
Sources of Variance in Human Tear Proteomic Samples: Statistical Evaluation, Quality Control, Normalization, and Biological Insight


Abstract
1. Introduction
2. Results and Discussion
2.1. Ophthalmological Examination
2.2. Global Parameters of Tear Samples
2.3. Proteomics Results
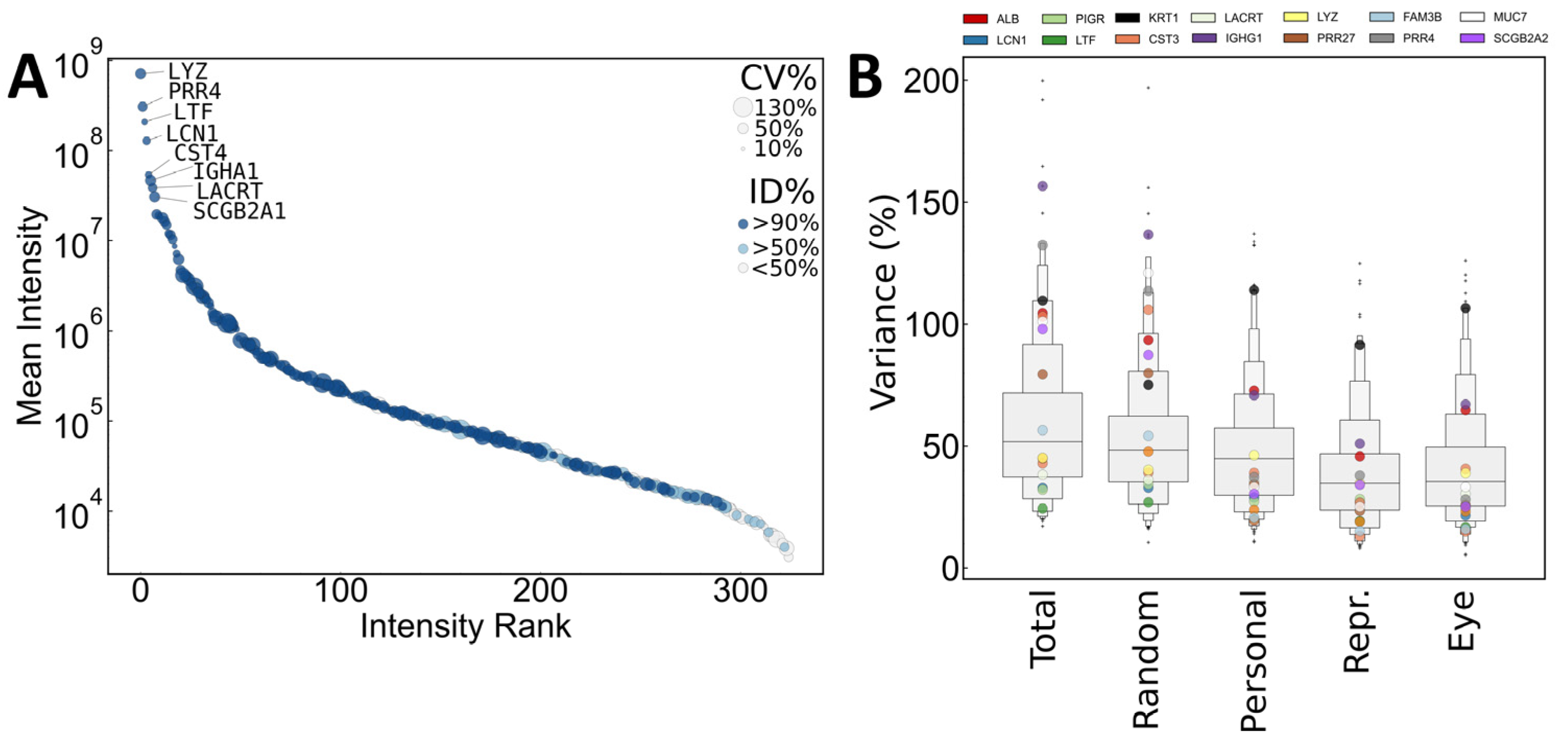
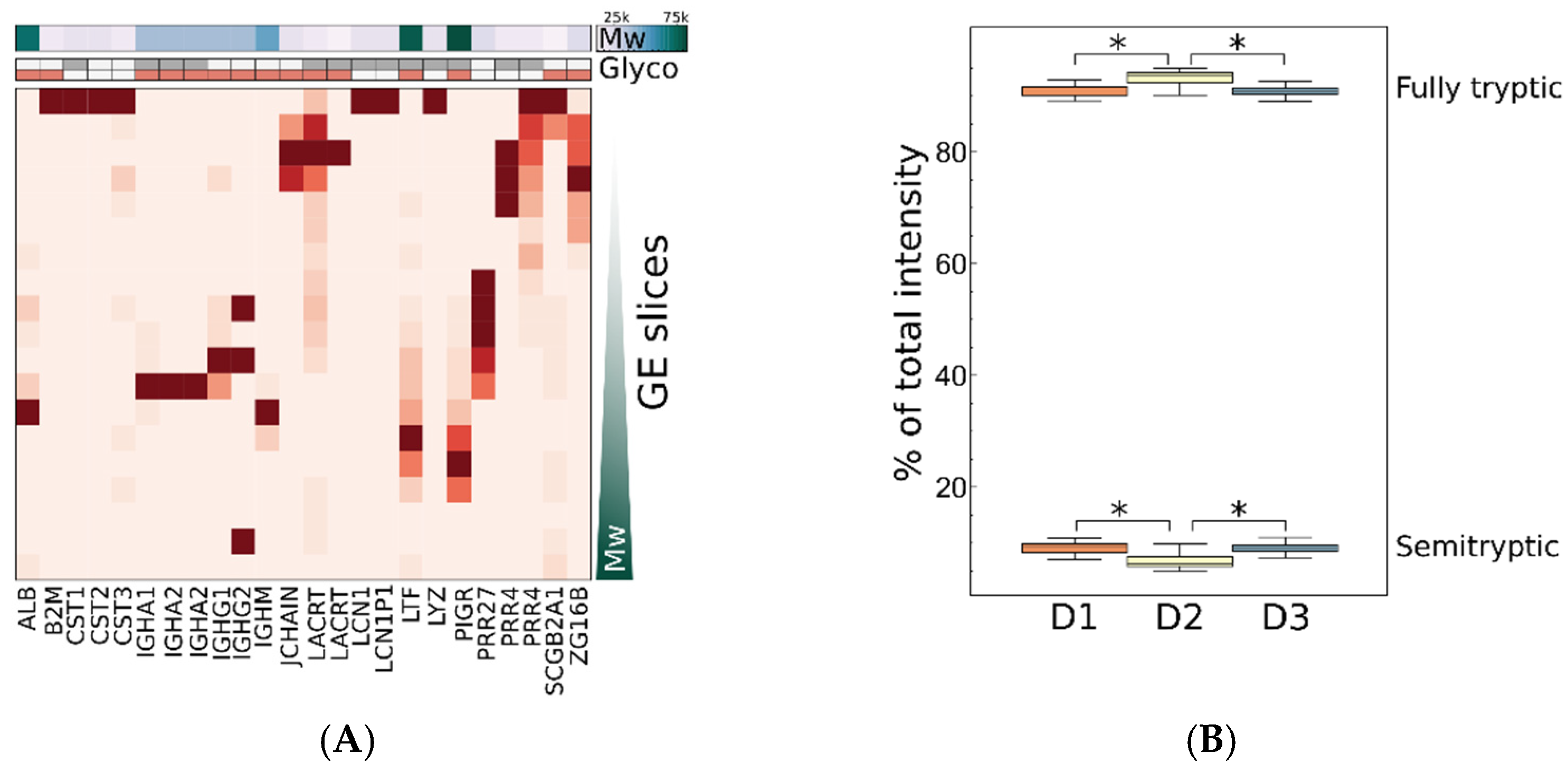
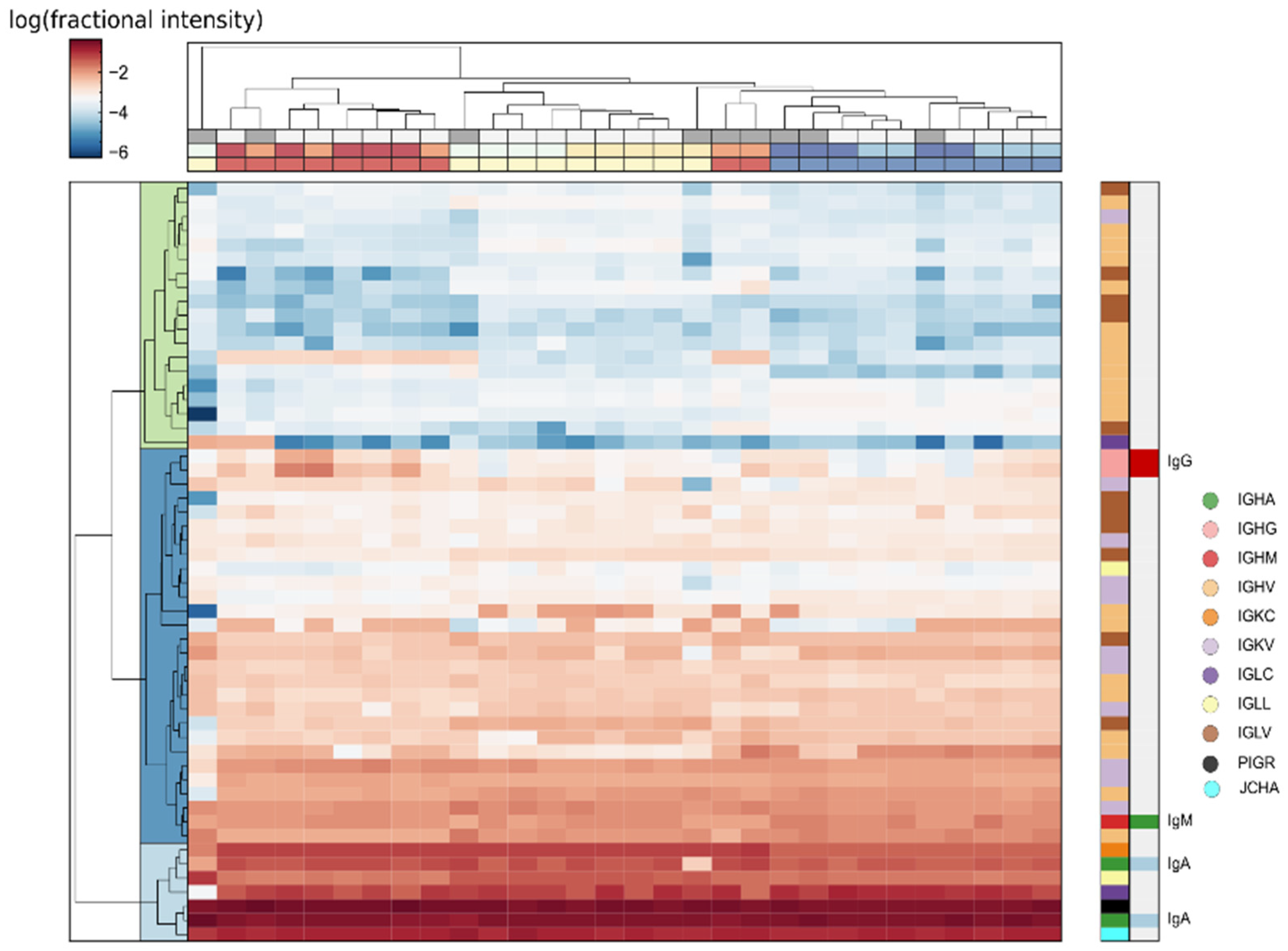
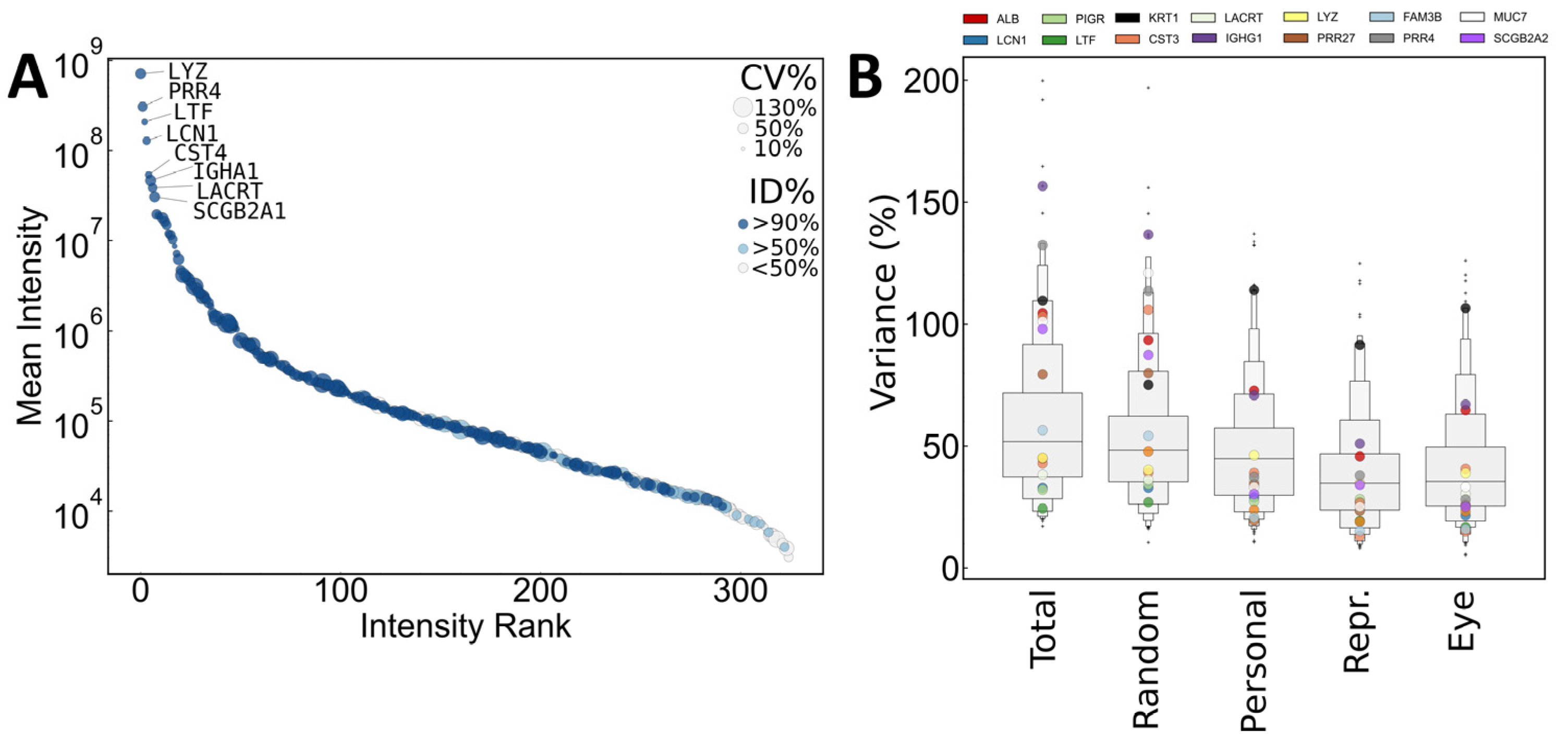
2.3.1. Composition of the Identified Tear Proteome
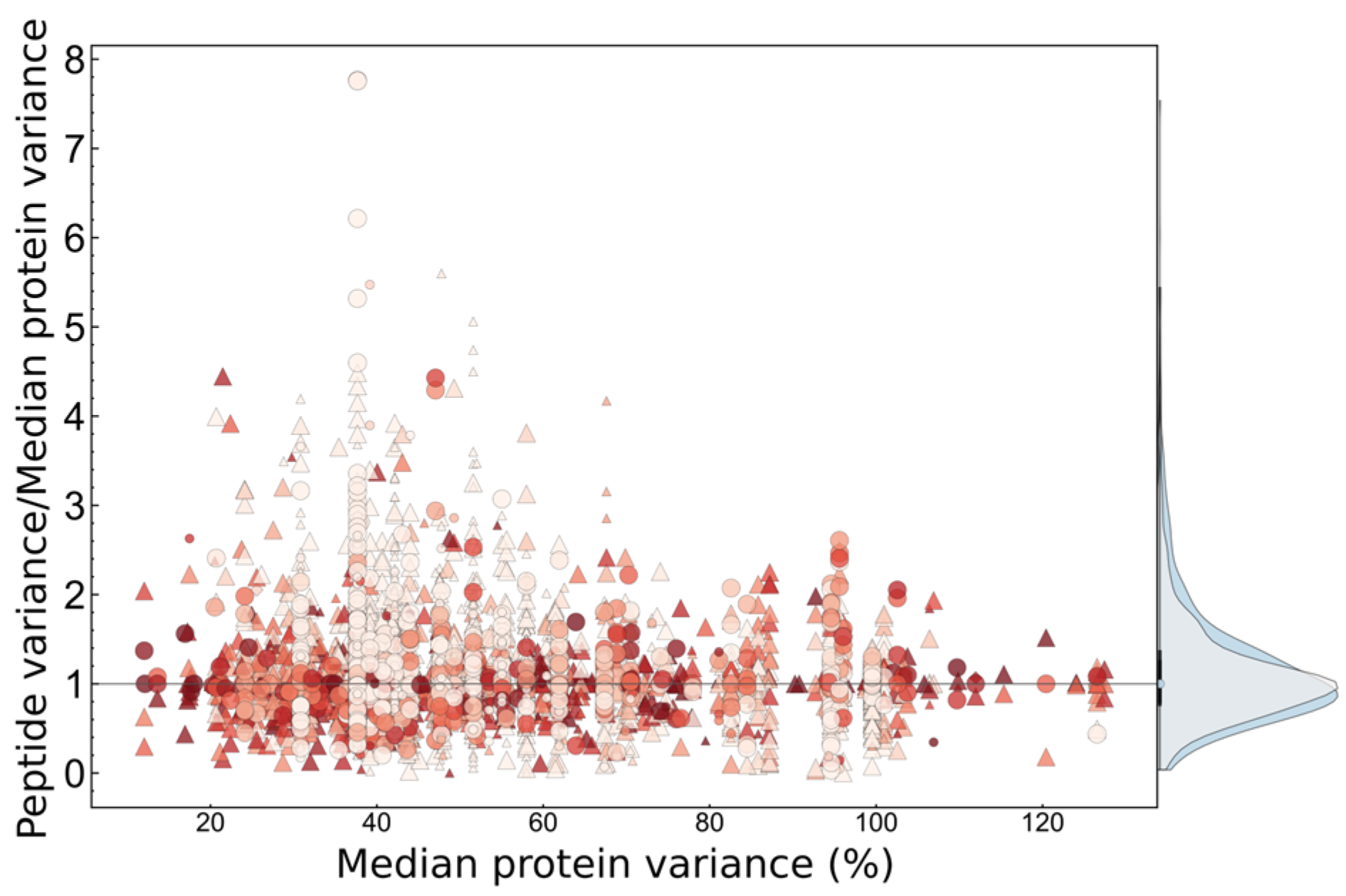
2.3.2. Quantitative Variance in the Composition of the Human Tear Proteome
2.3.3. Methods to Decrease Variance: Normalization and Selection of Representative Samples
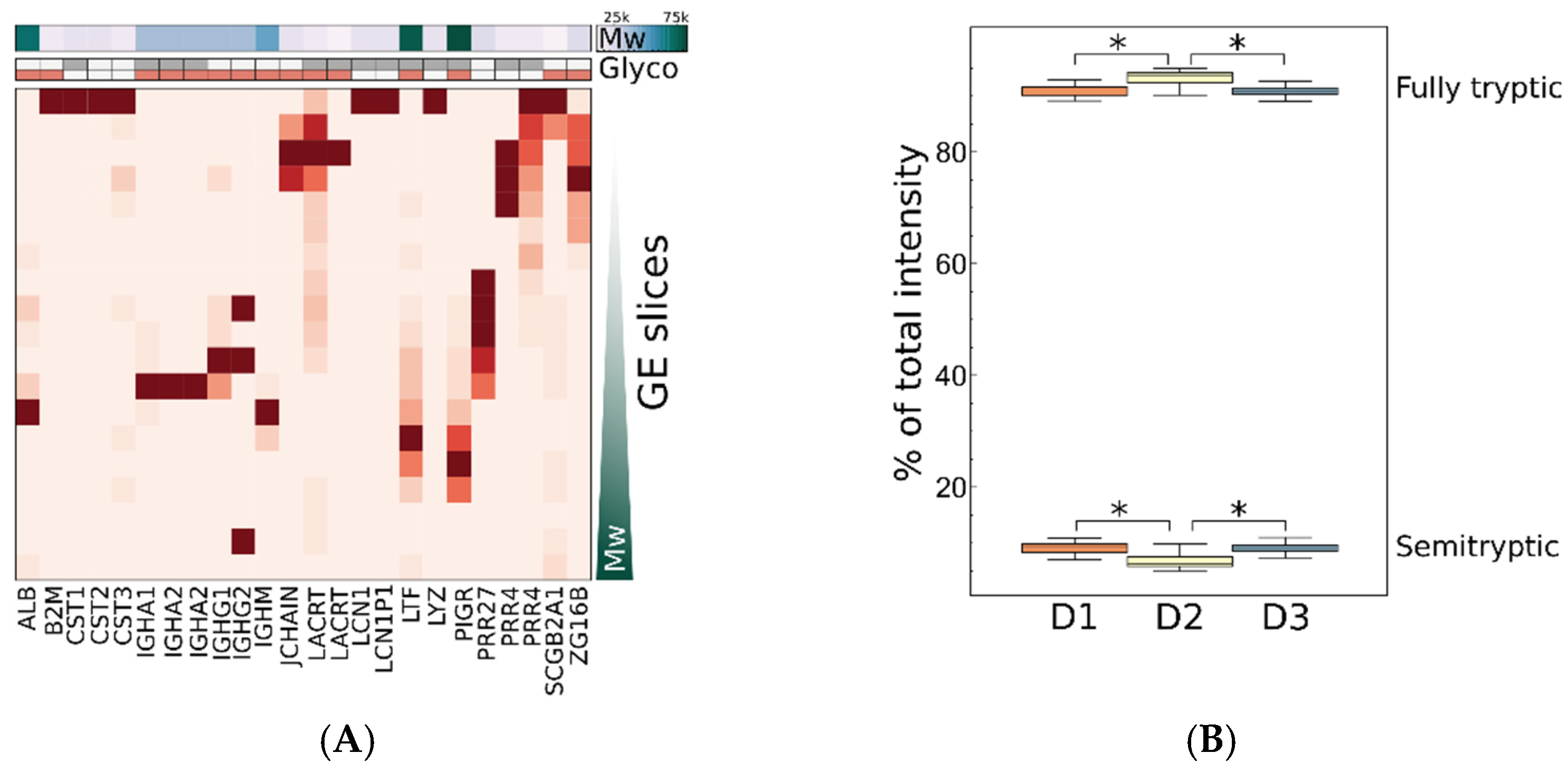
2.3.4. Contribution of Proteoforms to Observed Variance
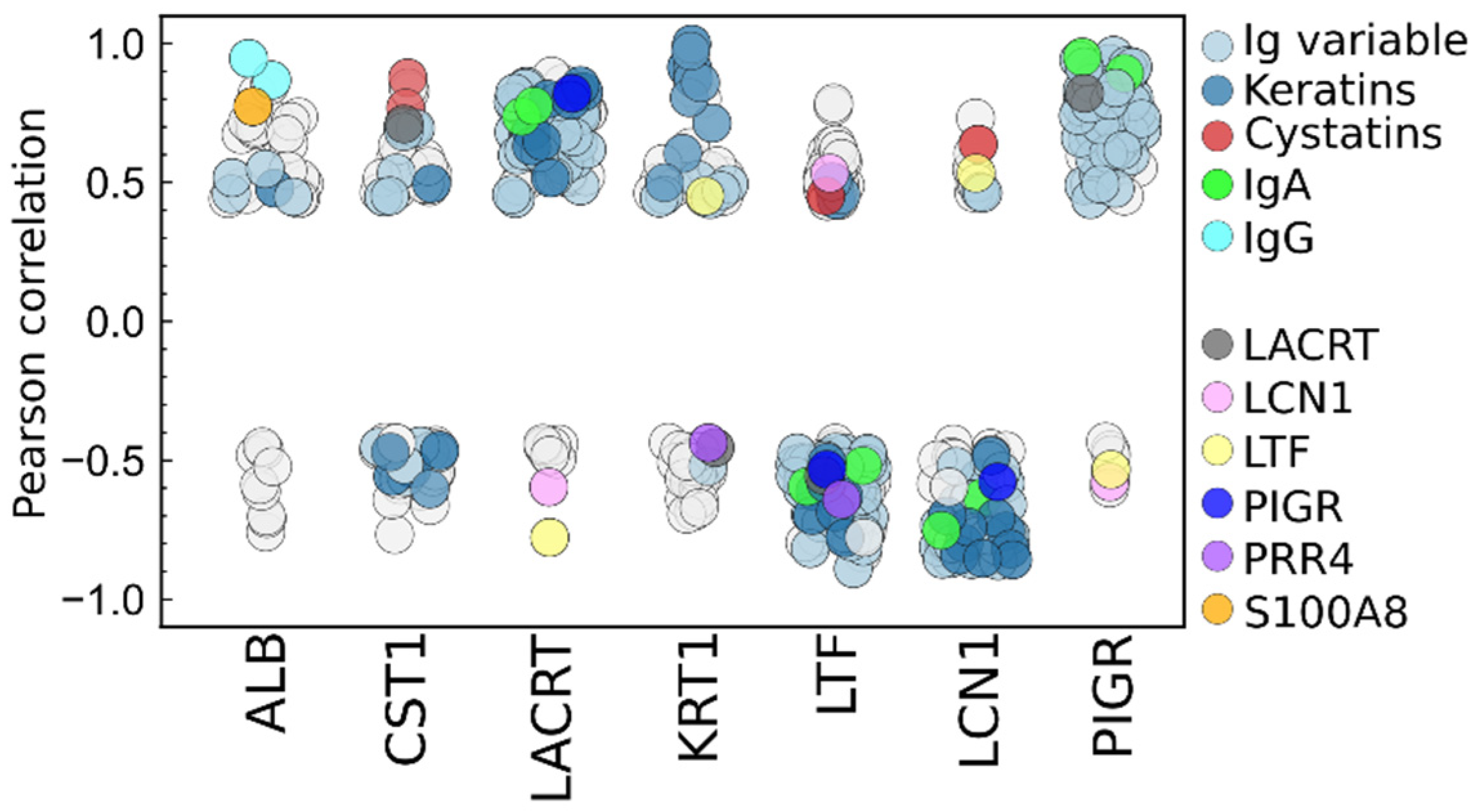
2.3.5. Statistical Determination of Sources of Biological Variance
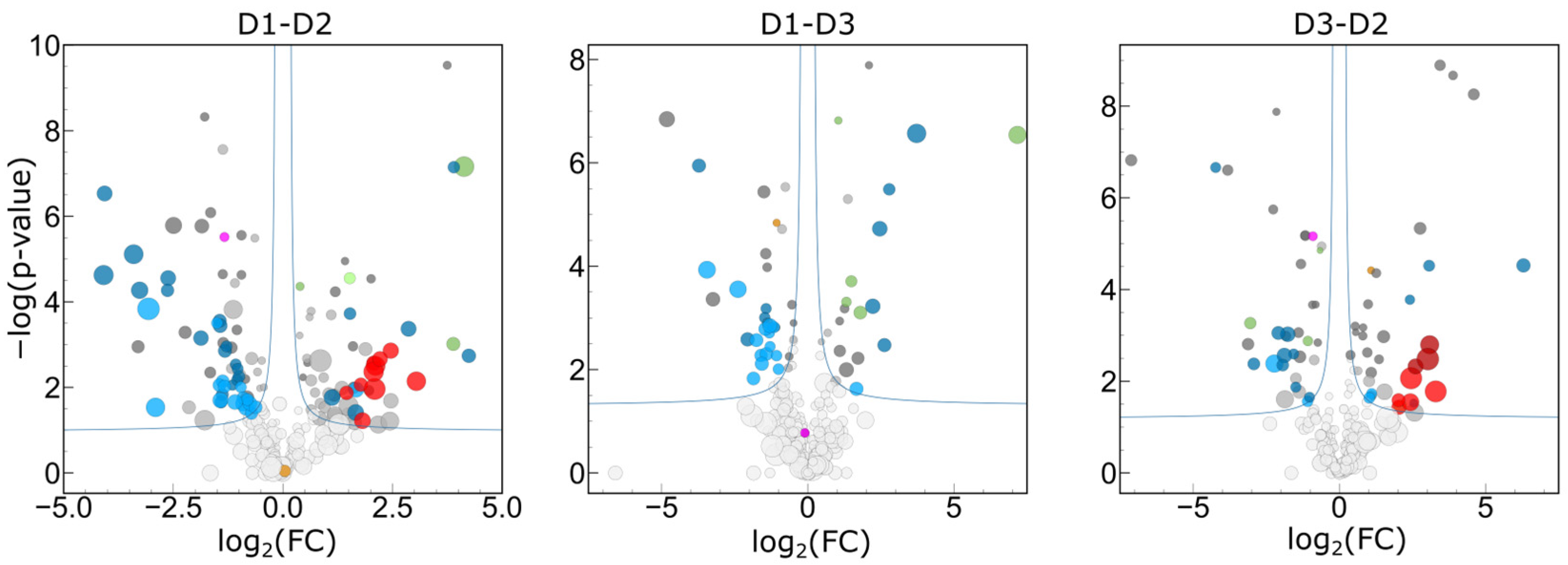
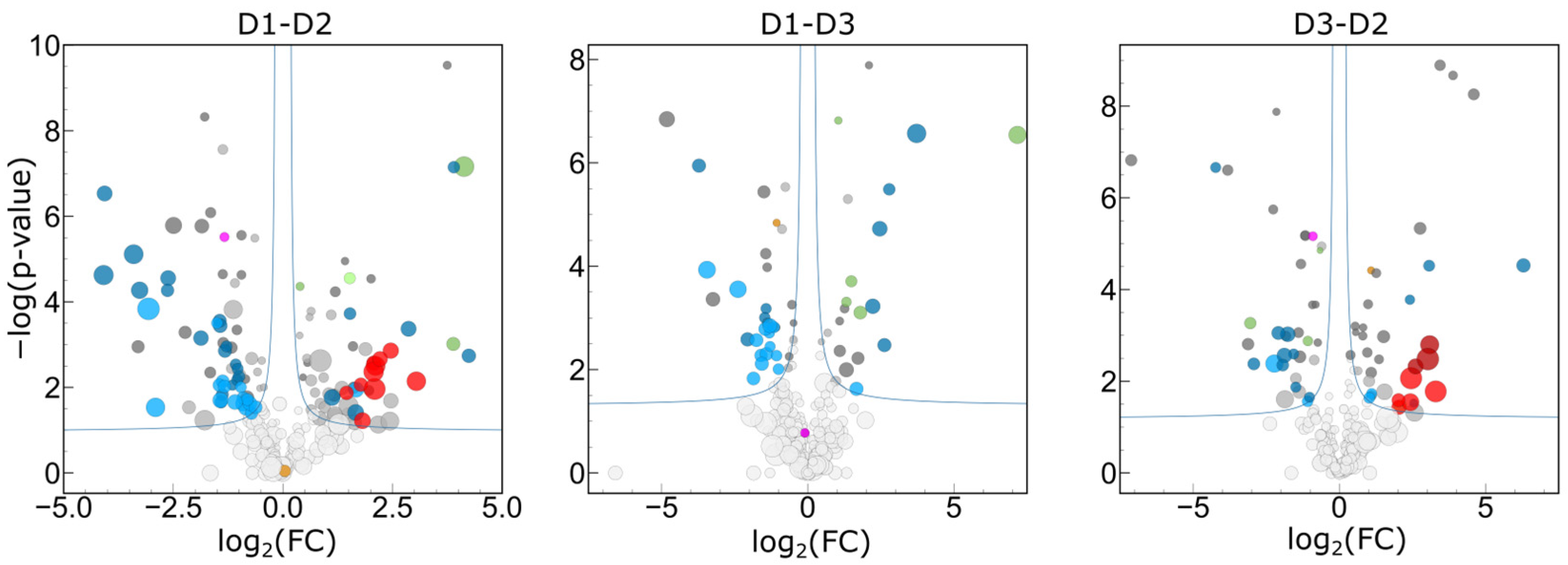
2.3.6. Interpersonal Comparisons Based on Replicate Personal Samples
3. Materials and Methods
3.1. Participants
3.2. Ophthalmological Examination
3.3. Sample Collection
3.4. Sample Processing
3.5. LC–MS Analysis
3.6. Data Analysis
3.7. Materials/Reagents
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGC | Automatic gain control |
| ALBU | Albumin |
| ANOVA | analysis of variance |
| BUT | break up time |
| CIS | clinically isolated syndrome |
| CV | coefficient of variation |
| D | Donor |
| DDA | data dependent acquisition |
| DIA | data independent acquisition |
| DTT | dithothreitol |
| EyeOME | The Human Eye Proteome Project |
| FDR | false discovery rate |
| HCD | Higher-energy collisional dissociation |
| IAA | iodoacetamide |
| Ig | immunoglobulin |
| LC/MS | Liquid chromatography–mass spectrometry |
| m/z | mass/charge |
| M | mol/L |
| MS/MS | tandem mass spectrometry |
| MS | mass spectrometry |
| NH4HCO3 | ammonium-hydorgencarbonate |
| OD | right eye (oculus dexter) |
| OS | left eye (oculus sinister) |
| Pers | personal |
| PIGR | Polymeric immunoglobulin receptor |
| SDS-PAGE | sodium dodecyl sulphate–polyacrylamide gel electrophoresis |
| Tot | total |
| TPC | total protein concentration |
| TRFL | lactotransferrin |
| UPLC | Ultra Performance Liquid Chromatography |
| v/v% | volume/volume % |
| Vol | volume |
References
- McDermott, A.M. Antimicrobial compounds in tears. Exp. Eye Res. 2013, 117, 53–61. [Google Scholar] [CrossRef]
- Holly, F.J.; Lemp, M.A. Tear physiology and dry eyes. Surv. Ophthalmol. 1977, 22, 69–87. [Google Scholar] [CrossRef]
- Rentka, A.; Koroskenyi, K.; Harsfalvi, J.; Szekanecz, Z.; Szucs, G.; Szodoray, P.; Kemeny-Beke, A. Evaluation of commonly used tear sampling methods and their relevance in subsequent biochemical analysis. Ann. Clin. Biochem. 2017, 54, 521–529. [Google Scholar] [CrossRef]
- Csősz, É.; Kalló, G.; Márkus, B.; Deák, E.; Csutak, A.; Tőzsér, J. Quantitative body fluid proteomics in medicine—A focus on minimal invasiveness. J. Proteom. 2017, 153, 30–43. [Google Scholar] [CrossRef]
- Hagan, S.; Enríquez-de-Salamanca, M.E. Tear fluid biomarkers in ocular and systemic disease: Potential use for predictive, preventive and personalised medicine. EPMA J. 2016, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Beuerman, R.W.; Chan, C.M.; Zhao, S.Z.; Li, X.R.; Yang, H.; Tong, L.; Liu, S.; Stern, M.E.; Tan, D. Identification of tear fluid biomarkers in dry eye syndrome using iTRAQ quantitative proteomics. J. Proteome Res. 2009, 8, 4889–4905. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, X.; Chen, Z.; Huang, Q.; Wu, K. Quantification of tear proteins and sPLA2-IIa alteration in patients with allergic conjunctivitis. Mol. Vis. 2010, 16, 2084–2091. [Google Scholar] [PubMed]
- Hida, R.Y.; Ohashi, Y.; Takano, Y.; Dogru, M.; Goto, E.; Fujishima, H.; Saito, I.; Saito, K.; Fukase, Y.; Tsubota, K. Elevated levels of human alpha-defensin in tears of patients with allergic conjunctival disease complicated by corneal lesions: Detection by SELDI ProteinChip system and quantification. Curr. Eye Res. 2005, 30, 723–730. [Google Scholar] [CrossRef]
- Koo, B.S.; Lee, D.Y.; Ha, H.S.; Kim, J.C.; Kim, C.W. Comparative analysis of the tear protein expression in blepharitis patients using two-dimensional electrophoresis. J. Proteome Res. 2005, 4, 719–724. [Google Scholar] [CrossRef]
- Acera, A.; Suarez, T.; Rodriguez-Agirretxe, I.; Vecino, E.; Duran, J.A. Changes in tear protein profile in patients with conjunctivochalasis. Cornea 2011, 30, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Evans, V.; Vockler, C.; Friedlander, M.; Walsh, B.; Willcox, M.D. Lacryglobin in human tears, a potential marker for cancer. Clin. Exp. Ophthalmol. 2001, 29, 161–163. [Google Scholar] [CrossRef]
- Kalló, G.; Emri, M.; Varga, Z.; Ujhelyi, B.; Tőzsér, J.; Csutak, A.; Csősz, É. Changes in the chemical barrier composition of tears in Alzheimer’s disease reveal potential tear diagnostic biomarkers. PLoS ONE 2016, 11, e0158000. [Google Scholar] [CrossRef]
- Markoulli, M.; Papas, E.; Petznick, A.; Holden, B. Validation of the flush method as an alternative to basal or reflex tear collection. Curr. Eye Res. 2011, 36, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Fullard, R.J.; Snyder, C. Protein levels in nonstimulated and stimulated tears of normal human subjects. Investig. Ophthalmol. Vis. Sci. 1990, 31, 1119–1126. [Google Scholar]
- Perumal, N.; Funke, S.; Wolters, D.; Pfeiffer, N.; Grus, F.H. Characterization of human reflex tear proteome reveals high expression of lacrimal proline-rich protein 4 (PRR4). Proteomics 2015, 15, 3370–3381. [Google Scholar] [CrossRef]
- Posa, A.; Brauer, L.; Schicht, M.; Garreis, F.; Beileke, S.; Paulsen, F. Schirmer strip vs. capillary tube method: Non-invasive methods of obtaining proteins from tear fluid. Ann. Anat. 2013, 195, 137–142. [Google Scholar] [CrossRef]
- Nättinen, J.; Aapola, U.; Jylhä, A.; Vaajanen, A.; Uusitalo, H. Comparison of capillary and schirmer strip tear fluid sampling methods using swath-ms proteomics approach. Transl. Vis. Sci. Technol. 2020, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- de Souza, G.A.; Godoy, L.M.; Mann, M. Identification of 491 proteins in the tear fluid proteome reveals a large number of proteases and protease inhibitors. Genome Biol. 2006, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Aass, C.; Norheim, I.; Eriksen, E.F.; Thorsby, P.M.; Pepaj, M. Single unit filter-aided method for fast proteomic analysis of tear fluid. Anal. Biochem. 2015, 480, 1–5. [Google Scholar] [CrossRef]
- Runström, G.; Mann, A.; Tighe, B. The fall and rise of tear albumin levels: A multifactorial phenomenon. Ocul. Surf. 2013, 11, 165–180. [Google Scholar] [CrossRef]
- Ahmad, M.T.; Zhang, P.; Dufresne, C.; Ferrucci, L.; Semba, R.D. The Human Eye Proteome Project: Updates on an emerging proteome. Proteomics 2018, 18, e1700394. [Google Scholar] [CrossRef]
- Markoulli, M.; Gokhale, M.; You, J. Substance P in flush tears and Schirmer strips of healthy participants. Optom. Vis. Sci. 2017, 94, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Markoulli, M.; Papas, E.; Cole, N.; Holden, B.A. The diurnal variation of matrix metalloproteinase-9 and its associated factors in human tears. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-F.; Zhang, Y.; Rittenhouse, K.D.; Pickering, E.H.; McDowell, M.T. Evaluations of tear protein markers in dry eye disease: Repeatability of measurement and correlation with disease. Investig. Opthalmol. Vis. Sci. 2012, 53, 4564. [Google Scholar] [CrossRef] [PubMed]
- González, N.; Iloro, I.; Durán, J.A.; Elortza, F.; Suárez, T. Evaluation of inter-day and inter-individual variability of tear peptide/protein profiles by MALDI-TOF MS analyses. Mol. Vis. 2012, 18, 1572–1582. [Google Scholar] [PubMed]
- Dammeier, S.; Martus, P.; Klose, F.; Seid, M.; Bosch, D.; D‘Alvise, J.; Ziemssen, F.; Dimopoulos, S.; Ueffing, M. Combined targeted analysis of metabolites and proteins in tear fluid with regard to clinical applications. Transl. Vis. Sci. Technol. 2018, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Ponzini, E.; Ami, D.; Duse, A.; Santambrogio, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Pezzoli, F.; Natalello, A.; Tavazzi, S.; et al. Single-Tear Proteomics: A Feasible Approach to Precision Medicine. Int. J. Mol. Sci. 2021, 22, 10750. [Google Scholar] [CrossRef]
- Lépine, M.; Zambito, O.; Sleno, L. Targeted Workflow Investigating Variations in the Tear Proteome by Liquid Chromatography Tandem Mass Spectrometry. ACS Omega 2023, 8, 31168–31177. [Google Scholar] [CrossRef]
- Bachhuber, F.; Huss, A.; Senel, M.; Tumani, H. Diagnostic biomarkers in tear fluid: From sampling to preanalytical processing. Sci. Rep. 2021, 11, 10064. [Google Scholar] [CrossRef]
- Zhan, X.; Li, J.; Guo, Y.; Golubnitschaja, O. Mass spectrometry analysis of human tear fluid biomarkers specific for ocular and systemic diseases in the context of 3P medicine. EPMA J. 2019, 12, 449–475. [Google Scholar] [CrossRef]
- Hsiao, Y.-C.; Chu, L.J.; Chen, Y.-T.; Chi, L.-M.; Chien, K.-Y.; Chiang, W.-F.; Chang, Y.-T.; Chen, S.-F.; Wang, W.-S.; Chuang, Y.-N.; et al. Variability assessment of 90 salivary proteins in intraday and interday samples from healthy donors by multiple reaction monitoring-mass spectrometry. Proteom. Clin. Appl. 2018, 12, 1700039. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhao, S.Z.; Koh, S.K.; Chen, L.; Vaz, C.; Tanavde, V.; Li, X.R.; Beuerman, R.W. In-depth analysis of the human tear proteome. J. Proteom. 2012, 75, 3877–3885. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Zhou, X.Y.; Jylha, A.; Aapola, U.; Liu, D.N.; Koh, S.K.; Tian, D.; Quah, J.; Uusitalo, H.; Beuerman, R.W.; et al. Quantitation of 47 human tear proteins using high resolution multiple reaction monitoring (HR-MRM) based-mass spectrometry. J. Proteom. 2015, 115, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Kulak, N.A.; Pichler, G.; Holdt, L.M.; Teupser, D.; Mann, M. Plasma proteome profiling to assess human health and disease. Cell Syst. 2016, 2, 185–195. [Google Scholar] [CrossRef]
- Ben-Aryeh, H.; Fisher, M.; Szargel, R.; Laufer, D. Composition of whole unstimulated saliva of healthy children: Changes with age. Arch. Oral Biol. 1990, 35, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Willforss, J.; Chawade, A.; Levander, F. NormalyzerDE: Online Tool for Improved Normalization of Omics Expression Data and High-Sensitivity Differential Expression Analysis. J. Proteome Res. 2019, 18, 732–740. [Google Scholar] [CrossRef]
- Willcox, M.D.P.; Argüeso, P.; Georgiev, G.A.; Holopainen, J.M.; Laurie, G.W.; Millar, T.J.; Papas, E.B.; Rolland, J.P.; Schmidt, T.A.; Stahl, U.; et al. TFOS DEWS II Tear Film Report. Ocul. Surf. 2017, 15, 366–403. [Google Scholar] [CrossRef]
- Koopmans, F.; Li, K.W.; Klaassen, R.V.; Smit, A.B. MS-DAP Platform for Downstream Data Analysis of Label-Free Proteomics Uncovers Optimal Workflows in Benchmark Data Sets and Increased Sensitivity in Analysis of Alzheimer’s Biomarker Data. J. Proteome Res. 2022, 22, 374–386. [Google Scholar] [CrossRef]
- Schmelter, C.; Brueck, A.; Perumal, N.; Qu, S.; Pfeiffer, N.; Grus, F.H. Lectin-Based Affinity Enrichment and Characterization of N-Glycoproteins from Human Tear Film by Mass Spectrometry. Molecules 2023, 28, 648. [Google Scholar] [CrossRef]
- Azkargorta, M.; Soria, J.; Ojeda, C.; Guzmán, F.; Acera, A.; Iloro, I.; Suárez, T.; Elortza, F. Human Basal Tear Peptidome Characterization by CID, HCD, and ETD Followed by in Silico and in Vitro Analyses for Antimicrobial Peptide Identification. J. Proteome Res. 2015, 14, 2649–2658. [Google Scholar] [CrossRef]
- Georgiev, G.A.; Sharifian, G.M.; Romano, J.; Teixeira, K.L.D.; Struble, C.; Ryan, D.S.; Sia, R.K.; Kitt, J.P.; Harris, J.M.; Hsu, K.-L.; et al. Lacritin proteoforms prevent tear film collapse and maintain epithelial homeostasis. J. Biol. Chem. 2021, 296. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.W.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.T.; van Bijsterveld, O.P. Origin and biosynthesis of human tear fluid proteins. Investig. Ophthalmol. Vis. Sci. 1983, 24, 623–630. [Google Scholar] [PubMed]
- Karnati, R.; Laurie, D.E.; Laurie, G.W. Lacritin and the tear proteome as natural replacement therapy for dry eye. Exp. Eye Res. 2013, 117, 39–52. [Google Scholar] [CrossRef]
- Muttuvelu, D.L.; Cehofski, L.J.; Holtz, J.; Utheim, T.P.; Chen, X.; Vorum, H.; Heegaard, S.; Rasmussen, M.L.R.; Khan, A.M.; Abduljabar, A.B.; et al. Meibomian Gland Dysfunction Is Associated with Low Levels of Immunoglobulin Chains and Cystatin-SN. Int. J. Mol. Sci. 2023, 24, 15115. [Google Scholar] [CrossRef]
- McGill, J.I.; Liakos, G.M.; Goulding, N.; Seal, D.V. Normal tear protein profiles and age-related changes. Br. J. Ophthalmol. 1984, 68, 316–320. [Google Scholar] [CrossRef]
- McKay, T.B.; Serjersen, H.; Hjortdal, J.; Zieske, J.D.; Karamichos, D. Characterization of Tear Immunoglobulins in a Small-Cohort of Keratoconus Patients. Sci. Rep. 2020, 10, 9426. [Google Scholar] [CrossRef]
- Janssen, N.A.F.; Grondman, I.; de Nooijer, A.H.; Boahen, C.K.; Koeken, V.A.C.M.; Matzaraki, V.; Kumar, V.; He, X.; Kox, M.; Koenen, H.J.P.M.; et al. Dysregulated Innate and Adaptive Immune Responses Discriminate Disease Severity in COVID-19. J. Infect. Dis. 2021, 223, 1322–1333. [Google Scholar] [CrossRef]
- Turiak, L.; Ozohanics, O.; Marino, F.; Drahos, L.; Vekey, K. Digestion protocol for small protein amounts for nano-HPLC-MS(MS) analysis. J. Proteom. 2011, 74, 942–947. [Google Scholar] [CrossRef]
- Trudgian, D.C.; Fischer, R.; Guo, X.; Kessler, B.M. GOAT—A simple LC-MS/MS gradient optimization tool. Proteomics 2014, 14, 1467–1471. [Google Scholar] [CrossRef]
- Kelstrup, C.D.; Young, C.; Lavallee, R.; Nielsen, M.L.; Olsen, J.V. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J. Proteome Res. 2012, 11, 3487–3497. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. msFragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Nolte, H.; MacVicar, T.D.; Tellkamp, F.; Krüger, M. Instant Clue: A Software Suite for Interactive Data Visualization and Analysis. Sci. Rep. 2018, 8, 12648. [Google Scholar] [CrossRef]
- Allansmith, M.R.; Radl, J.; Haaijman, J.J.; Mestecky, J. Molecular forms of tear IgA and distribution of IgA subclasses in human lacrimal glands. J. Allergy Clin. Immunol. 1985, 76, 569–576. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age/Gender | Schirmer I Test OD/OS | BUT OD/OS | Corneal Fluorescein Punctate Staining | |
|---|---|---|---|---|
| D1 | 26/male | 25/25 | 25/25 | no |
| D2 | 27/male | 20/20 | 20/25 | no |
| D3 | 33/female | 20/20 | 25/23 | no |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruszel, B.; Tóth-Molnár, E.; Janáky, T.; Szabó, Z. Sources of Variance in Human Tear Proteomic Samples: Statistical Evaluation, Quality Control, Normalization, and Biological Insight. Int. J. Mol. Sci. 2024, 25, 1559. https://doi.org/10.3390/ijms25031559
Bruszel B, Tóth-Molnár E, Janáky T, Szabó Z. Sources of Variance in Human Tear Proteomic Samples: Statistical Evaluation, Quality Control, Normalization, and Biological Insight. International Journal of Molecular Sciences. 2024; 25(3):1559. https://doi.org/10.3390/ijms25031559
Chicago/Turabian StyleBruszel, Bella, Edit Tóth-Molnár, Tamás Janáky, and Zoltán Szabó. 2024. "Sources of Variance in Human Tear Proteomic Samples: Statistical Evaluation, Quality Control, Normalization, and Biological Insight" International Journal of Molecular Sciences 25, no. 3: 1559. https://doi.org/10.3390/ijms25031559
APA StyleBruszel, B., Tóth-Molnár, E., Janáky, T., & Szabó, Z. (2024). Sources of Variance in Human Tear Proteomic Samples: Statistical Evaluation, Quality Control, Normalization, and Biological Insight. International Journal of Molecular Sciences, 25(3), 1559. https://doi.org/10.3390/ijms25031559