biology of metalloproteinases. Rom. J. Morphol. Embryol. 2007, 48, 323–334. [Google Scholar] [PubMed]
- Selman, M.; Ruiz, V.; Cabrera, S.; Segura, L.; Ramirez, R.; Barrios, R.; Pardo, A. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L562–L574. [Google Scholar] [CrossRef] [PubMed]
- Ghezelbash, B.; Rostami, M.; Heidarvand, M.; Mafi, A.; Chegni, H.; Eskandari, N. Correlation of Expression of MMP-2, ACE2, and TMPRSS2 Genes with Lymphopenia for Mild and Severity of COVID-19. Iran J. Allergy Asthma Immunol. 2023, 22, 91–98. [Google Scholar] [CrossRef]
- Avila-Mesquita, C.D.; Couto, A.E.S.; Campos, L.C.B.; Vasconcelos, T.F.; Michelon-Barbosa, J.; Corsi, C.A.C.; Mestriner, F.; Petroski-Moraes, B.C.; Garbellini-Diab, M.J.; Couto, D.M.S.; et al. MMP-2 and MMP-9 levels in plasma are altered and associated with mortality in COVID-19 patients. Biomed. Pharmacother. 2021, 142, 112067. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Herazo-Maya, J.D.; Slade, M.; Chu, J.H.; Deiuliis, G.; Ryu, C.; Li, Q.; Sakamoto, K.; Ibarra, G.; Pan, H.; et al. Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis. Respirology 2017, 22, 486–493. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Raghu, G.; Selman, M. Nintedanib and pirfenidone. New antifibrotic treatments indicated for idiopathic pulmonary fibrosis offer hopes and raises questions. Am. J. Respir. Crit. Care Med. 2015, 191, 252–254. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, D.; Kong, X.; Wei, C.; LvQiu, S.; Wang, L.; Lin, Y.; Yin, Z.; Zhou, Z.; Luo, H. Case Report: Pirfenidone in the Treatment of Post-COVID-19 Pulmonary Fibrosis. Front. Med. 2022, 9, 925703. [Google Scholar] [CrossRef]
- Bazdyrev, E.; Panova, M.; Brachs, M.; Smolyarchuk, E.; Tsygankova, D.; Gofman, L.; Abdyusheva, Y.; Novikov, F. Efficacy and safety of Treamid in the rehabilitation of patients after COVID-19 pneumonia: A phase 2, randomized, double-blind, placebo-controlled trial. J. Transl. Med. 2022, 20, 506. [Google Scholar] [CrossRef]
- Cesarone, M.R.; Hu, S.; Belcaro, G.; Cornelli, U.; Feragalli, B.; Corsi, M.; Bombardelli, E.; Cotellese, R.; Hosoi, M.; Rosenkvist, L. Pycnogenol(R)-Centellicum(R) supplementation improves lung fibrosis and post-COVID-19 lung healing. Minerva. Med. 2022, 113, 135–140. [Google Scholar] [CrossRef]
- Park, S.-A.; Kim, M.-J.; Park, S.-Y.; Kim, J.-S.; Lee, S.-J.; Woo, H.A.; Kim, D.-K.; Nam, J.-S.; Sheen, Y.Y. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-β/Smad and ROS signaling. Cell Mol. Life Sci. 2015, 72, 2023–2039. [Google Scholar] [CrossRef]
- Fernandes, R.S.; Dias, H.B.; de Souza Jaques, W.A.; Becker, T.; Rigatto, K. Assessment of Alamandine in Pulmonary Fibrosis and Respiratory Mechanics in Rodents. J. Renin. Angiotensin. Aldosterone Syst. 2021, 2021, 9975315. [Google Scholar] [CrossRef] [PubMed]
- Colunga Biancatelli, R.M.L.; Solopov, P.; Dimitropoulou, C.; Gregory, B.; Day, T.; Catravas, J.D. The Heat Shock Protein 90 Inhibitor, AT13387, Protects the Alveolo-Capillary Barrier and Prevents HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis. Cells 2022, 11, 1046. [Google Scholar] [CrossRef]
- Sheng, H.; Lin, G.; Zhao, S.; Li, W.; Zhang, Z.; Zhang, W.; Yun, L.; Yan, X.; Hu, H. Antifibrotic Mechanism of Piceatannol in Bleomycin-Induced Pulmonary Fibrosis in Mice. Front. Pharmacol. 2022, 13, 771031. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Fernandez Perez, E.R.; Costabel, U.; Albera, C.; Lederer, D.J.; Flaherty, K.R.; Ettinger, N.; Perez, R.; Scholand, M.B.; Goldin, J.; et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2020, 8, 25–33. [Google Scholar] [CrossRef]
- Liu, Z.W.; Zhao, M.Y.; Su, X.L.; Ye, T.H.; Zhuang, Y.J.; Zhang, Y.; Zhang, Z.Z.; Yang, J.L.; Chen, L.J.; Long, C.F.; et al. The antifibrotic effect and mechanism of a novel tyrosine kinase inhibitor, ZSP1603, in preclinical models of pulmonary fibrosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1481–1491. [Google Scholar] [CrossRef]
- Soltani Hekmat, A.; Javanmardi, K. Alamandine: Potential Protective Effects in SARS-CoV-2 Patients. J. Renin. Angiotensin. Aldosterone Syst. 2021, 2021, 6824259. [Google Scholar] [CrossRef]
- Hussein, R.M.; Arafa, E.A.; Raheem, S.A.; Mohamed, W.R. Thymol protects against bleomycin-induced pulmonary fibrosis via abrogation of oxidative stress, inflammation, and modulation of miR-29a/TGF-beta and PI3K/Akt signaling in mice. Life Sci. 2022, 314, 121256. [Google Scholar] [CrossRef]
- Ebrahimpour, A.; Ahir, M.; Wang, M.; Jegga, A.G.; Bonnen, M.D.; Eissa, N.T.; Montesi, S.B.; Raghu, G.; Ghebre, Y.T. Combination of esomeprazole and pirfenidone enhances antifibrotic efficacy in vitro and in a mouse model of TGFbeta-induced lung fibrosis. Sci. Rep. 2022, 12, 20668. [Google Scholar] [CrossRef]
- Wilkinson, A.L.; John, A.E.; Barrett, J.W.; Gower, E.; Morrison, V.S.; Man, Y.; Pun, K.T.; Roper, J.A.; Luckett, J.C.; Borthwick, L.A.; et al. Pharmacological characterisation of GSK3335103, an oral alphavbeta6 integrin small molecule RGD-mimetic inhibitor for the treatment of fibrotic disease. Eur. J. Pharmacol. 2021, 913, 174618. [Google Scholar] [CrossRef]
- Medina-De la Garza, C.E.; Salvador Flores-Torres, A.; Garcia-Hernandez, M.; de Los Angeles Castro-Corona, M. Diethylcarbamazine as potential treatment of COVID-19 lung fibrosis. Med. Hypotheses 2022, 160, 110774. [Google Scholar] [CrossRef]
- Sgalla, G.; Biffi, A.; Richeldi, L. Idiopathic pulmonary fibrosis: Diagnosis, epidemiology and natural history. Respirology 2016, 21, 427–437. [Google Scholar] [CrossRef]
- Kaarteenaho, R.; Lappi-Blanco, E. Tissue is an issue in the search for biomarkers in idiopathic pulmonary fibrosis. Fibrogenesis Tissue Repair 2015, 8, 3. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Kaminski, N. Biomarkers in idiopathic pulmonary fibrosis. Curr. Opin. Pulm. Med. 2012, 18, 441–446. [Google Scholar] [CrossRef]
- Guiot, J.; Moermans, C.; Henket, M.; Corhay, J.L.; Louis, R. Blood Biomarkers in Idiopathic Pulmonary Fibrosis. Lung 2017, 195, 273–280. [Google Scholar] [CrossRef]
- Drakopanagiotakis, F.; Wujak, L.; Wygrecka, M.; Markart, P. Biomarkers in idiopathic pulmonary fibrosis. Matrix Biol. 2018, 68–69, 404–421. [Google Scholar] [CrossRef]
- Siekacz, K.; Kumor-Kisielewska, A.; Milkowska-Dymanowska, J.; Pietrusinska, M.; Bartczak, K.; Majewski, S.; Stanczyk, A.; Piotrowski, W.J.; Bialas, A.J. Soluble ITGaM and ITGb2 Integrin Subunits Are Involved in Long-Term Pulmonary Complications after COVID-19 Infection. J. Clin. Med. 2023, 12, 342. [Google Scholar] [CrossRef]
- Wu, Y.; Zhong, L.; Qiu, L.; Dong, L.; Yang, L.; Chen, L. A potential three-gene-based diagnostic signature for idiopathic pulmonary fibrosis. Front. Genet 2022, 13, 985217. [Google Scholar] [CrossRef]
- Pulito-Cueto, V.; Genre, F.; Lopez-Mejias, R.; Mora-Cuesta, V.M.; Iturbe-Fernandez, D.; Portilla, V.; Sebastian Mora-Gil, M.; Ocejo-Vinyals, J.G.; Gualillo, O.; Blanco, R.; et al. Endothelin-1 as a Biomarker of Idiopathic Pulmonary Fibrosis and Interstitial Lung Disease Associated with Autoimmune Diseases. Int. J. Mol. Sci. 2023, 24, 1275. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Chen, S.; Wang, Q.; Yang, Y.; Shen, D.; Ma, J.; Wen, Z.; Ning, S.; Chen, H. S100A12 as Biomarker of Disease Severity and Prognosis in Patients with Idiopathic Pulmonary Fibrosis. Front. Immunol. 2022, 13, 810338. [Google Scholar] [CrossRef]
- Spagnolo, P.; Oldham, J.M. On Target: CYFRA 21-1 as an Idiopathic Pulmonary Fibrosis Biomarker. Am. J. Respir. Crit. Care Med. 2022, 205, 1376–1377. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Fahy, W.A.; Byrne, A.J.; Braybrooke, R.; Saunders, P.; Toshner, R.; Albers, G.; Chua, F.; Renzoni, E.A.; Wells, A.U.; et al. CYFRA 21-1 Predicts Progression in Idiopathic Pulmonary Fibrosis: A Prospective Longitudinal Analysis of the PROFILE Cohort. Am. J. Respir. Crit. Care Med. 2022, 205, 1440–1448. [Google Scholar] [CrossRef]
- Dai, X.; Yang, Z.; Zhang, W.; Liu, S.; Zhao, Q.; Liu, T.; Chen, L.; Li, L.; Wang, Y.; Shao, R. Identification of diagnostic gene biomarkers related to immune infiltration in patients with idiopathic pulmonary fibrosis based on bioinformatics strategies. Front. Med. 2022, 9, 959010. [Google Scholar] [CrossRef]
- De Vitis, C.; D’Ascanio, M.; Sacconi, A.; Pizzirusso, D.; Salvati, V.; Mancini, M.; Scafetta, G.; Cirombella, R.; Ascenzi, F.; Bruschini, S.; et al. B4GALT1 as a New Biomarker of Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2022, 23, 15040. [Google Scholar] [CrossRef]
- Zheng, J.; Dong, H.; Zhang, T.; Ning, J.; Xu, Y.; Cai, C. Development and Validation of a Novel Gene Signature for Predicting the Prognosis of Idiopathic Pulmonary Fibrosis Based on Three Epithelial-Mesenchymal Transition and Immune-Related Genes. Front. Genet 2022, 13, 865052. [Google Scholar] [CrossRef]
- Yu, Y.Z.; Gui, X.H.; Yu, M.; Huang, W.; Peng, L.Y.; Dai, J.H.; Xu, Q.Q.; Zhao, T.T.; Xie, W.P.; Xiao, Y.L.; et al. Soluble ST2 in serum predicts the prognosis of idiopathic pulmonary fibrosis: A retrospective study. Ann. Transl. Med. 2022, 10, 797. [Google Scholar] [CrossRef]
- Cao, M.; Gu, L.; Guo, L.; Liu, M.; Wang, T.; Zhang, J.; Zhang, H.; Zhang, Y.; Shi, Y.; Zhao, Y.; et al. Elevated Expression of Growth Differentiation Factor-15 Is Associated with Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Front. Immunol. 2022, 13, 891448. [Google Scholar] [CrossRef]
- Huang, T.; Zhou, H.F. A Novel 5-Methylcytosine- and Immune-Related Prognostic Signature Is a Potential Marker of Idiopathic Pulmonary Fibrosis. Comput. Math Methods Med. 2022, 2022, 1685384. [Google Scholar] [CrossRef]
- Tian, M.; Meng, K.; Gao, Y.; Zhang, J.; Xie, M.; Tian, Y.; Liu, X.; Ma, M.; Cai, Y.; Wu, H.; et al. Elevated serum human epididymis protein 4 is associated with disease severity and worse survival in idiopathic pulmonary fibrosis: A cohort study. Ann. Transl. Med. 2022, 10, 992. [Google Scholar] [CrossRef]
- Wang, E.; Wang, Y.; Zhou, S.; Xia, X.; Han, R.; Fei, G.; Zeng, D.; Wang, R. Identification of three hub genes related to the prognosis of idiopathic pulmonary fibrosis using bioinformatics analysis. Int. J. Med. Sci. 2022, 19, 1417–1429. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Ref. | Indication | Mechanism of Action | Phase/ID | Results or Conclusions |
|---|---|---|---|---|---|
| Pirfenidone | [77] | COVID-19 pneumonia | Inhibits TGF-β. | NA/NA | FEV1, FEV1%, FVC, and FVC% values were higher in the methylprednisolone group together with pirfenidone than in the methylprednisolone group. |
| Nintedanib | [75] | Fibrosis lung post-COVID-19 | Tyrosine kinase inhibitor. | NA/TCTR20220426001 | There was no improvement in oxygenation, chest X-ray findings, or mortality at 60 days after admission. However, the nintedanib group had a significantly greater difference in SpO2/FiO2 ratio after treatment than the controls. |
| Nimotuzumab | [76] | Inflammation and PF due to COVID-19 | Modulates the dimerization of EGFR, which inhibits kinase activity. | I/II/RPCEC00000369 | It is a safe antibody that could reduce IL-6 and PAI-1 and prevent fibrosis in patients with severe and moderate COVID-19 at high risk of worsening. |
| Treamid | [130] | Patients after COVID-19 pneumonia | Derived from bisamide of dicarboxylic acid; a metal chelator. | II/NCT04527354 | Clinically significant improvement in FVC and/or DLCO was achieved, as well as a decrease in dyspnea. |
| GB0139 | [78] | COVID-19 pneumonitis | Participates in the inhibition of galectin-3. | Ib/IIa/NCT04473053 EurdraCT: 2020002230-32 | It decreases circulating concentrations of galectin-3 and may also reduce inflammation. |
| Collagen-Polyvinylpyrrolidone | [79] | Hyper-inflammation of hospitalized patients with COVID-19 | Downregulates pro-inflammatory cytokines, in addition to several adhesion molecules (ELAM-1, VCAM-1 and ICAM-1), as well as the expression of COX-1 and collagenase. | I/NCT04517162 | Decreases IP-10, IL-8, and M-CSF and shortened the duration of symptoms; in addition, a higher mean oxygen saturation value and a higher proportion of patients retaining oxygen saturation values ≥ 92% were observed. |
| Tranilast | [80] | Severe pneumonia after COVID-19 | Modulates the suppression of the expression and/or action of the TGF-β pathway. | Case report/NA | Reduced PF and improved respiratory function. |
| Pycnogenol-Centellicum | [131] | Post-COVID-19 lung disease | Pycnogenol controls and decreases edema; Centellicum, by modulating the position of collagen, slows irregular scarring, keloid scarring, and fibrosis. | NA/NA | The progression from edema to fibrosis appears to be slower or attenuated with this combination of supplements both in patients with idiopathic IPF and patients with lung disease. |
| Anakinra | [81] | PF and persistent hypoxemia in COVID-19 | Antagonist of the IL-1 receptor. | Case Report/NA | Improved respiratory and clinical parameters, which allowed an early hospital discharge. During follow-up, the evolution was favorable with resolution of fibrosis. |
| Drug | Ref. | Indication | Mechanism of Action | Phase | Results or Conclusions |
|---|---|---|---|---|---|
| Pamrevlumab (FG-3019) | [136] | IPF | Modulates the inhibition of action of connective tissue growth factor (CTGF). | II | Pamrevlumab attenuated the progression of IPF and was well tolerated, reducing the decrease in the predicted FVC percentage by 60; 3% at week 48. |
| EW-7197 | [132] | IPF | Blocks the catalytic activity of ALK5 by competitively blocking ATP from binding to ALK5. | Preclinical | In fibrosis, inhibited TGF-β-/Smad2/3 and ROS signaling. |
| ZSP1603 | [137] | PF | Blocks PDGF-Rβ and ERK. It also inhibited primary human PF differentiation by reducing the expression of TGF-β1, TIMP-1, and COL1A1. | Preclinical | Significantly attenuated lung damage, inflammation, and fibrosis in vivo in an animal model. |
| Piceatannol | [135] | Possible use in PF | An analogue of the polyphenolic compounds of resveratrol that inhibits non-receptor tyrosine kinase-Syk. | Preclinical | In an in vitro model, it modulates fibroblast activation induced by the TGF-β1 pathway and ECM production. |
| Alamandine | [133,138] | PF | A peptide with protective effects on the cardiovascular system, in addition to vasodilator and antifibrotic effects. | Preclinical | Attenuated collagen deposition and improved ventilatory mechanics in PF in a Wistar rat model. |
| Thymol | [139] | PF | Participates in modulating oxidative stress, inflammation, expression of miR-29a/TGF-β, and PI3K/phospho-Akt signaling in PF. | Preclinical | Fibrotic markers were reduced: α-SMA and fibronectin, inflammatory mediators; TNF-α, IL-1β, IL-6, and NF-κB and biomarkers of oxidative stress; MDA, GSH, and SOD. Prevented bleomycin-induced PF. |
| Esomeprazole and pirfenidone | [140] | PF | Esomeprazole has anti-inflammatory and antifibrotic activity.Pirfenidona inhibits TGF-β. | Preclinical | Mixing esomeprazole with pirfenidone improves the antifibrotic efficacy of pirfenidone. |
| AT13387 | [134] | Chronic lung injury and PF | Heat shock protein inhibitor 90. | Preclinical | Treatment with AT13387 15 mg/kg reduced alveolar inflammation, fibrosis, and NLRP3 staining and blocked activation of ERK and HSP90. It also reduces collagen deposition, chronic lung injury, and airway hyper-responsiveness. |
| GSK3335103 | [141] | Fibrotic disease | A novel inhibitor of integrin αvβ6 mimetic of RGD. | In vitro and preclinical | Attenuates TGF-β signaling in vitro and in vivo with a well-defined pharmacokinetic/pharmacodynamic relationship. That is, it reduces collagen deposition in vivo. |
| Diethylcarbamazine | [142] | Antiparasitic and antifibrotic | Immunomodulatory, anti-inflammatory, and antifibrotic activities. | NA | Still without results; however, reduces the production of fibrotic factors and collagen. |
| Title | Status | Conditions | Molecule | Phase | NTC Number (Accessed November 2023) |
|---|---|---|---|---|---|
| Pirfenidone compared to placebo in PF post-COVID-19 | Recruiting | PF post-COVID-19 infection | Pirfenidone | Phase 2 | NCT04607928 |
| Colchicine and PF post-COVID-19 | Active, not recruiting |
| Colchicine | Phase 4 | NCT04818489 |
| Treatment of PF due to COVID-19 with Fuzheng Huayu | Completed | PF due to COVID-19 |
| Phase 2 | NCT04279197 |
| Safety and effectiveness of EV Pure + WJ-Pure Treatment on PF secondary to COVID-19 | Recruiting |
|
| Phase 1 | NCT05387239 |
| Anti-inflammatory and anti-fibrotic drugs in PF post- COVID-19 | Completed |
|
| Not Applicable | NCT05648734 |
| Short term low dose corticosteroids for management of PF post-COVID-19 | Completed | COVID-19 | Drug: 20 Mg Prednisone for 14 days | Not Applicable | NCT04551781 |
| Mineralocorticoid receptor antagonist and PF in COVID-19 | Recruiting | COVID-19 | Drug: Canrenoate potassium | Phase 4 | NCT04912011 |
| Efficacy and safety of nintedanib in the treatment of PF in patients with moderate to severe COVID-19 | Unknown status | COVID-19 | Nintedanib | Phase 2 | NCT04338802 |
| Detection of integrin avb6 in IPF, PSC, and COVID-19 using PET/CT | Recruiting |
| Drug: FP-R01-MG-F2 | Early Phase 1 | NCT03183570 |
| Intramuscular effect of polymerized type I collagen on the cytokine storm in COVID-19 patients | Recruiting |
| Collagen-Polyvinylpyrrolidone |
| NCT04517162 |
| Assessing the efficacy of sirolimus in patients with COVID-19 pneumonia for prevention of post-COVID fibrosis | Recruiting |
| Drug: Sirolimus |
| NCT04948203 |
| Study to assess efficacy and safety of treamid for patients with reduced exercise tolerance after COVID-19 | Not yet recruiting |
| Drug: Treamid |
| NCT05516550 |
| The MONACO cell therapy study: monocytes as an antifibrotic treatment after COVID-19 | Recruiting |
| Biological: MON002 |
| NCT04805086 |
| Study of longidaze in the prevention and treatment of PF, interstitial lung disease caused by COVID-19 | Recruiting | PF | Drug: Bovhyaluronidase azoxymer | Not Applicable | NCT04645368 |
| Safety and effectiveness of cyclosporin in the management of COVID-19 ARDS patients in Alexandria University Hospital | Not yet recruiting |
| Drug: Cyclosporine | Phase 3 | NCT04979884 |
| BIO 300 oral suspension in previously hospitalized long COVID-19 patients | Recruiting |
| Drug: BIO 300 Oral Suspension | Phase 2 | NCT04482595 |
| The study of the use of nintedanib in slowing lung disease in patients with fibrotic or non-fibrotic interstitial lung disease related to COVID-19 | Recruiting |
| Drug: Nintedanib | Phase 4 | NCT04619680 |
| Pirfenidone vs. nintedanib for fibrotic lung disease after coronavirus disease-19 pneumonia | Active, not recruiting | Novel coronavirus-induced lung fibrosis |
| Phase 4 | NCT04856111 |
| Pilot study to assess efficacy and safety of Treamid in the rehabilitation of patients after COVID-19 pneumonia | Completed |
| Drug: Treamid | Phase 2 | NCT04527354 |
| Use of cSVF via IV deployment for residual lung damage after symptomatic COVID-19 infection | Enrolling by invitation |
|
| Early Phase 1 | NCT04326036 |
| APX-115 use in hospitalized patients with confirmed mild to moderate COVID-19 | Recruiting | COVID-19 | Drug: APX-115 | Phase 2 | NCT04880109 |
| Biomarker and Matrix | Ref. | Biological Function | Type of Biomarker | Findings |
|---|---|---|---|---|
| sITGaM and sITGb2 Samples of serum | [148] | Integrins participate in the inflammatory response by contributing to the anchoring of various cells, such as leukocytes, to the endothelium, thus allowing their diapedesis. | Predictor Therapeutic | Elevation of soluble integrin subunits was reported in the (+) group; therefore, they may be biomarkers for predicting pulmonary complications and, thus, a potential therapeutic target in post-COVID-19 patients. R = 0.42, p = 0.01. |
| IL13RA2, CDH3 and COMP Lung tissue | [149] | IL-13Rα2: acts as a non-signaling decoy receptor. CDH3: a classic cell-to-cell adhesion molecule; regulates multiple cellular homeostatic processes in normal tissue. COMP: an ECM glycoprotein; participates in fibrillogenesis and collagen secretion. | Diagnostic | Elevation of IL13RA2, CDH3, and COMP could serve as a diagnostic signature for IPF and could offer new insights into the underlying diagnosis of IPF. The area under the curve reported for the three-gene group was 0.98. |
| Endothelin-1 (ET-1). Samples of serum | [150] | A molecule produced by the vascular endothelium, which is involved in the homeostasis of vascular tone. | Diagnostic | Serum levels of ET-1 were found to be elevated, so it may be useful as a biomarker of PID, but it could not help in the differential diagnosis between IPF and ILD associated with autoimmune diseases (AD-ILD). In addition, ET-1 levels may be associated with the severity of ILD. The area under the curve reported was 0.803 (95% CI: 0.728–0.878). |
| Calcium binding protein S100 A12. Lung tissue, Blood and Bronchoalveolar lavage. | [151] | S100A12 is involved in the adhesion and migration of leukocytes and in the production of cytokines and chemokines. | Prognosis/Severity | Down-expression of S100 A12 and the composite variable may be a more effective predictive index. |
| CYFRA 21-1 Samples of serum. | [152,153] | Keratins are part of the cytoskeleton of epithelial cells. Cytokeratin-19 is expressed by airway epithelial cells. | Diagnostic and Prognosis Therapeutic | It predicted short-term progression and long-term survival when assessed cross-sectionally and at serial time points, notably beyond 3 months. Because it is a marker of epithelial damage and turnover, it may have potential utility as a prognostic and therapeutic biomarker in people with IPF. CYFRA 21-1 demonstrated an association with overall mortality in both the discovery (HR, 1.13 [95% CI, 0.02–1.25]; p = 0.023) and validation cohorts (HR = 1.12 [95% CI, 0.02–1.25]; p = 0.023) 1.06–1.19]; p = 0.0001). |
| RBM11, RIC3, TRAF5, ZNF14 and RBM47. Related genes with m6A. Peripheral blood | [154] | TRAF5: an important signal transducer for a wide range of TNF receptor superfamily members, and it mainly mediates the activation of NF-κB pathway. RBM47: involved in EMT. RIC3: a chaperone protein. RBM11: involved in cellular response to oxidative stress and regulation of alternative splicing. ZNF14: has a zinc finger and a Kruppel-associated box (KRAB) domain. This domain is involved in the transcriptional repression of several zinc finger proteins. | Prognosis | Survival analysis showed that high expression of RBM11, RIC3, TRAF5, and ZNF14 was associated with a good prognosis of IPF, while high expression of RBM47 was associated with a poor prognosis. The AUC value in the first year was low (AUC at 1 year = 0.63), and the AUC value gradually increased with time (AUC at 2 years = 0.77, AUC at 3 years = 0.85, AUC at 4 years = 0.95); this shows the prediction utility. |
| CRTAC1, COL10A1, COMP, IGFL2, NECAB1, SCG5, SLC6A4 and SPP1. Lung tissue | [154] | COMP: a non-collagenous glycoprotein component of extracellular matrix (ECM) that accentuates TGF-β1 signaling and is associated with extracellular matrix polymerization and stiffness. SPP1: a protein formerly related to PF and COPD in lung process in mice. SLC6A4: a serotonin transporter gene. COL10A1: members of the collagen family. CRTAC1: regarded as an opponent of nogo receptor-1. IGFL2, NECAB1, SCG5: found to play a role in PF. | Diagnostic | These genes were expressed at high levels and associated with monocytes, plasma cells, neutrophils, and regulatory T cells (T reg), suggesting their use as diagnostic biomarkers of IPF. Their diagnostic ability showed an advantageous diagnostic value, with an AUC of 0.943 (95% CI 0.883–0.986) in CRTAC1, AUC of 0.886 (95% CI 0.778–0.970) in COL10A1, AUC of 0.984 (95% CI 0.956–1.000) in COMP, AUC of 0.633 (95% CI 0.469–0.782) in RPS4Y1, AUC of 0.936 (95% CI 0.873–0.980) in IGFL2, AUC of 0.925 (95% CI 0.841–0.987) in NECAB1, AUC of 0.967 (95% CI 0.923–0.995) in SCG5, AUC of 0.763 (95% CI 0.625–0.882) in SLC6A4, and AUC of 0.886 (95% CI 0.790–0.957) in SPP1. |
| B4-galactosyltransferase (B4GALT1). Lung tissue | [155] | B4GALTs are involved in the expression of biologically active carbohydrates through glycosylated glycans. | Diagnostic | High levels of B4GALT1, both in mRNA and protein, were found in 4 patients with IPF and in the primary human cells derived from IPF. A positive correlation was found between B4GALT and genes belonging to the EMT pathway (p = 0.01). |
| IL1R2, S100A12 and CCL8. Peripheral blood | [156] | IL1R2: through competitive binding with IL-1β, prevents it from binding to IL1R1, blocking the signal transduction of IL-1β in inflammatory diseases and acting as a bait receptor. S100A12: has a proinflammatory cytokine function; is related to the fibrotic process of skin scars. CCL8: expressed by monocytes/macrophages in inflammatory tissues, stimulated by T cells and accessory pathways of IFN and other pro-inflammatory cytokines or by innate mechanisms. | Prognosis | Increased expression of IL1R2, S100A12, and CCL8 could predict survival at 1, 2, and 3 years. The AUCs reported for predicting 1-, 2-, and 3-year survival were greater than 0.7. |
| sST2 Serum | [157] | ST2: belongs to the interleukin-1 (IL-1) receptor family, which exists in transmembrane (ST2L) and soluble (sST2) isoforms. | Prognosis | High levels of serum sST2 have been found in patients with PF, and this level helped predict greater deterioration and poorer outcomes in these patients. The overexpression of sST2 increased the hazard ratio to 1.005 (95% CI: 1.001–1.010). |
| Growth differentiation factor-15 (GDF-15). Serum | [158] | Also known as macrophage inhibitory cytokine-1 (MIC-1) and is a member of the TGF-β family. | Diagnostic and Prognosis | Elevated expression of GDF-15 could be a promising biomarker for the occurrence and survival of acute exacerbations (AEs) in patients with IPF. The areas under the ROC curves of serum GDF-15 levels in patients with AE-IPF or S-IPF showed significance (ROC: 0.738, p < 0.001, 95% CI: 0.529–0.809, cut-off value 989.3 pg/mL). |
| m5CPS Bronchoalveolar lavage | [159] | Affects a series of biological functions, such as RNA stabilization and translation. | Prediction and Prognosis | Expression of m5CPS was positively associated with active mast cell infiltration levels in all training, testing, and validation cohorts. Therefore, the results suggest important roles of mast cells in IPF. In terms of the results, m5CPS could predict the 1-, 3-, and 5-year survival rates of IPF patients with high accuracy (AUC = 0.803–0.973). |
| Human epididymis protein 4 (HE4). Serum | [160] | The epididymis-specific protein can be found in other tissues, such as the respiratory tract, and its main function is to inhibit the activity of several proteases, such as MMP2 and MMP9, which contribute to the progression of fibrosis. | Severity and prognosis | High levels of HE4 were found in patients with IPF, especially in patients with AE-IPF, and in statistical analysis, serum levels of HE4 [hazard ratio (HR) = 1.004, p = 0.042] and GAP index (HR = 1.374, p = 0.010) were associated with worse survival in patients with IPF. |
| TPST1, MRVI1 and TM4SF1 Bronchoalveolar lavage | [161] | TPST1: a member of a family of sulfotransferase proteins, involved in the sulfidation of tyrosine residues. MRVI1: the cGMP kinase substrate associated with the inositol 1,4,5-trisphosphate receptor; it participates in the regulation of intracellular calcium induced by IP3 through a NO/PRKG1-dependent mechanism. TM4SF1: a tumor-associated protein that is widely expressed in multiple human cancers, is localized at the surface of the cell membrane and late endocytic organelles, and plays a vital role in cell motility. | Prognosis | The discharge expression of these biomarkers showed that inflammation and immunological processes significantly affected the prognosis of IPF patients. The areas under the curve reported for 1-, 2-, and 3-year survival rates were 0.862, 0.885, and 0.833 (TPST1, MRVI1, and TM4SF1), respectively. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Favila, A.; Garza-Veloz, I.; Hernandez-Marquez, L.d.S.; Gutierrez-Vela, E.F.; Flores-Morales, V.; Martinez-Fierro, M.L. Antifibrotic Drugs against Idiopathic Pulmonary Fibrosis and Pulmonary Fibrosis Induced by COVID-19: Therapeutic Approaches and Potential Diagnostic Biomarkers. Int. J. Mol. Sci. 2024, 25, 1562. https://doi.org/10.3390/ijms25031562
Perez-Favila A, Garza-Veloz I, Hernandez-Marquez LdS, Gutierrez-Vela EF, Flores-Morales V, Martinez-Fierro ML. Antifibrotic Drugs against Idiopathic Pulmonary Fibrosis and Pulmonary Fibrosis Induced by COVID-19: Therapeutic Approaches and Potential Diagnostic Biomarkers. International Journal of Molecular Sciences. 2024; 25(3):1562. https://doi.org/10.3390/ijms25031562
Chicago/Turabian StylePerez-Favila, Aurelio, Idalia Garza-Veloz, Lucia del Socorro Hernandez-Marquez, Edgar Fernando Gutierrez-Vela, Virginia Flores-Morales, and Margarita L. Martinez-Fierro. 2024. "Antifibrotic Drugs against Idiopathic Pulmonary Fibrosis and Pulmonary Fibrosis Induced by COVID-19: Therapeutic Approaches and Potential Diagnostic Biomarkers" International Journal of Molecular Sciences 25, no. 3: 1562. https://doi.org/10.3390/ijms25031562
APA StylePerez-Favila, A., Garza-Veloz, I., Hernandez-Marquez, L. d. S., Gutierrez-Vela, E. F., Flores-Morales, V., & Martinez-Fierro, M. L. (2024). Antifibrotic Drugs against Idiopathic Pulmonary Fibrosis and Pulmonary Fibrosis Induced by COVID-19: Therapeutic Approaches and Potential Diagnostic Biomarkers. International Journal of Molecular Sciences, 25(3), 1562. https://doi.org/10.3390/ijms25031562