

The Relationship Between Non-Transferrin-Bound Iron (NTBI), Labile Plasma Iron (LPI), and Iron Toxicity
 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
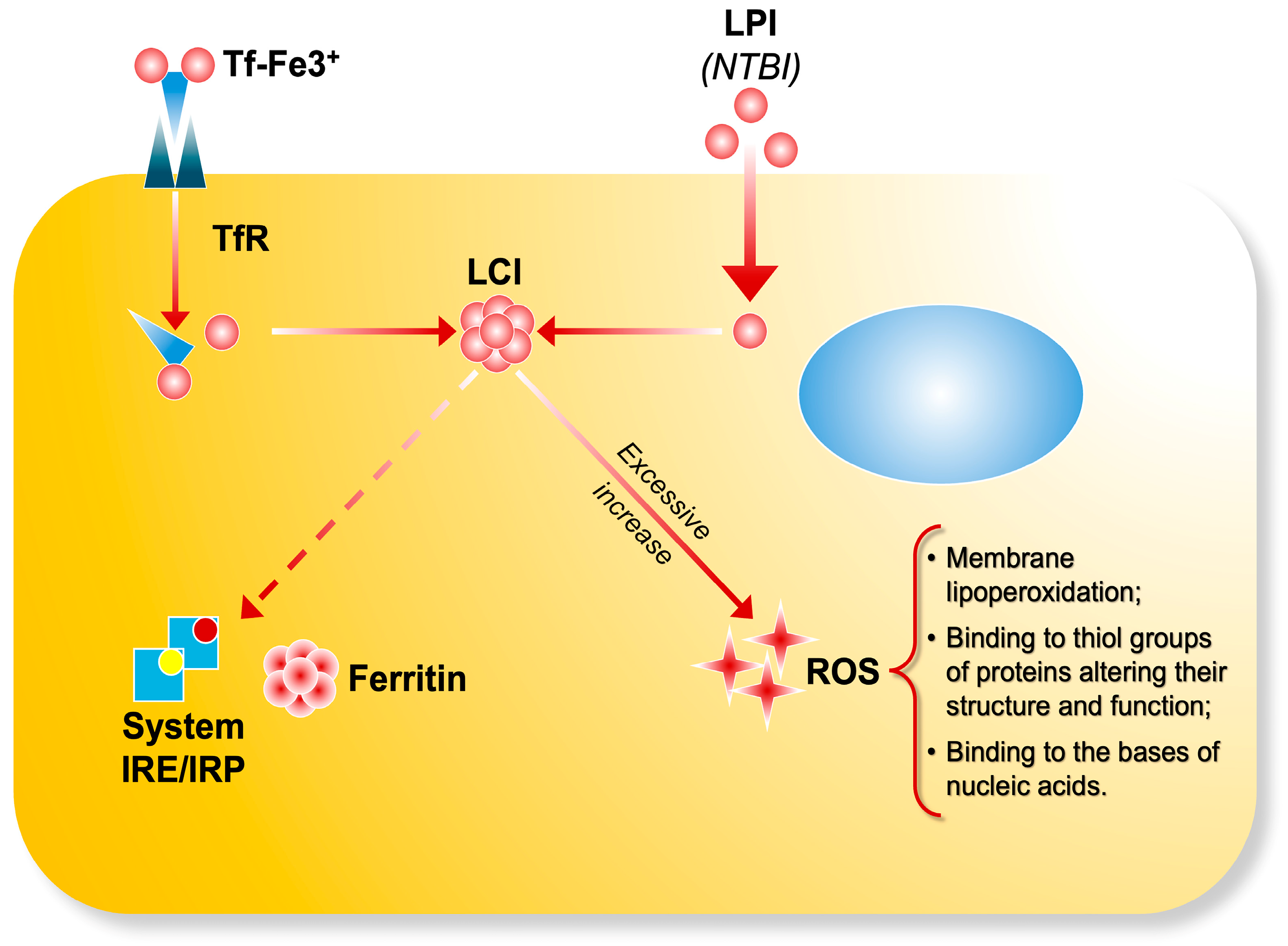
2. Iron Overload, Reactive Oxygen Species, and Organ Damage
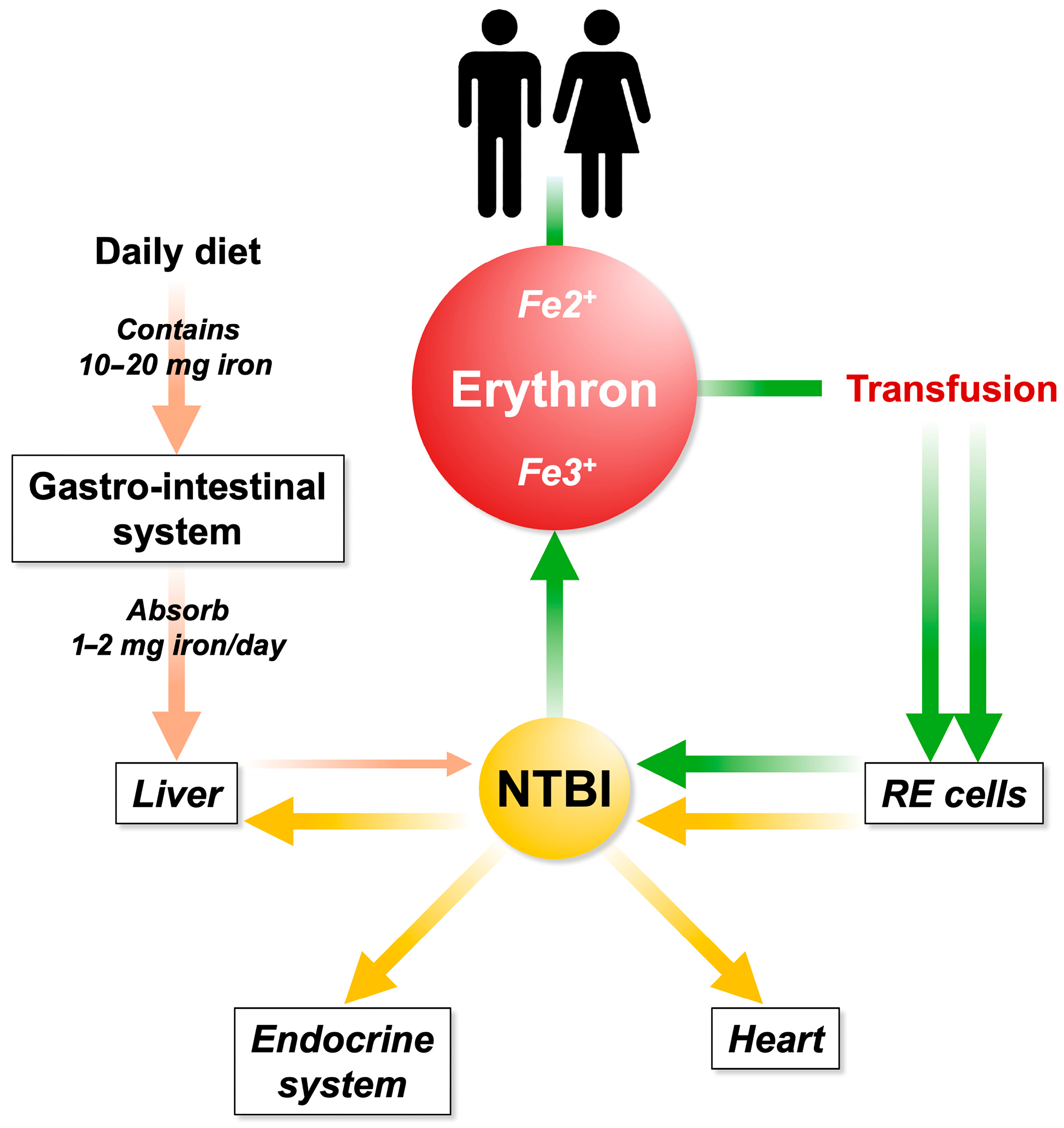
3. NTBI and LPI: Key Factors in the Pathogenesis of Iron-Related Diseases
3.1. Hemolytic Anemia
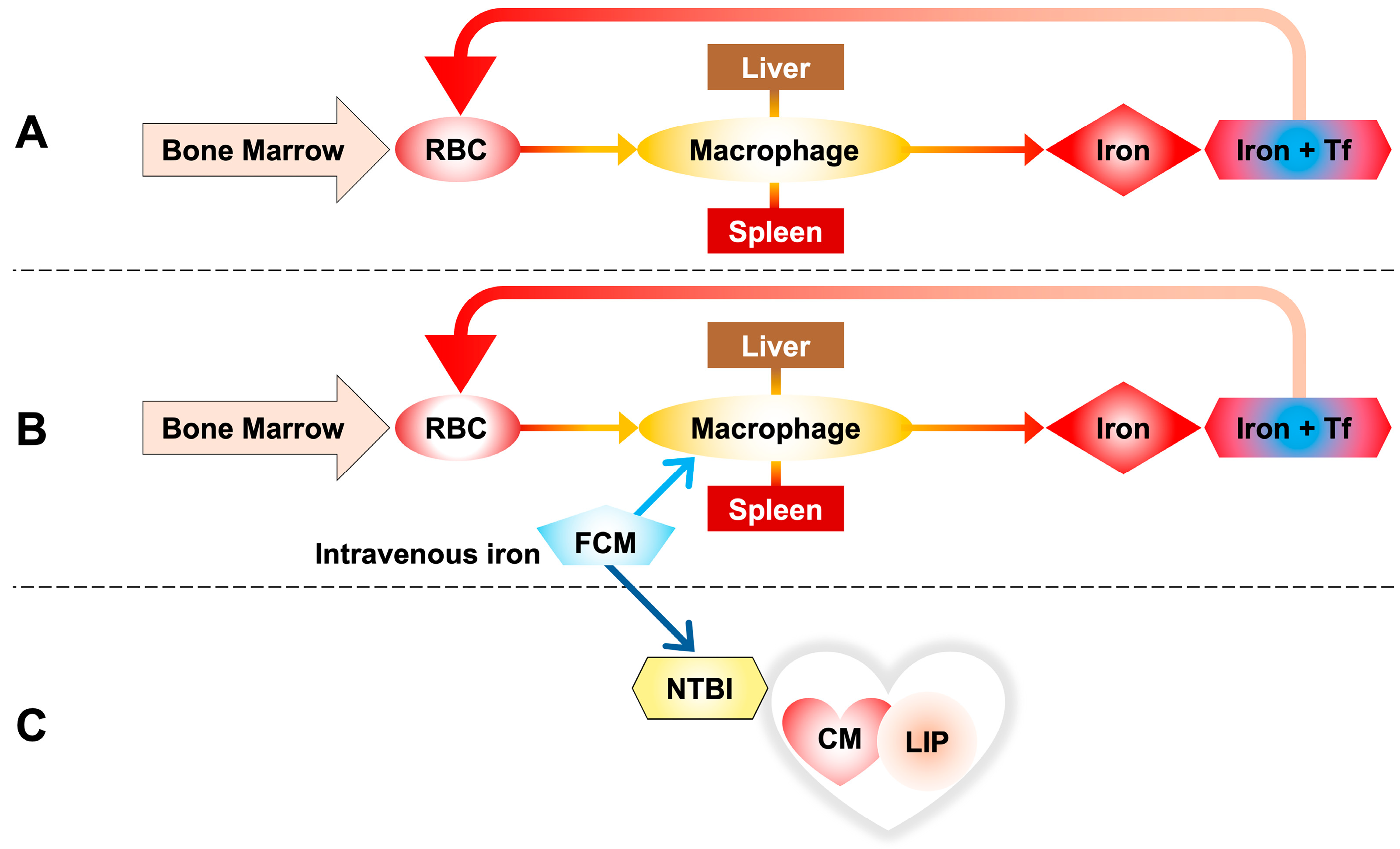
3.2. Iron Metabolism in Hemodialysis Patients Treated by Intravenous Iron Supplementation: Labile Plasma Iron in Parenteral Iron Formulations and Its Potential Generation of Non-Transferrin-Bound Iron
4. NTBI and LPI in Patients with Iron Overload
4.1. Effects of Iron Chelation
4.2. Potential Role of Antioxidants
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NTBI | Non-transferrin-bound iron |
| LPI | Labile plasma iron |
| LCI | Labile cell iron |
| ROS | Reactive oxygen species |
| TDT | Transfusion-dependent thalassemia |
| NTDT | Non-transfusion-dependent thalassemia |
| MDS | Myelodysplastic syndrome |
| DFO | Deferoxamine |
| DFP | Deferiprone |
| DFX | Deferasirox |
| IO | Iron overload |
References
- Soares, M.P.; Hamza, I. Macrophages and Iron metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45–55. [Google Scholar] [CrossRef]
- Silva, A.M.N. The (Bio) Chemistry of Non-Transferrin-Bound Iron. Molecules 2022, 27, 1784–1797. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Kong, X.; Parkin, M.C.; Cammack, R.; Hider, R.C. Iron (III) citrate speciationin aqueous solution. Dalton Trans. 2009, 40, 8616–8625. [Google Scholar] [CrossRef] [PubMed]
- Pierre, J.L.; Gautier-Luneau, I. Iron and citric acid: A fuzzy chemistry of ubiquitous biological relevance. Biometals 2000, 13, 91–96. [Google Scholar] [CrossRef]
- Garbowski, M.W.; Cabantchik, I.; Hershko, C.; Hider, R.; Porter, J.B. The clinical relevance of detectable plasma iron species in iron overload states and subsequent to intravenous iron-carbohydrate administration. Am. J. Hematol. 2022, 98, 533–540. [Google Scholar] [CrossRef]
- Ma, Y.; Podinovskaia, M.; Evans, P.J.; Emma, G.; Schaible, E.; Porter, J.; Hider, C. Anovel method for non-transferrin-bound iron quantification by chelatable fluorescent beads based on flow cytometry. Biochem. J. 2014, 463, 351–362. [Google Scholar] [CrossRef]
- de Swart, L.; Hendriks, J.C.M.; van de Vorm, L.N.; Cabantchik, Z.I.; Evans, P.J.; Hod, E.A.; Brittenham, G.M.; Furman, Y.; Wojczyk, B.; Janssen, M.C.H.; et al. Second International round robin for the quantification of serum non-transferrin-bound iron and labile plasma iron in patients with iron-overload disorders. Haematologica 2016, 101, 38–45. [Google Scholar] [CrossRef]
- Healy, J.; Tipton, K. Ceruloplasmin and what it might do. J. Neural Transm. 2007, 114, 777–781. [Google Scholar] [CrossRef]
- Breuer, W.; Hershko, C.; Cabantchik, Z.I. The importance of non-transferrin iron in of iron metabolism. Transfus. Sci. 2000, 23, 185–192. [Google Scholar] [CrossRef]
- Esposito, B.P.; Epsztejn, S.; Breuer, W.; Sirankapracha, P.; Pootrakul, P.; Hershko, C.; Cabantchik, Z.I. Labile plasma iron in iron overload: Redox activity and susceptibility to chelation. Blood 2003, 102, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Kaklkon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and partecipation in cellular processes. Free Raic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Napier, I.; Ponka, P.; Richardoson, D.R. Iron trafficking in the mitochondrion: Novel pathways revealed by disease. Blood 2005, 105, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Shang, P. The significance, trafficking and determination of labile iron cytosol, mitochondria and lysosomes. Metallomics 2018, 10, 899–916. [Google Scholar] [CrossRef]
- Fibach, E.; Rachmilewitz, E.A. Iron overload in hematological disorders. Presse Med. 2017, 46, 296–305. [Google Scholar] [CrossRef]
- Hershko, C. Pathogenesis and management of iron toxicity in thalassemia. Ann. N. Y. Acad. Sci. 2010, 1202, 1–9. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Hiang, R.; Wan, D.; Yang, G. Regulation of iron homeostasis and related diseases. Mediat. Inflamm. 2020, 2020, 6062094. [Google Scholar] [CrossRef]
- Fibach, E.; Rachmilewitz, E. The role of oxidative stress in hemolytic anemia. Curr. Mol. Med. 2008, 8, 609–619. [Google Scholar] [CrossRef]
- Pilo, F.; Cilloni, D.; Della Porta, M.G.; Forni, G.L.; Piperno, A.; Santini, V.; Angelucci, E. Iron-mediated tissue damage in acquired ineffective erytropoiesis disease: It’s more amatter of burden or more of exposure to toxic iron form? Leuk. Res. 2022, 114, 106792. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. Beta-thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Taher, A.T.; Verma, A.; Shah, A.; Hermine, O. Erythropoiesis in lower-risk myelodysplastic syndromes and beta-thalassemia. Blood Rev. 2023, 59, 101039. [Google Scholar] [CrossRef] [PubMed]
- Kattamis, A.; Kwiatkowski, J.L.; Aydinok, Y. Thalassemia. Lancet 2022, 399, 2310–2324. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Wood, J.C.; Taher, A.T. Iron overload in non-transfusion-dependent thalassemia: A clinical perspective. Blood Rev. 2012, 26 (Suppl. S1), S16–S19. [Google Scholar] [CrossRef]
- Taher, A.T.; Porter, J.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Galanello, R.; Karakas, Z.; Lawniczek, T.; et al. Deferasinox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood 2012, 120, 970–977. [Google Scholar] [CrossRef]
- Musallam, K.M.; Cappellini, M.D.; Daar, S.; Karimi, M.; Graziadei, G.; Magestro, M.; Wulff, J.; Pietri, G.; Taher, A.T. Serum ferritin level and morbidity risk in transfusion-independent patients with beta-thalassemia intermedia: The ORIENT study. Haematologica 2014, 99, e218–e221. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, G.; Wang, M.; Wei, X.; Pan, L.; Liu, J.; Lei, Y.; Peng, L.L.; Lai, Y.; Liu, R. Iron overload status in patients whit non-transfusion-dependent thalassemia in China. Ther. Adv. Hematol. 2022, 13, 20406207221084639. [Google Scholar] [CrossRef] [PubMed]
- Heriatmo, N.L.; Estuningtyas, A.; Soetikno, V. Iron-overload conditions: Manifestations to the kidney organs- A review. Borneo J. Pharm. 2023, 6, 360–369. [Google Scholar] [CrossRef]
- Jha, J.C.; Chow, B.S.M.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and kidney disease: Role of oxidative stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef]
- Erichsen, K.; Ulvik, R.J.; Grimstad, T.; Berstad, A.; Berge, R.K.; Hausken, T. Effects of ferrous silphate and non-ionic iron-polymaltose complex on markers of oxidative tissue damage in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2005, 22, 831–838. [Google Scholar] [CrossRef]
- Pennell, D.J. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymtomatic myocardial siderosis. Blood 2006, 107, 3738–3744. [Google Scholar] [CrossRef]
- Garbowski, M.W.; Bansal, S.; Porter, J.B.; Mori, C.; Burckhardt, S.; Hider, R.C. Intravenous iron preparations transiently generate non-transferrin-bound iron from two proposed pathways. Haematologica 2021, 106, 2885–2896. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.B.; Walter, P.B.; Neumayr, L.D.; Evans, P.; Bansal, S.; Garbowski, M.; Weyhmiller, M.G.; Harmatz, P.R.; Wood, J.C.; Miller, J.L.; et al. Mechanisms of plasma non-transferrin bound iron generation: Insights from comparing transfused diamond blackfan anaemia with sickle cell and thalassaemia patients. Br. J. Haematol. 2014, 167, 692–696. [Google Scholar] [CrossRef]
- Prakash, M.; Upadhya, S.; Prabhu, R. serum non-transferrin bound iron in hemodialysis patients not receiving intravenous iron. Clin. Chim. Acta 2005, 360, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Dresow, B.; Petersen, D.; Fischer, R.; Nielsen, P. Non-transferrin-bound iron in plasma following administration of oral iron drugs. Biometals 2008, 21, 273–276. [Google Scholar] [CrossRef]
- Malyszko, J.; Koc-Zorawska, E.; Levin-Iaina, N.; Slotki, I.; Matuszkiewicz-Rowinska, J.; Glowinska, I.; Malyszko, J.S. Iron metabolism in hemodialyzed patient- a story half told? Clin. Res. 2014, 10, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Esposito, B.P.; Breuer, W.; Slotki, I.; Cabantchik, Z.I. Labile iron in parenteral iron formulations and its potential for gene rating plasma nontransferrin-bound iron in dialysis patients. Eur. J. Clin. Invest. 2002, 32 (Suppl. S1), 42–49. [Google Scholar] [CrossRef]
- Bnaya, A.; Shavit, L.; Malyszko, J.S.; Malyszko, J.; Slotki, I. Labile plasma iron levels in chronic hemodialysis patients treated by intravenous iron supplementation. Ther. Apher. Dial. 2020, 24, 416–422. [Google Scholar] [CrossRef]
- Belotti, A.; Duca, L.; Borin, L.; Realini, S.; Renso, R.; Parma, M.; Pioltelli, P.; Pogliani, E.; Cappellini, M.D. Non transferrin bound iron (NTBI) in acute leucemia throughout conventional intensive chemiotherapy: Kinetic of its appearance and potential predictive role in infectious complications. Leuk. Res. 2015, 39, 88–91. [Google Scholar] [CrossRef]
- Camaschella, C.; Girelli, D. The changing landscape of iron deficiency. Mol. Asp. Med. 2020, 75, 100861. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Lüscher, T.F. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef]
- Vera-Aviles, M.; Kabir, S.N.; Shah, A.; Polzella, P.; Lim, D.Y.; Buckley, P.; Ball, C.; Swinkels, D.; Matlung, H.; Blans, C.; et al. Intravenous iron therapy results in rapid and sustained rise in myocardial iron content through a novel pathway. Eur. Heart J. 2024, 45, 4497–4508. [Google Scholar] [CrossRef]
- Mentz, R.J.; Garg, J.G.; Rockhold, F.W.; Butler, J.; De Pasquale, C.G.; Ezekowitz, J.A.; Lewis, G.D.; O’Meara, E.; Ponikowski, P.; Troughton, R.W.; et al. Ferric carboxymaltose in heart failure with iron deficiency. N. Engl. J. Med. 2023, 389, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15. [Google Scholar] [CrossRef]
- Cabantchik, Z.l.; Breuer, W.; Zanninelli, G.; Cianciulli, P. LPI-labile plasma iron in ironoverload. Best Pract. Res. Clin. Haematol. 2005, 18, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Coates, T.D. Management of iron overload: Lessons from transfusion-dependent hemoglobinopathies. Blood 2025, 145, 359–371. [Google Scholar] [CrossRef]
- Bruzzese, A.; Martino, E.A.; Mendicino, F.; Lucia, E.; Olivito, V.; Bova, C.; Filippelli, G.; Capodanno, I.; Neri, A.; Morabito, F. Ironchelationtherapy. Eur. J. Haematol. 2023, 110, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Porter, J.B.; Kattamis, A.; Viprakasit, V.; Cappellini, M.D. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with nontransfusion-dependent thalassemia syndrome. Drug Des. Dev. Ther. 2016, 10, 4073–4078. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; El-Beshlawy, A.; Karimi, M.; Daar, S.; Belhoul, K.; Saned, M.S.; Graziadei, G.; Cappellini, M.D. Age-related complications in treatment-naive patients with thalassemia intermedia. Br. J. Haematol. 2010, 150, 486–489. [Google Scholar] [CrossRef]
- Coates, T.D.; Carson, S.; Wood, J.C.; Berdoukas, V. Management of iron overload in hemoglobinopathies: What is the appropriate target iron level? Ann. N. Y. Acad. Sci. 2016, 1368, 95–106. [Google Scholar] [CrossRef]
- Taher, A.T.; Saliba, A.N. Iron overload in thalassemia: Different organs at different rates. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 265–271. [Google Scholar] [CrossRef]
- Hod, E.A.; Zhang, N.; Sokol, S.A.; Wojczyk, B.S.; Francis, R.O.; Ansaldi, D.; Francis, K.P.; Della-Latta, P.; Whittier, S.; Sheth, S.; et al. Transfusion of red blood cells after prolonged storage produces harmful effects that are mediated by iron and inflammation. Blood 2010, 115, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Porter, J.B.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Origa, R.; Karakas, Z.; Habr, D.; et al. Defining serum ferritin thresholds to predict clinically relevant liver iron concentrations for guiding deferasirox therapy when MRI is unavailable in patients with non-transfusion-dependent thalassaemia. Br. J. Haematol. 2015, 168, 284–290. [Google Scholar] [CrossRef]
- Vinchi, F.; Hell, S.; Platzbecker, U. Controversies on the consequences of iron overload and chelation in MDS. HemaSphere 2020, 4, e357. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.P.; He, T.; Kirk, P.; Roughton, M.; Anderson, L.J.; de Noronha, S.V.; Sheppard, M.N.; Porter, J.B.; Walker, M.; Wood, J.C. On T2* magnetic resonance and cardiac iron. Circulation 2011, 123, 1519–1528. [Google Scholar] [CrossRef]
- Steensma, D.P.; Gattermann, N. When is iron overload deleterious, and when and low should iron chelation therapy be administered in myelodysplastic sindrome? Best Pract. Res. Clin. Haematol. 2013, 26, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Lyle, L.; Hirose, A. Iron overload in myelodysplastic syndrome: Pathophysiology, consequences, diagnosis, and treatment. J. Adv. Pract. Oncol. 2018, 9, 392–405. [Google Scholar]
- Angelucci, E.; Urru, S.A.M.; Pilo, F.; Piperno, A. Myelodysplastic syndromes and iron chelation therapy. Mediterr. J. Hematol. Infect. Dis. 2017, 9, e2017021. [Google Scholar] [CrossRef]
- Rose, C.; Fitoussi, O.; Gyan, E.; Hacini, M.; Amé, S.; Corront, B.; Beyne-Rauzy, O.; Adiko, D.I.; Loppinet, E.A.; Ali-Ammar, N.; et al. Prospective evaluation of the effect of deferasinox on hematologic response in transfusion-dependent patients with low-risk MDS and iron overload: The rythmex study. Blood 2016, 128, 2008. [Google Scholar] [CrossRef]
- Forni, G.L.; Gianesin, B.; Musallam, K.M.; Longo, F.; Rosso, R.; Lisi, R.; Gamberini, M.R.; Pinto, V.M.; Graziadei, G.; Vitucci, A.; et al. Overall and complication-free survival in a large cohort of patients with β-thalassemia major followed over 50 years. Am. J. Hematol. 2023, 98, 381–387. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Rugolotto, S.; De Stefano, P.; Zhao, H.Q.; Cappellini, M.D.; Del Vecchio, G.C.; Romeo, M.A.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004, 89, 1187–1193. [Google Scholar]
- Davis, B.A.; Porter, J.B. Results of long term iron chelation treatment with deferoxamine. Adv. Exp. Med. Biol. 2002, 509, 91–125. [Google Scholar] [CrossRef] [PubMed]
- Hershko, C.; Cappellini, M.D.; Galanello, R.; Piga, A.; Tognoni, G.; Masera, G. Purging iron from the heart. Br. J. Haematol. 2004, 125, 545–551. [Google Scholar] [CrossRef]
- Porter, J.B.; Rafique, R.; Srichairatanakool, S.; Davis, B.A.; Shah, F.T.; Hair, T.; Evans, P. Recent insights into interactions of deferoxamine with cellular and plasma iron pools: Implications for clinical use. Ann. N. Y. Acad. Sci. 2005, 1054, 155–168. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Cappellini, M.D.; De Stefano, P.; Del Vecchio, G.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; Piga, A.; Romeo, M.A.; Zhao, H.; et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood 2006, 107, 3733–3737. [Google Scholar] [CrossRef] [PubMed]
- Piga, A.; Gaglioti, C.; Fogliacco, E.; Tricta, F. Comparative effects of deferiprone and deferoxamine on serviva and cardiac disease in patients with thalassemia major: A retrospective analisys. Haematologica 2003, 88, 489–496. [Google Scholar]
- Glickstein, H.; Ben Ei, R.; Link, G.; Breuer, W.; Konijn, A.M.; Hershko, C.; Nick, H.; Cabantchik, Z.I. Action of chelators in iron-loaded cardiac cells: Accessibility to intracellular labile iron and functional consequences. Blood 2006, 108, 3195–3203. [Google Scholar] [CrossRef] [PubMed]
- Garbowski, M.W.; Vlachodimitropoulou, E.; Hider, R.; Porter, J.B. Residual erythropoiesis protects against myocardial hemosiderosis in transfusion-dependent thalassemia by lowering labile plasma iron via transient generation of apotransferrin. Haematologica 2017, 102, 1640–1649. [Google Scholar] [CrossRef]
- Evans, P.; Kayyali, R.; Hider, R.C.; Eccleston, J.; Porter, J.B. Mechanisms for the shuttling of plasma non-transferrin-bound iron (NTBI) onto deferoxamine by deferiprone. Trans. Res. 2010, 156, 55–67. [Google Scholar] [CrossRef]
- Koren, A.; Fink, D.; Admoni, O.; Tennenbaum-Rakover, Y.; Levin, C. Non-transferrin bound labile plasma iron and iron overload in sickle cell diseases: A comparative study between sickle cell disease and b thalassemic patients. Eur. J. Haematol. 2010, 84, 72–78. [Google Scholar] [CrossRef]
- Grange, C.; Lux, F.; Brichart, T.; David, L.; Couturier, A.; Leaf, D.E.; Allaouchiche, B.; Tillement, O. Iron as an emerging therapeutic target in critically ill patients. Crit. Care 2023, 27, 475–488. [Google Scholar] [CrossRef]
- Kumfu, S.; Chattipakorn, S.; Chinda, K.; Fucharoen, S.; Chattipakorn, N. T-type calcium channel blockade improves survival and cardiovascular function in thalassemic mice. Eur. J. Haematol. 2012, 88, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhabyeyev, P.; Wang, S.; Oudit, G.Y. Role of iron metabolism in heart failure: From iron deficiency to iron overload. Biochim. Biophys. ActaMol. Bas. Dis. 2019, 1865, 1925–1937. [Google Scholar] [CrossRef]
- De Franceschi, L.; Turrini, F.; Honczarenko, M.; Ayi, K.; Rivera, A.; Fleming, M.D.; Law, T.; Mannu, F.; Kuypers, F.A.; Bast, A.; et al. In vivo reduction of erythrocytes oxidant stress in a murine model of β-thalassemia. Hematologica 2004, 89, 1287–1298. [Google Scholar]
- Piga, A.; Longo, F.; Duca, L.; Roggero, S.; Vinciguerra, T.; Calabrese, R.; Hershko, C.; Cappellini, M.D. High nontransferrin bound iron levels and heart disease in thalassemia major. Am. J. Hematol. 2009, 84, 29–33. [Google Scholar] [CrossRef]
- Cighetti, G.; Duca, L.; Bortone, L.; Sala, S.; Nava, I.; Fiorelli, G.; Cappellini, M.D. Oxidative status and malondialdehyde in β-thalassemia. Eur. J. Clin. Investig. 2002, 32 (Suppl. S1), 55–60. [Google Scholar] [CrossRef]
- Rachmilewitz, E.A.; Weizwe-Sten, O.; Adamsky, K.; Amariglio, N.; Rechavi, G.; Breda, L.; Rivella, S.; Cabantchik, Z.I. Role of iron in inducing oxidative stress in thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 118–123. [Google Scholar] [CrossRef]
- Koren, E.; Kohen, R.; Ginsburg, I. Polyphenols enhance total oxidant-scavenging capacities of human blood by binding to red blood cells. Exp. Biol. Med. 2010, 235, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Badria, F.A.; Ibrahim, A.S.; Badria, A.F.; Elmarakby, A.A. Curcumin attenuates iron accumulation and oxidative stress in the liver and spleen of chronic iron-overloaded rats. PLoS ONE 2015, 10, e0134156. [Google Scholar] [CrossRef] [PubMed]
- Prus, E.; Fibach, E. The antioxidant effect of fermented papaya preparation involves iron chelation. J. Biol. Regul. Homeost. Agents 2012, 26, 203–210. [Google Scholar]
- Coesang, T.; Li, H.; Dussiot, M.; Maciel, T.; Breda, L.; Garcia Santos, D.; Chen, H.; Feola, M.; Bao, W.; Pham, P.; et al. Exogenous apo-transferrin increases monoferric transferrin, decreasing cytosolic iron uptake and heme and globin synthesis in β-thalassemic mice. Blood 2014, 124, 4037. [Google Scholar] [CrossRef]
- Sturm, B.; Laggner, H.; Ternes, N.; Goldenberg, H.; Scheiber-Mojdehkar, B. Intravenous iron preparations and ascorbic acid: Effects on chelatable and bioavailable iron. Kidney Int. 2005, 67, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duca, L.; Di Pierro, E.; Scaramellini, N.; Granata, F.; Graziadei, G. The Relationship Between Non-Transferrin-Bound Iron (NTBI), Labile Plasma Iron (LPI), and Iron Toxicity. Int. J. Mol. Sci. 2025, 26, 6433. https://doi.org/10.3390/ijms26136433
Duca L, Di Pierro E, Scaramellini N, Granata F, Graziadei G. The Relationship Between Non-Transferrin-Bound Iron (NTBI), Labile Plasma Iron (LPI), and Iron Toxicity. International Journal of Molecular Sciences. 2025; 26(13):6433. https://doi.org/10.3390/ijms26136433
Chicago/Turabian StyleDuca, Lorena, Elena Di Pierro, Natalia Scaramellini, Francesca Granata, and Giovanna Graziadei. 2025. "The Relationship Between Non-Transferrin-Bound Iron (NTBI), Labile Plasma Iron (LPI), and Iron Toxicity" International Journal of Molecular Sciences 26, no. 13: 6433. https://doi.org/10.3390/ijms26136433
APA StyleDuca, L., Di Pierro, E., Scaramellini, N., Granata, F., & Graziadei, G. (2025). The Relationship Between Non-Transferrin-Bound Iron (NTBI), Labile Plasma Iron (LPI), and Iron Toxicity. International Journal of Molecular Sciences, 26(13), 6433. https://doi.org/10.3390/ijms26136433