2.1. Synthesis and Characteristics of Calcium Phosphate Nanoparticles
Since CaPs could serve as carriers of a wide range of substances, both thermally stable and labile, we compared two different methods of CaP synthesis, differed by the temperature and pH of the synthesis medium, as well as the concentration of a stabilizing agent, sodium citrate. We always synthesized the particles with ultrasound exposure so that they did not have a tendency to form a precipitate, but instead formed a stable suspension. This is extremely important for their further use in both medicine and agriculture, so that they can be applied as suspensions by instillation, dripping, or spraying.
It is known that sodium citrate can serve as a stabilizing agent for CaPs [
34], as it forms a charged layer around the nanoparticles and prevents their coagulation. So, we varied sodium citrate concentration to find the proper conditions of CaP formation. These experiments were carried out at fixed concentrations of stock solutions of potassium phosphate and calcium chloride and at fixed power and time of ultrasonic treatment without any precautions, that is, without any cooling of the reaction mixture under ultrasonic treatment. We have shown that the presence of sodium citrate is absolutely necessary for proper CaP formation, as in the absence of citrate or at low citrate concentrations, only large particles with substantial precipitation occurred, likely due to aggregates formation (
Table 1). The increase in the sodium citrate concentration up to 7.8 mM allowed us to obtain the nanosized particles with a mean hydrodynamic diameter of about 210 nm (
Table 1), whereas a further increase to 15.6 mM sodium citrate led to a further significant decrease in the mean hydrodynamic diameter (about 80 nm) of the obtained CaPs measured by dynamic light scattering (DLS). Higher concentrations of sodium citrate did not further decrease CaP size, while ζ-potential of CaPs was virtually the same at any sodium citrate concentration (
Table 1).
Then, we synthesized CaPs at 15.6 mM sodium citrate but at two different temperature conditions: (a) without cooling as above; in these conditions the temperature of the system rose from room temperature to 60–70 °C at the end of a constant ultrasonication; (b) with ice cooling of a reaction mixture during all ultrasonication time, which resulted in a temperature rise of 20–23 °C towards the end of the treatment. Additionally, we varied the pH values of the stock solution of potassium chloride in both cases. We showed that the synthesis without cooling led to the formation of a stable suspension of CaPs. The mean hydrodynamic diameter of CaPs measured by DLS was lower at higher pH values, and the polydispersity index of the system indicated more uniform CaP formation (
Table 2). However, CaPs synthesized at low temperature were characterized by low stability, as some precipitation was observed at all pH values of the reaction medium (
Table 2).
We showed that increasing the concentration of the stabilizing agent citrate allowed us to increase the stability of the system when obtaining CaP particles without cooling (
Table 1). Therefore, we hypothesized that increasing the citrate content would also affect the stability of CaPs synthesized upon cooling. Indeed, a two- and three-fold increase in the initial concentration of sodium citrate helped to obtain the stable suspension of CaPs with appropriate sizes without any precipitate (
Table 3). Note that pH values were checked at each stage of the synthesis. The different concentrations of sodium citrate used in this study did not influence the final pH value of the system. It is interesting, however, that at higher pH of the stock solution of potassium phosphate, the mean hydrodynamic diameter of the CaPs was higher, in contrast to what was observed when CaPs were obtained without cooling (see
Table 2).
As a result, for further experiments, we selected two methods for the synthesis of CaPs (
Figure 1) bearing in mind that a method without cooling could be further used for the inclusion of thermostable active agents in the coprecipitation stage, while a method with cooling could be useful for the inclusion of less stable biological molecules, i.e., nucleic acids and proteins. It should be especially noted that we use the term “coprecipitation” for the process where a substance can be included into CaPs during particles formation, i.e., in situ. Despite the fact that these systems did not form real precipitates in the chosen conditions, they formed stable suspensions. Additionally, we tried both types of CaPs as putative carriers of the substances which could not be included into the nanoparticles by coprecipitation but could be included by sorption.
It should be noted that CaPs obtained by the chosen methods were characterized by similar values of a mean hydrodynamic diameter and ζ-potential; but, the polydispersity index of the particles obtained with cooling was two times higher, that is, CaPs obtained with cooling were characterized by broader distribution in size (
Figure 1). Additionally, CaP suspensions obtained under different conditions had different CaP content, determined by weight after dialysis: CaP concentration obtained without cooling was about 1 mg/mL, CaP concentration obtained with cooling was two-fold less: approximately 0.5 mg/mL. Apparently, this is due to the solubility of calcium phosphate, as the higher the pH of the system, the lower the solubility of calcium phosphate, and, accordingly, the more effective the precipitation of the salt at higher pH (method without cooling).
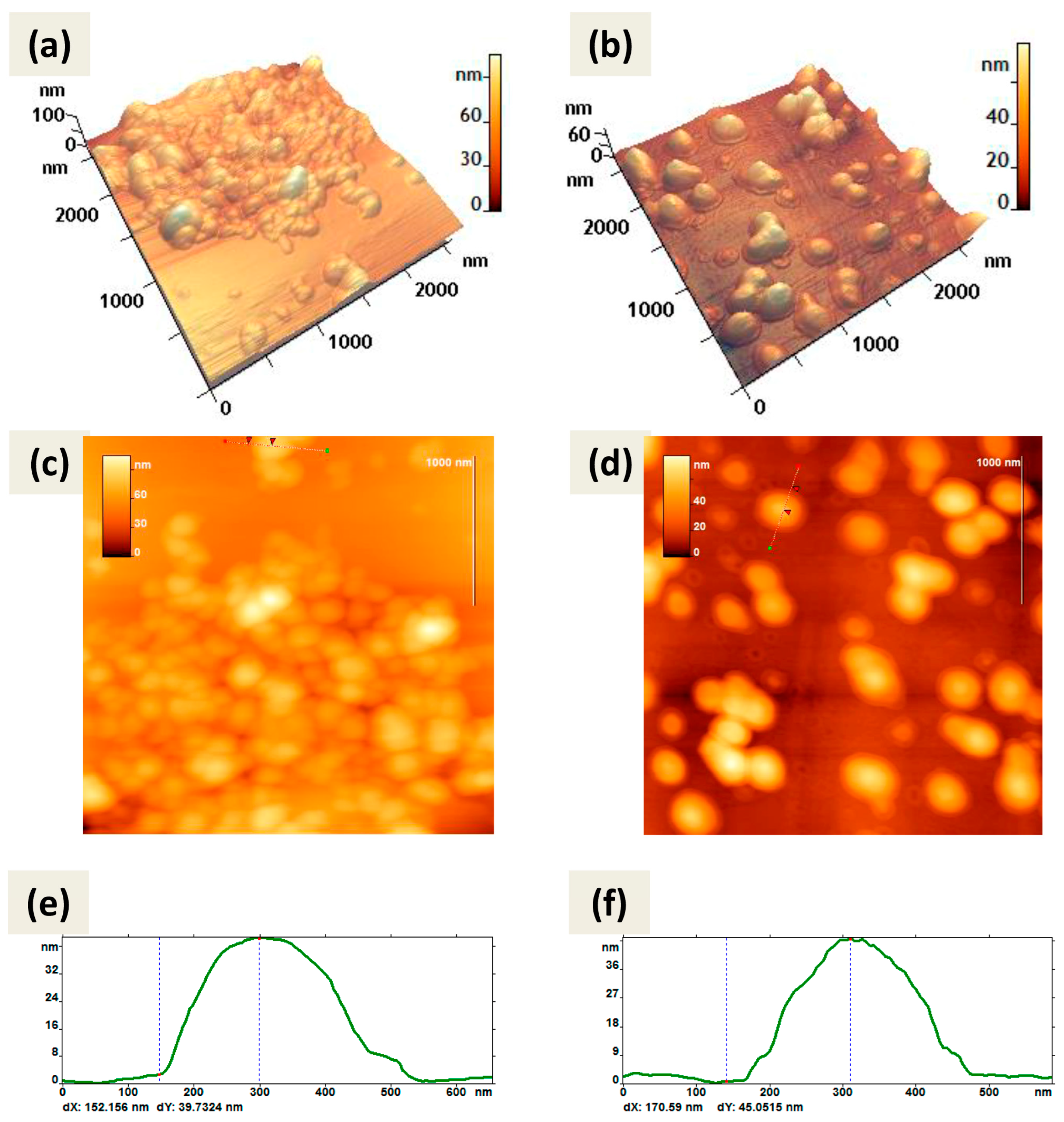
Both types of CaPs were characterized by scanning electron microscopy (SEM) (
Figure 1a,b) and scanning transmission electron microscopy (STEM) (
Figure 1c,d) methods. CaPs obtained without cooling were represented by a rounded shape with a size range of 20–150 nm (
Figure 1a,c). In addition, the formation of CaP aggregates was observed upon drying on a carbon matrix. The average hydrodynamic diameter measured with dynamic light scattering (DLS) was consistent with the particle sizes in SEM and STEM images. STEM and SEM images of CaPs with cooling showed agglomerates consisting of nanocrystallites of 10–20 nm in size (
Figure 1b,d). Apparently, the smaller size of separate particles with lower synthesis temperature could be observed due to the slower growth of the particles than at a high temperature. Despite the differences in morphology, the hydrodynamic diameters obtained by DLS were similar and corresponded to 80 nm for both particle types. Since the PDI value was twice as high for CaPs with cooling, it can be assumed that they exist in suspension as agglomerates of different sizes consisting of nanocrystallites. It is worth noting that we could observe a bright halo around individual CaPs obtained both with and without cooling, likely formed by the stabilizing agent, sodium citrate.
Atomic force microscopy (AFM) also showed predominantly spherical shapes of CaPs obtained by both methods (
Figure 2). Both types of particles were not prone to destruction when exposed to a cantilever and were quite elastic and did not aggregate; when drying on graphite or mica they tend to form a monolayer.
Note that the method of CaP synthesis with cooling included increased amounts of the stabilizing agent, sodium citrate. So, we can attribute the observed in
Figure 2b,d border to the citrate shell on the surface of CaPs. Some CaPs obtained without cooling at lower concentration of sodium citrate also showed such borders (
Figure 2a) but not so obvious.
Using AFM, a detailed morphometric analysis of CaPs was carried out; for each sample of particles the parameter values were determined using FemtoScan Online software version 2.4.26.
Table 4 shows the average values of CaP parameters obtained during the AFM study.
As can be seen from
Figure 2 and
Table 4, CaPs obtained with cooling had larger values of perimeter, area, and surface roughness. Based on the form factor values, both types of particles had an approximately similar shape; not ideally spherical, but rather oval. This may partly be a consequence of the peculiarities of adsorption on the surface. When comparing the value of the diameter and the average height, CaPs obtained with cooling tended to spread out more over the surface of the substrate.
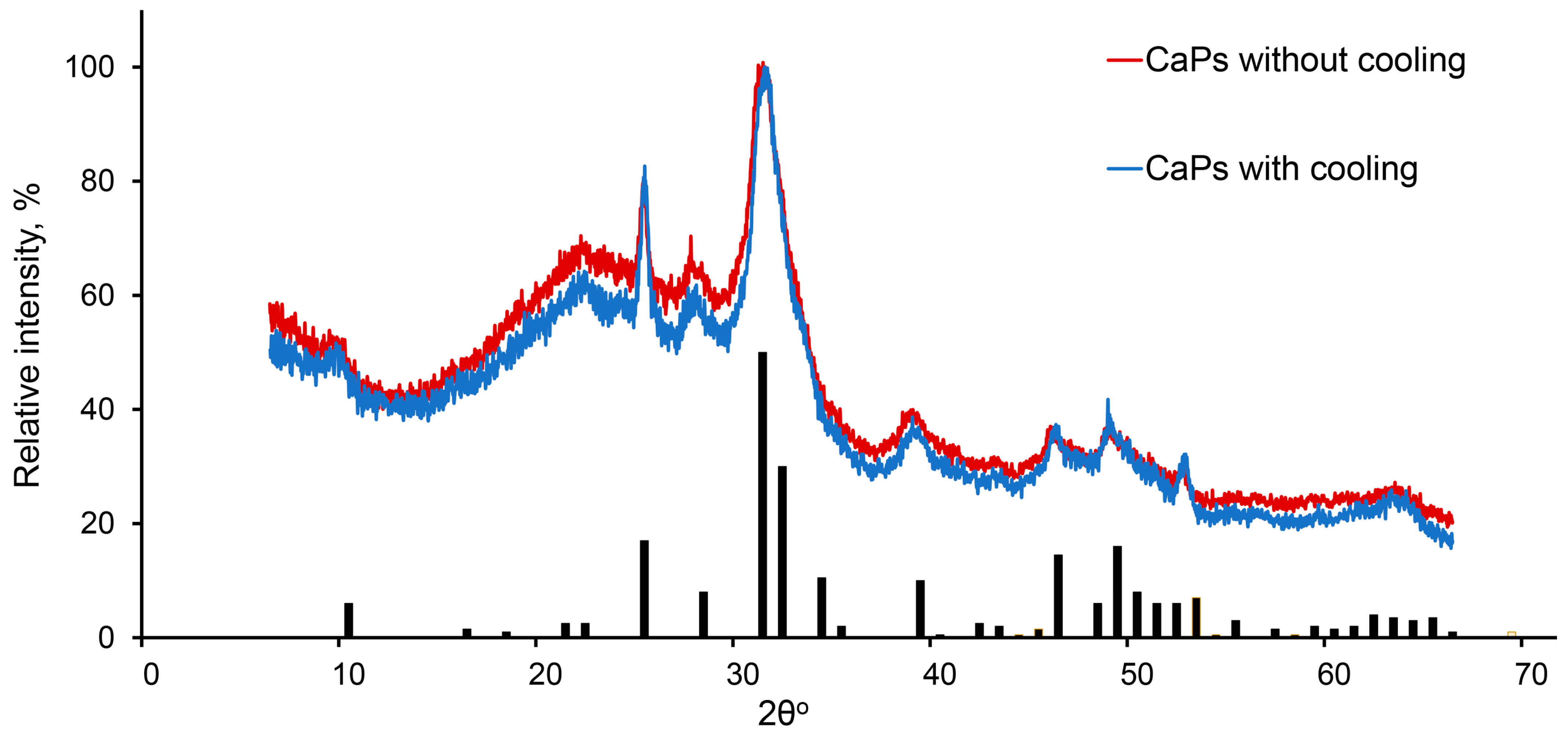
It was shown previously that at higher pH values and higher temperature, hydroxyapatite is formed [
35], while in a more acidic environment and lower temperature amorphous, calcium phosphate tends to be formed [
36]. In our conditions, the phase composition of CaP samples obtained by two chosen methods turned out to be the same. They consisted of both hexagonal hydroxyapatite (Ca
10(PO
4)
6(OH)
2) and amorphous calcium phosphate (Ca
x(PO
4)
y·zH
2O), which could be determined by the broadening of the peaks corresponding to the crystalline hydroxyapatite phase (
Figure 3).
Thus, we applied two methods of CaP synthesis—the first method was without cooling during ultrasonication, with a higher pH value of the stock solution of potassium phosphate and lower concentrations of sodium citrate; the second method was with ice cooling during ultrasonic treatment, with a lower pH value of the stock solution of potassium phosphate and higher concentrations of sodium citrate. Both methods led to a formation of nanoparticles which were characterized by similar phase composition, and similar mean hydrodynamic diameter and ζ-potential. Nevertheless, CaPs obtained at lower temperatures were smaller and more prone to aggregation.
Both types of CaPs, however, formed stable water suspensions which make them prospective for their use in different areas, including medicine and agriculture. So, CaPs appeared to be quite stable as suspensions in water and did not change the size and ζ-potential during storage for a month at 4 °C. The stability of CaPs in the physiological solution (0.15 M NaCl, pH 7.4) was lower, and the particles did not change their characteristics for two weeks; after that, aggregates began to form, although they were still soluble. Stability increased in the presence of albumin in the physiological solution, i.e., in a medium imitating tear fluid. Slow degradation of calcium phosphate in most human body fluids and cells is not a drawback but rather a merit, as this property does not allow the particles to accumulate somewhere in the organism with unexpected side-effects, while the stability of CaP suspensions in water provide a possibility of the use of such suspensions in the crop industry.
2.2. Inclusion of Different Substances into CaPs
2.2.1. Low Molecular Weight Substance Inclusion
To test the capacity of CaPs obtained by the two methods, we incorporated a model low-molecular-weight active substance, the angiotensin-converting enzyme (ACE) inhibitor enalaprilat (MW 348.4 Da), at the stage of CaP formation (referred to as coprecipitation), and by the sorption on the preliminarily obtained CaPs (
Table 5). We estimated the amount of enalaprilat bound with CaPs by a difference between the initial amount of enalaprilat and the amount found in filtrate after ultracentrifugation. As these values corresponded to the amounts of enalaprilat eluted from CaPs, we considered this approach as correct.
For the estimation of a weight load of enalaprilat, the corresponding amount of empty CaPs was washed with water, lyophilized, and weighed.
It turned out that the percentages of the inclusion of enalaprilat into CaPs obtained with and without cooling were different: 27 ± 4% and 40 ± 10%, respectively. Note that the contents of CaPs obtained with equal stock salt solutions, but using two methods described above, differed twice, with the content of CaPs with cooling being about two-fold lower. So, for the proper comparison of the capacities of two types of CaPs, we used equal ratios of enalaprilat/CaPs. The weight load of enalaprilat inclusion into CaPs without cooling was also higher (
Table 5). The difference in this enalaprilat capacity is likely due to the size and morphology of CaPs obtained in different conditions (
Figure 1), as the volume of individual particles obtained without any temperature precautions was larger than that of CaPs obtained with cooling.
The inclusion of enalaprilat into two types of CaPs provided a slight increase in hydrodynamic diameter to 110 ± 20 nm and a decrease in ζ-potential to −29 ± 2 mV. Enalaprilat-loaded particles remained completely stable (their hydrodynamic diameter and ζ-potential did not change) for two weeks, then they began to aggregate and increased their size to 200 nm after two months of storage. Meanwhile, their ζ-potential in absolute value decreased to −8 mV.
Unexpectedly, we did not detect any enalaprilat sorption on the preliminarily formed CaPs obtained with cooling, while the sorption on CaPs without cooling was rather high, although the percentage of the binding was much less (about 5-fold) than in the case of coprecipitation (
Table 5) and was less than 10% of the initial amount.
Thus, a low-molecular-weight enalaprilat is likely able to be included in the structure of CaPs as a result of coprecipitation by interacting with Ca2+, so the efficiency of its inclusion in CaPs in this way was quite high. However, the properties of CaP surfaces seem to be unfavorable for enalaprilat binding, which can be explained by a negative charge of enalaprilat and negative ζ-potential of CaPs themselves, besides the fact that the CaPs obtained with cooling required a larger amount of the stabilization agent, sodium citrate.
2.2.2. Protein Inclusion
To test the protein capacity of CaPs, we chose the enzyme, recombinant superoxide dismutase 1 (SOD1, MW 32.5 kDa), the inclusion of which could be followed both by protein and catalytic activity and tried to include this enzyme into CaPs obtained by the two methods: both by coprecipitation with CaPs and by sorption on the preliminarily obtained particles. As we expected, SOD1 failed to coprecipitate with CaPs without cooling: we observed intense foaming, and the enzyme irreversibly lost its catalytic activity due to the increase in temperature during ultrasonic treatment. However, SOD1 can be included into CaPs by coprecipitation with cooling, likely due to electrostatic coordination with calcium ions, although with a rather low yield of 11 ± 3%. The weight load of SOD1 into CaPs was equal to 6.6 ± 2 µg/mg particles. The loading of SOD1 into CaPs resulted in a slight increase in their hydrodynamic diameter from 80 nm to 110 ± 25 nm, and the ζ-potential remained unchanged.
The sorption of SOD1 on CaPs gave no result, likely because of the negative charge of SOD1 (pI about 4.8) under sorption conditions, as it was in the case of enalaprilat (see above). Thus, negative-charged enzyme SOD1 could only be included into CaPs by the coprecipitation technique.
SOD1-loaded CaPs remained stable (their hydrodynamic diameter and ζ-potential did not change) for at least two weeks, after which they started to aggregate, but they still retained suspension without precipitation. Loading SOD1 into CaPs allowed the enzyme to significantly prolong its activity during storage. SOD1 within CaPs reduced its activity to 95 ± 5% in a week, whereas SOD1 in an aqueous solution completely lost its activity by this time. Storing SOD1-loaded CaPs for two months reduced its enzymatic activity to 60 ± 10%.
2.2.3. DNA Inclusion
We showed that in the presence of DNA (MW 270–500 kDa from salmon milt) CaPs did not form at all; that is, likely large DNA molecules were just a formation of obstacle nanoparticles. So, DNA inclusion into CaPs by coprecipitation is highly unlikely.
However, we found that DNA was able to bind effectively to CaPs by sorption, especially in the presence of additional metal ions. The interaction of nucleic acids and CaPs is mediated by the electrostatic attractions between Ca
2+ and the phosphate groups of DNA or RNA [
7,
37,
38,
39], while additional ions can act as bridges between the nucleic acid and the surface of the carrier bearing a negative charge [
40,
41]. So, we chose a pair of divalent ions, Ca
2+ and Mg
2+, which were previously shown to enhance an adsorption of DNA to hydroxylapatite nanorods and a clay montmorillonite.
Thus, we studied the sorption of DNA on CaPs obtained without cooling in the presence of additional concentrations of Ca
2+ and Mg
2+. It appeared that at low-ions concentrations, less than 5 mM, the efficiency of DNA sorption was also low (
Figure 4).
Overall, the difference between the effects of Ca
2+ and Mg
2+ on DNA sorption on CaPs was statistically insignificant, with the exception of the concentrations around 2.5 mM, where Ca
2+ was more effective (
Figure 4). It should be noted that in these experiments we used a fixed DNA concentration of 50 μg/mL, and increasing it to 100 μg/mL reduced the relative efficiency of sorption (
Table 6). For further experiments, a concentration of 5 mM was chosen as a minimal additional divalent ion concentration when almost complete DNA binding to CaPs was observed.
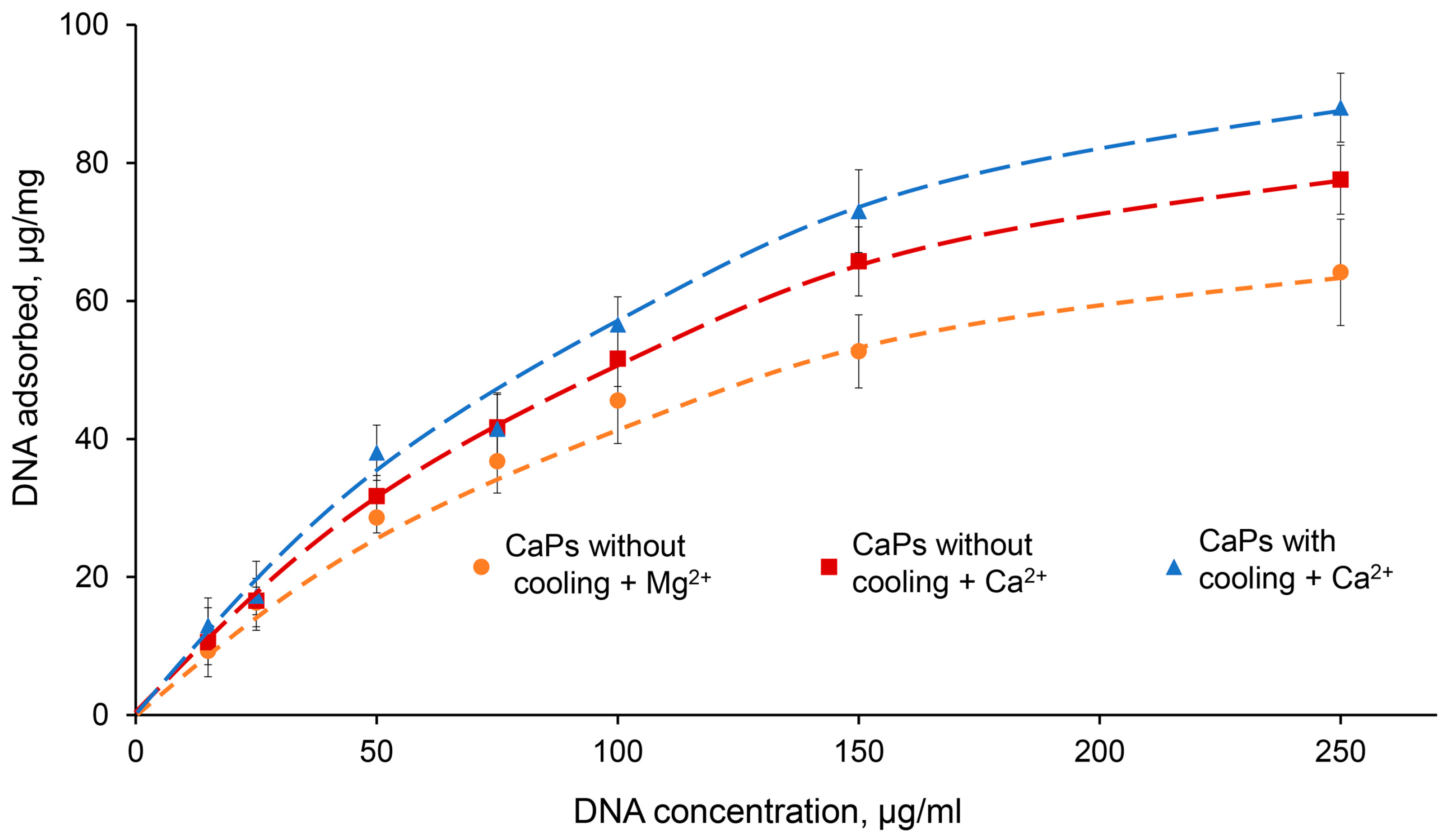
In order to characterize DNA sorption on CaPs more completely, we studied the isotherm of DNA sorption on CaPs obtained with and without cooling under optimal conditions, that is in the presence of 5 mM Ca
2+ or Mg
2+ (
Figure 5). Note that during the sorption of DNA on CaPs obtained with cooling in the presence of Mg
2+, it was not possible to precipitate the suspension properly by centrifugation and, therefore, we could not determine the amount of unbound DNA, and the sorption isotherm could not be obtained for this case.
As can be seen from the graph, at low DNA concentrations (up to 50 μg/mL) there was practically no difference between the curves obtained for CaPs with and without cooling, as well as for different ions; that is, the presence of a higher concentration of citrate required for the synthesis of CaPs with cooling was not important for DNA sorption. However, at higher DNA concentrations, 150 and 250 μg/mL, where the amount of sorbed DNA on the particles was also significantly higher, Ca
2+ demonstrated higher efficiency compared to Mg
2+. This effect of its action was more pronounced in the case of CaPs obtained with cooling (
Figure 5). The adsorption profile can be described by the Langmuir isotherm, which suggests monolayer adsorption. It is possible that, after sorption, large surface DNA may repel further incoming DNA molecules. Fitting the data to the Langmuir isotherm equation, X = X
∞KC/(1 + KC), where X is the amount of DNA adsorbed per unit mass of CaPs, X
∞ is the maximum amount of DNA that may be adsorbed, K is the Langmuir constant, C is the DNA concentration, and R
2 is the coefficient of determination; the sorption data were linearized in C/X—C coordinates (
Table 7).
Thus, CaPs obtained with cooling in the presence of 5 mM Ca2+ had the greatest adsorption capacity toward DNA.
2.3. A Release of Different Substances from CaPs
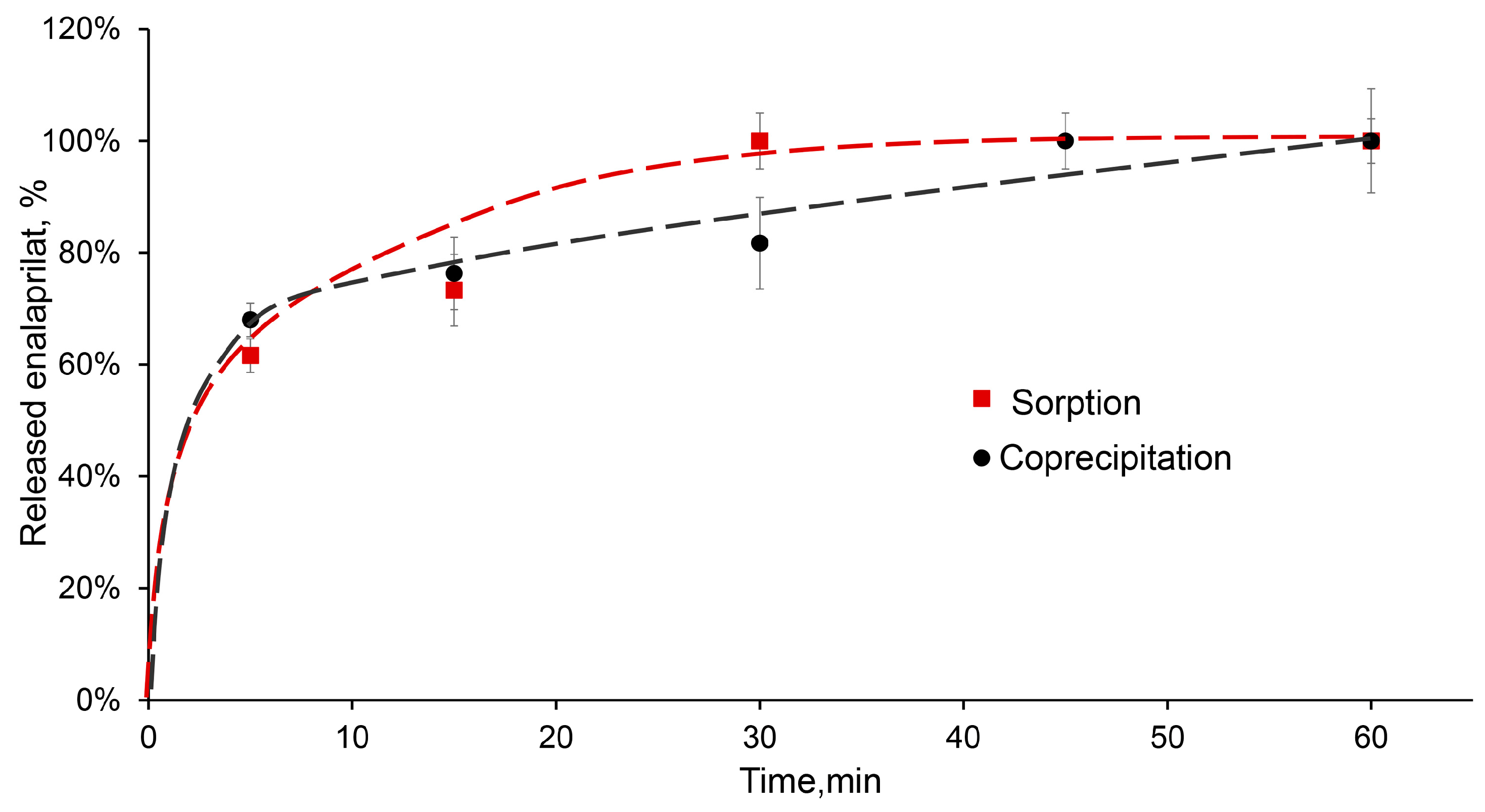
The desorption of low-molecular-weight substance, enalaprilat, from CaPs obtained without cooling was studied in a saline solution at pH 7.5. Enalaprilat included in CaPs by coprecipitation almost completely released from the particles in half an hour (
Figure 6). It is interesting that enalaprilat sorbed on CaPs released into the medium practically with the same kinetics, despite the different content of enalaprilat in these CaPs (
Table 5) and, likely, different enalaprilat distribution throughout the particle (
Figure 6).
The most suitable model for the release of enalaprilat from CaPs was the Korsmeyer–Peppas model (
Table 8). It takes into account the penetration of the medium into the matrix. The constant
n in the Korsmeyer–Peppas equation characterizes the type of diffusion and allows us to evaluate the mechanism of drug release. If the value of
n ≤ 0.5, then the release occurs due to diffusion, obeying Fick’s laws; a value in the range of 0.5 <
n < 1.0 indicates an anomalous transport that does not obey Fick’s laws. The release of enalaprilat from CaPs, whether coprecipitated or sorbed, was characterized by Fickian diffusion.
The release of the enzyme SOD1 coprecipitated with CaPs from the particles appeared to occur explosively; all the included SOD1 was released after 5 min of incubation in 0.15 M saline solution, which indicates low binding of the enzyme within CaPs.
The release of DNA sorbed on CaPs was studied in 0.01 M citric-phosphate buffer, distilled water, and saline solution. All solutions had a pH value of 6.5. DNA release was not observed in either distilled water or saline for 24 h, but the use of citric-phosphate buffer allowed DNA to be completely washed out from CaPs (
Figure 7). Apparently, this is ensured by the type of interaction between DNA and particles: the absence of the release during elution with saline indicates a non-electrostatic interaction, and the release in the presence of a phosphate anion is due to the replacement of the phosphate backbone of the DNA molecule in the complexes with PO
43− [
40].
This result is important for the putative usage of such complexes in the crop industry. There is no need to use acidic media or solutions with high salt concentrations in order to destroy the complexes; if necessary, the complexes would dissociate in the presence of a buffer, while being completely stable in water.
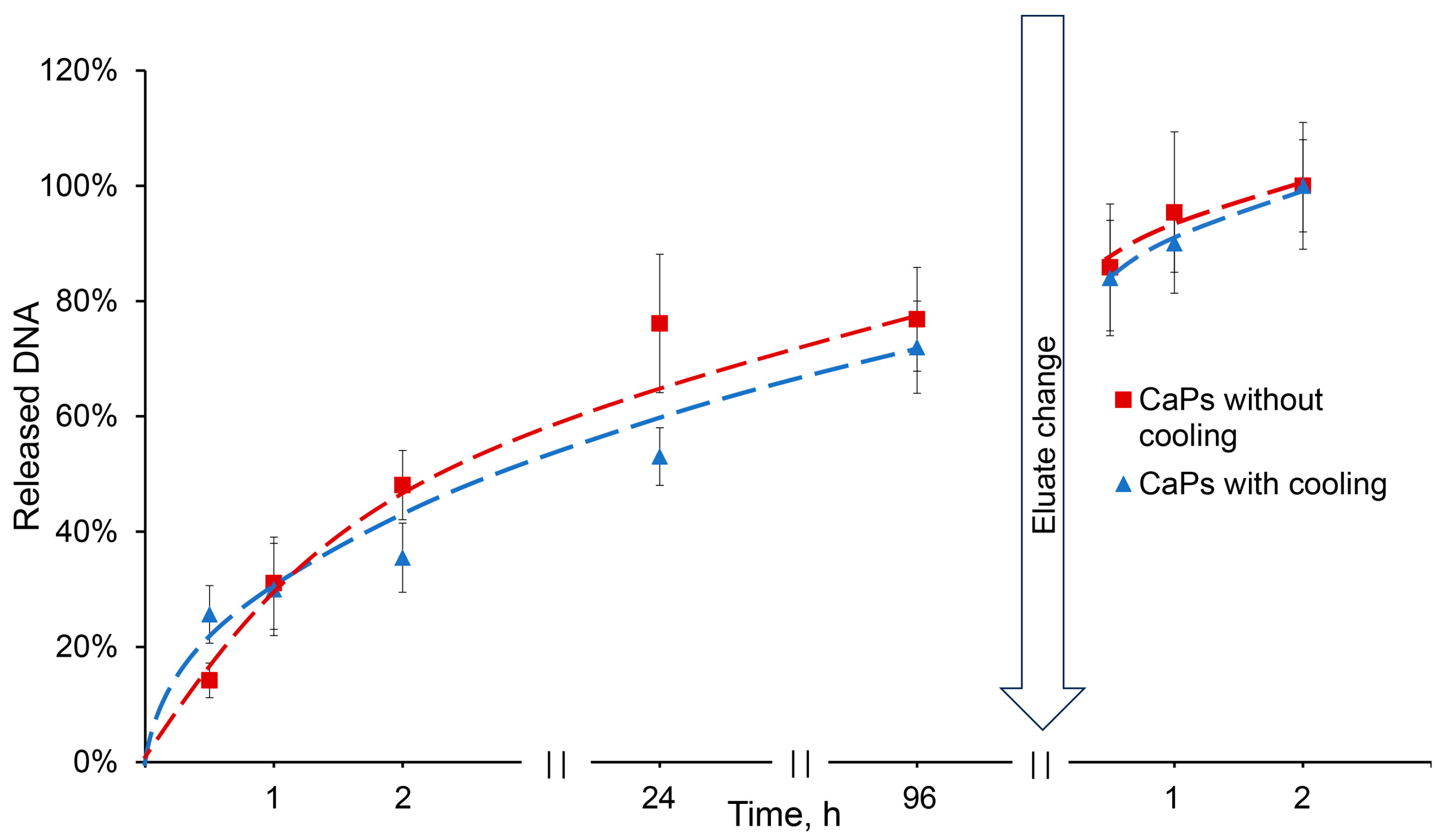
DNA release from CaPs obtained with/without cooling occurred in a similar manner. About 25% of the incorporated DNA was released in half an hour from CaPs obtained with cooling, while only about 15% was released from CaPs obtained without cooling. Then, the release of DNA slowed down, but after three days an equilibrium of 75% desorption was reached in both cases. After changing an elution buffer, the DNA was completely released from the particles (
Figure 7).
The release data were fitted to various models (
Table 9). The best-fitting model was assessed using the correlation coefficient (R
2). As can be seen from the table, the most suitable model is the same Korsmeyer–Peppas model; other models describe the release of DNA from CaP particles as much worse. A values of
n equal to 0.88 and 0.52 both indicate an anomalous DNA transport that does not obey Fick’s laws.
2.4. Studying of dsRNA Sorption on CaPs
The sorption of dsRNA on CaPs was carried out in the same way as for the DNA, in the presence of Ca
2+. The obtained complexes were analyzed with electrophoretic mobility shift assay (EMSA) and studied by AFM (
Figure 8 and
Figure 9).
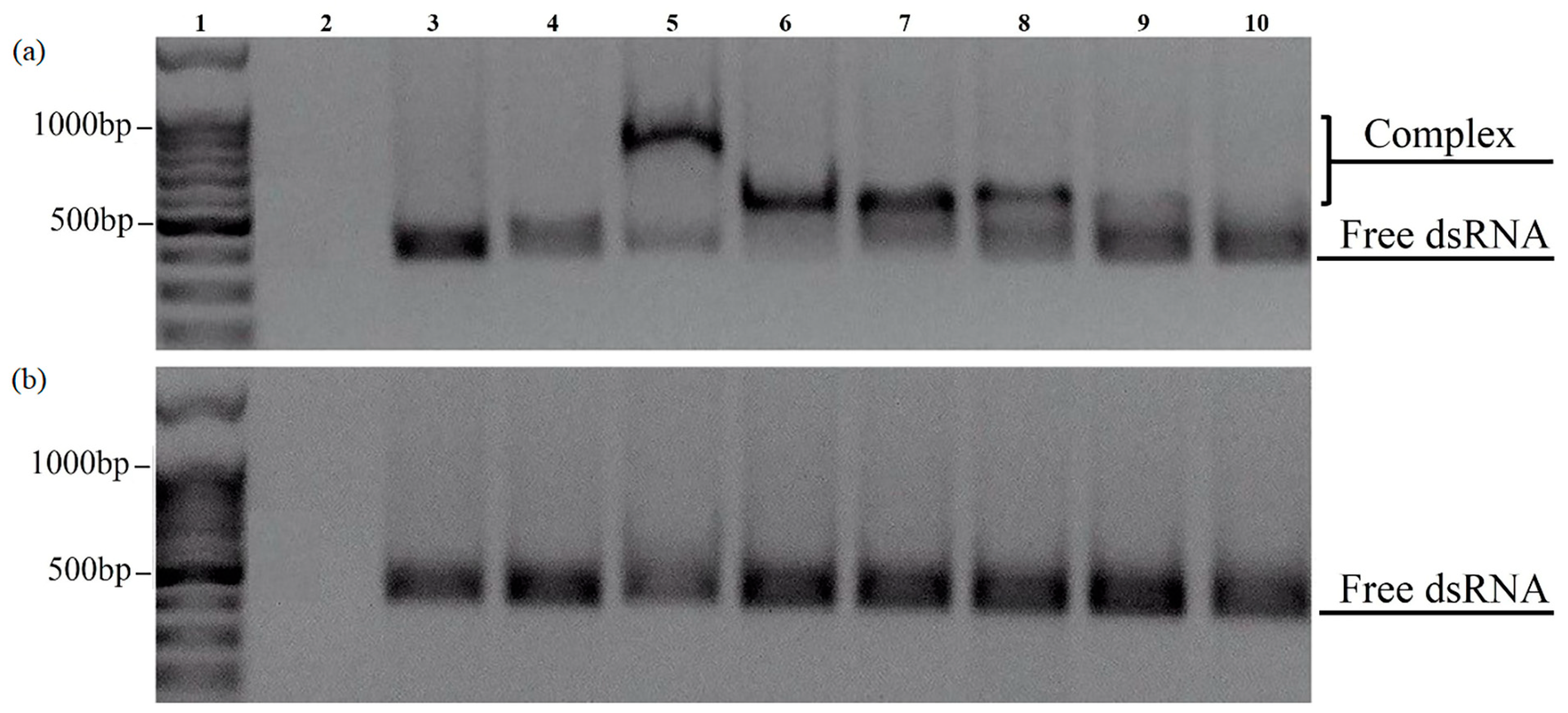
Electrophoresis showed that the CaPs pre-formed with cooling actually did not form complexes with dsRNA regardless of the presence or absence of Ca
2+, as at different CaPs:dsRNA ratios we only observed bands corresponding to free RNA (
Figure 8b), while DNA was able to bind to both types of CaPs. Thus, while the presence of a higher concentration of citrate at CaP synthesis with cooling did not affect DNA sorption to CaPs, it appeared to be critical for the sorption of RNA.
In contrast, CaPs pre-formed without cooling did form complexes with dsRNA but only in the presence of Ca
2+ (see bands 5–8 in
Figure 8a).
Moreover, an increase in the concentration of Ca2+ appears to significantly enhance affinity (binding) of dsRNA to preincubated CaPs, suggesting that Ca2+ are absolutely essential for CaPs-dsRNA binding.
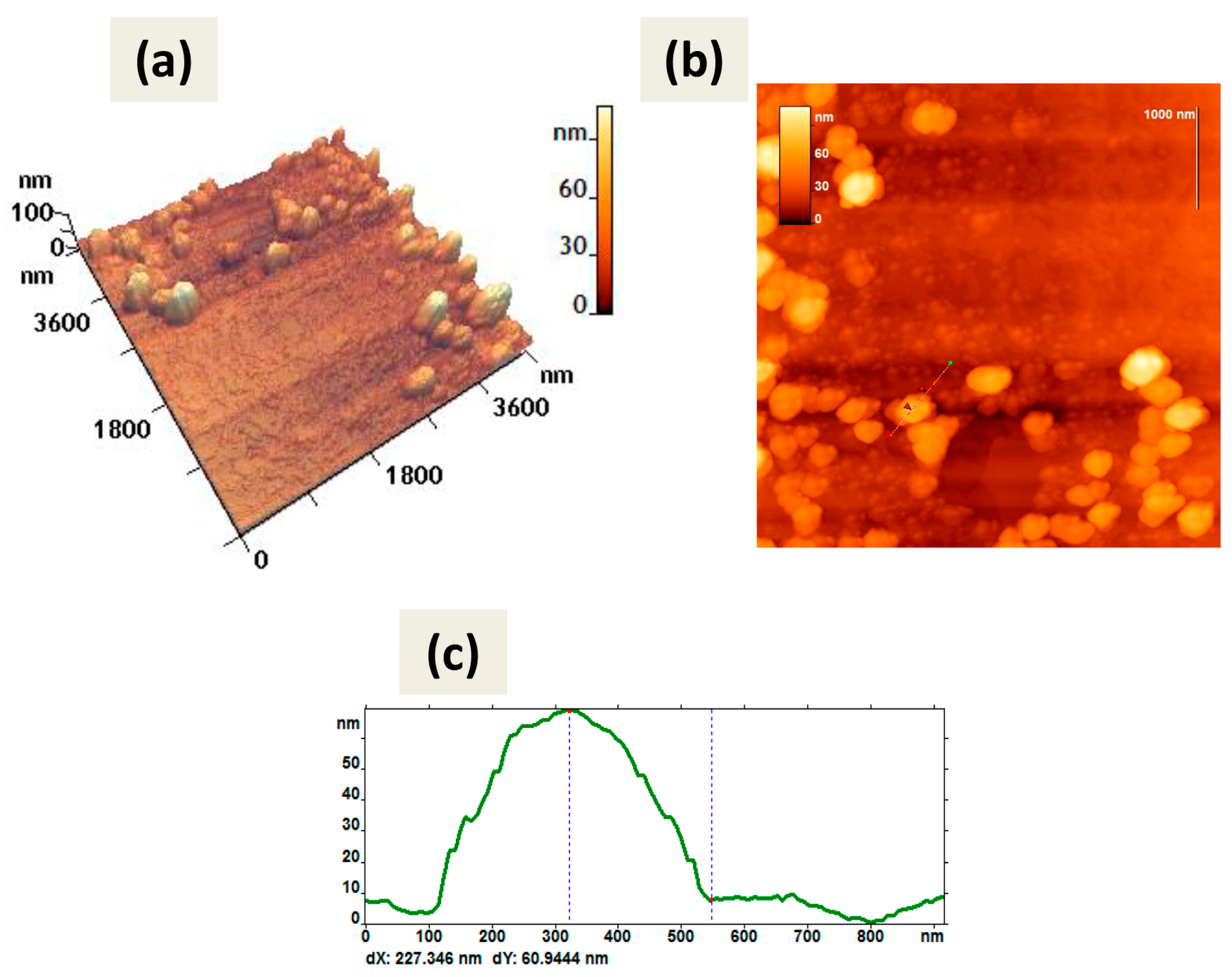
The inability of CaPs obtained with cooling to bind dsRNA was confirmed by AFM, as we observed only several accumulations of dsRNA molecules and CaPs in the AFM picture on graphite substrate, whereas CaPs obtained without cooling formed complexes with dsRNA, as we observed a noticeable change in the objects’ morphology (
Figure 9) compared with the original CaPs (
Figure 2).
The shape of the observed objects slightly changed; they became more rounded. All geometric parameters increased significantly: perimeter (increased by one third), area (more than doubled), and roughness (almost doubled) (
Table 10). The height of the observed objects also increased: the average height doubled from 35 nm to 60 nm; the maximum height increased by more than one-third. Moreover, the balls with a height of about 6–10 nm, likely belonging to dsRNA, were also visible on the surface of observed objects (
Figure 9).
Thus, CaPs obtained without cooling can adsorb dsRNA and, therefore, serve as its carriers.

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}