
A Comprehensive Functional Investigation of the Human Translocator Protein 18 kDa (TSPO) in a Novel Human Neuronal Cell Knockout Model

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
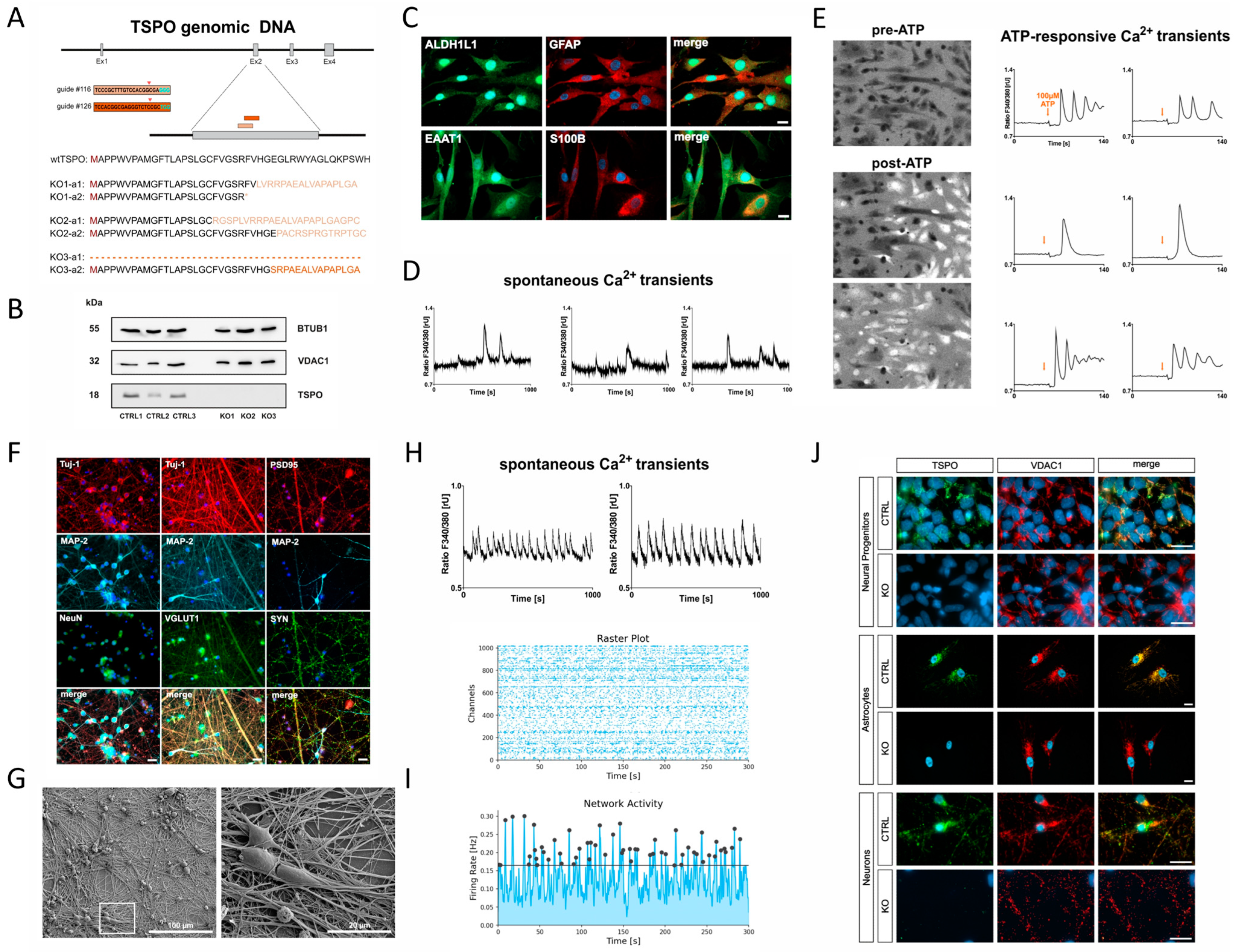
2.1. Confirmation of TSPO Knockout in hiPSCs
2.2. Differentiation of hiPSC-Derived NPCs into Astrocytes
2.3. Differentiation of hiPSC-Derived NPCs into Neurons
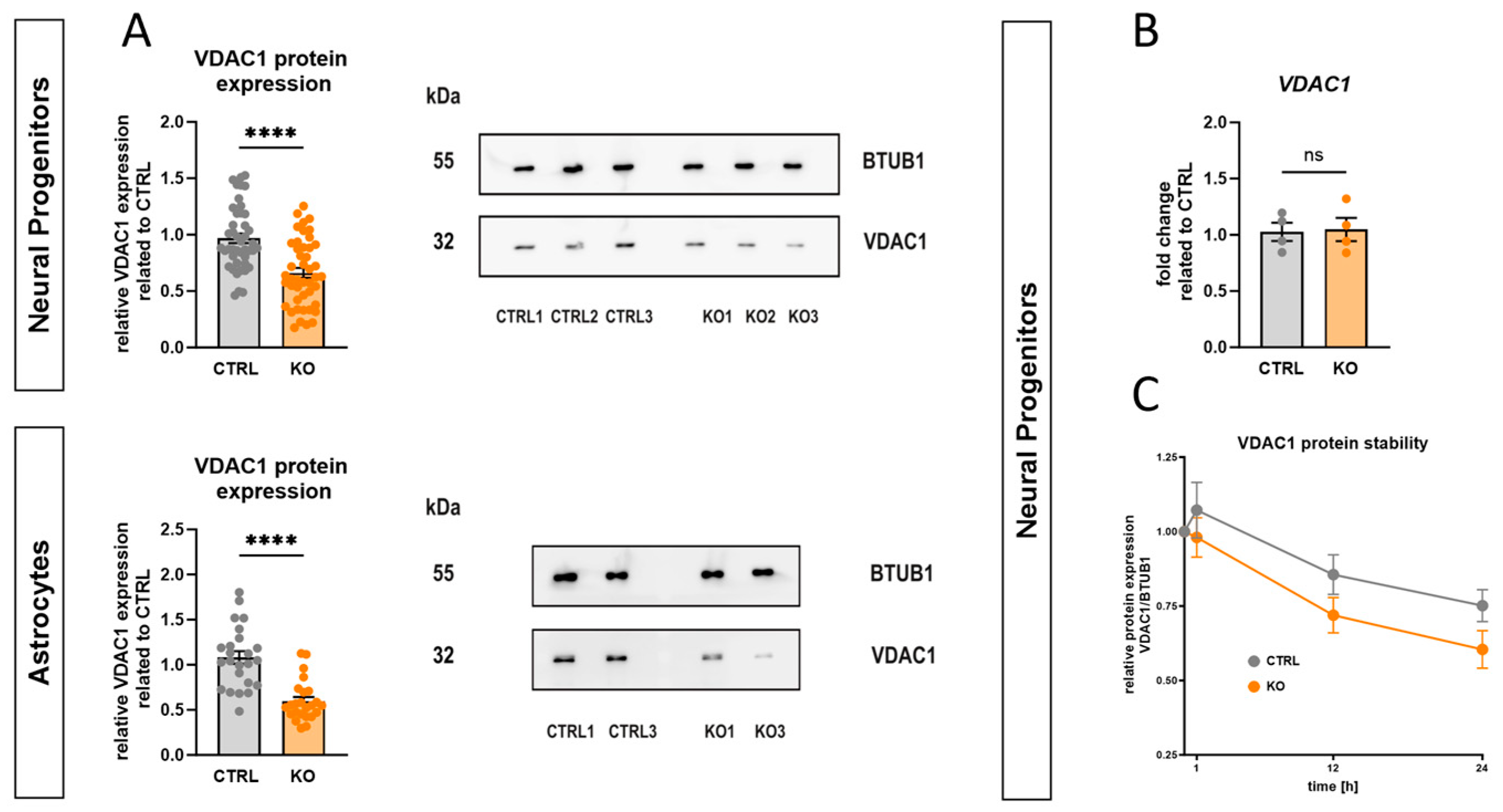
2.4. Confirmation of TSPO Knockout in NPCs, Astrocytes, and Neurons
2.5. Effect of TSPO Expression on Steroid Synthesis in hiPSC-Derived Astrocytes
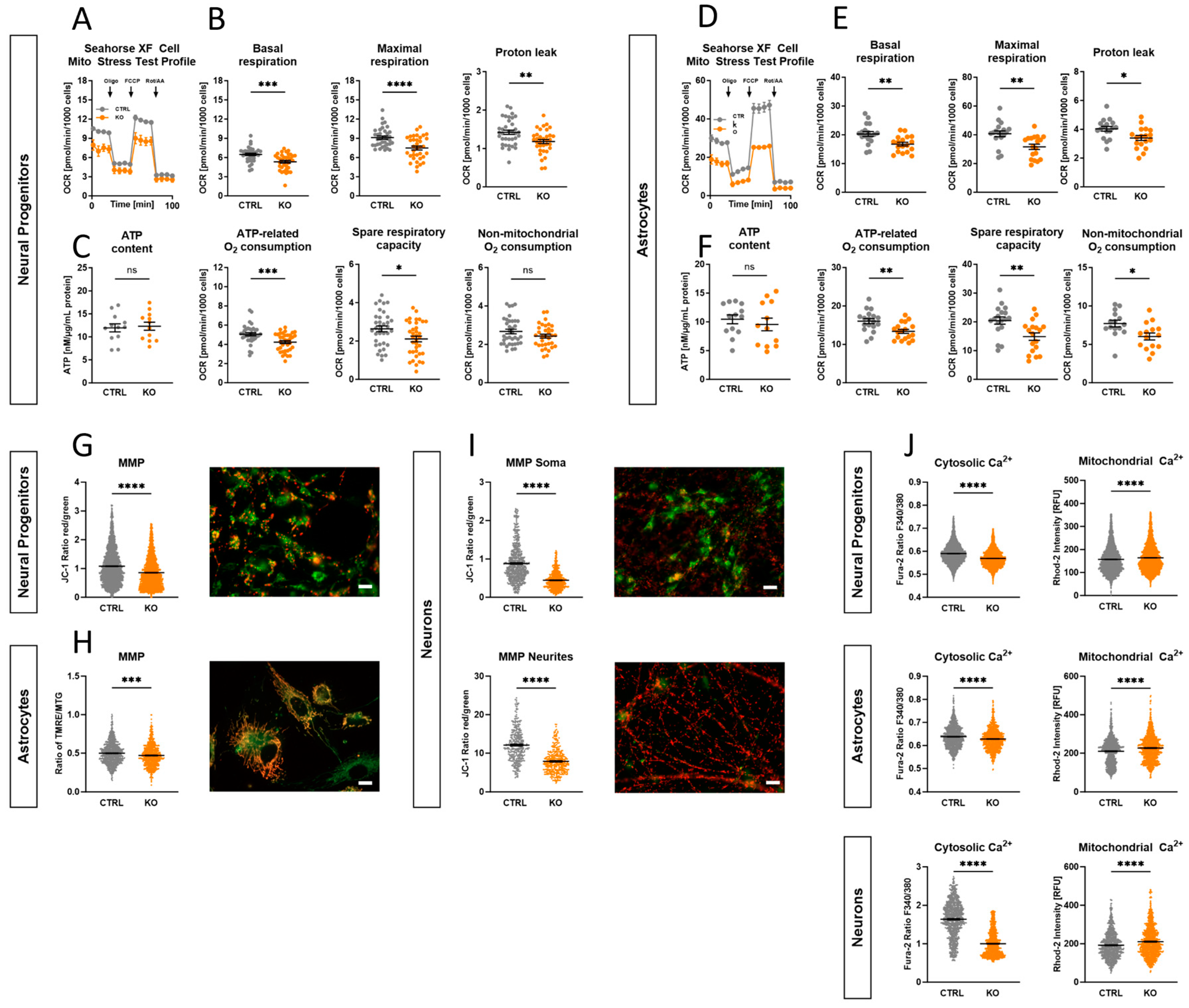
2.6. Impact of TSPO Expression on Mitochondrial Respiration
2.7. Role of TSPO in the Modulation of the Mitochondrial Membrane Potential
2.8. Involvement of TSPO in Ca2+ Homeostasis
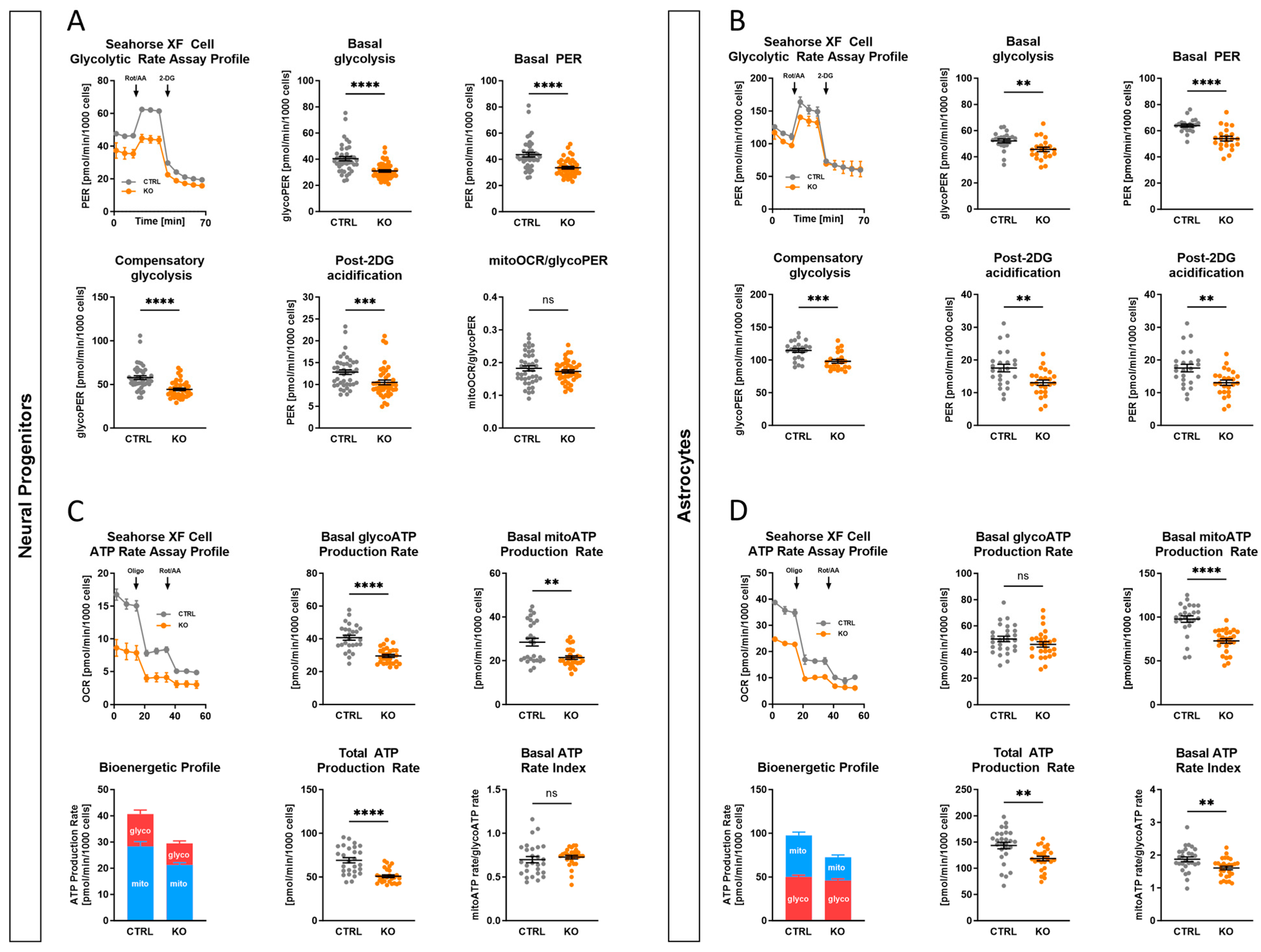
2.9. Impact of TSPO on Cellular Bioenergetics and Glycolysis
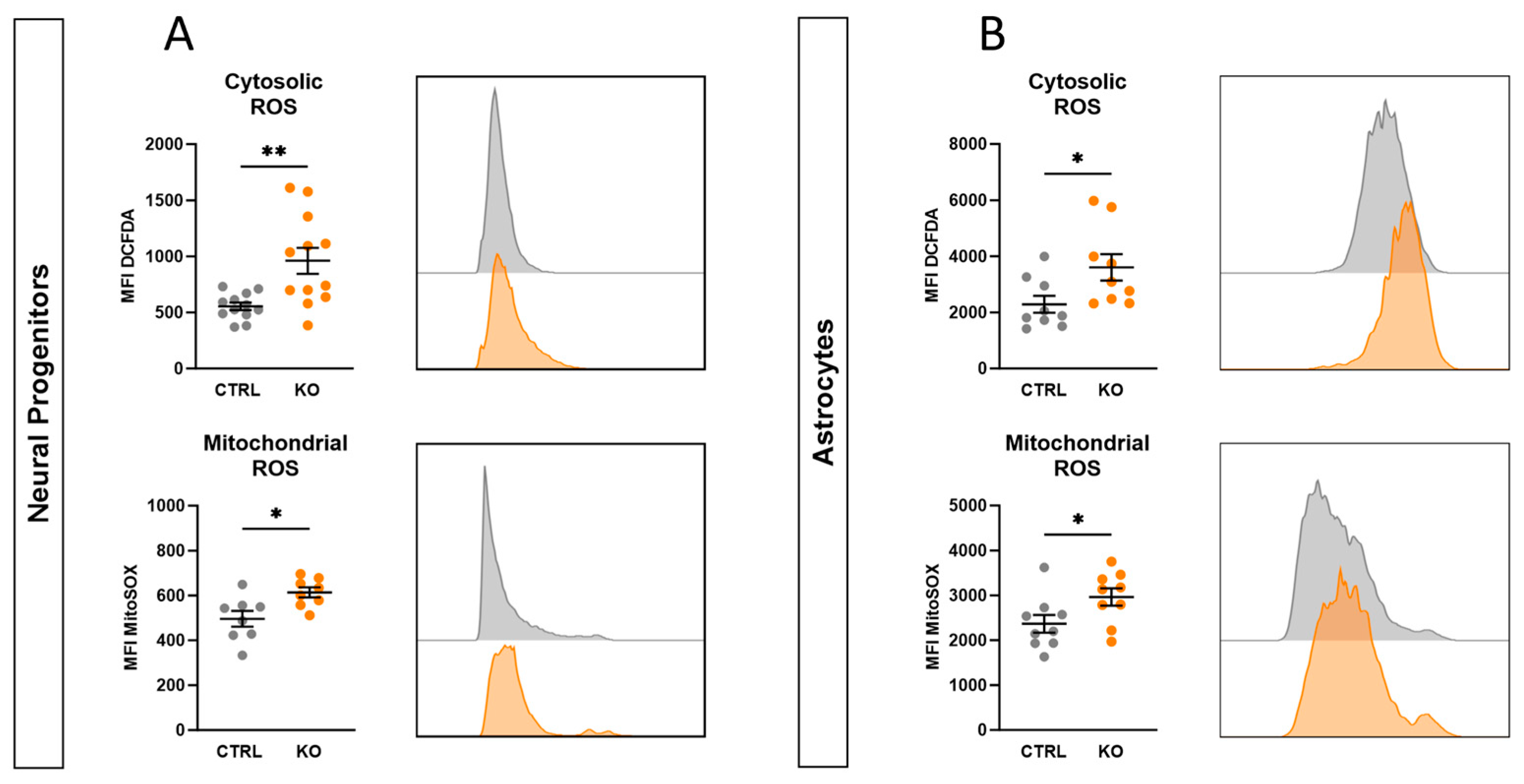
2.10. TSPO-Deficient Neural Progenitors and Astrocytes Show Oxidative Stress
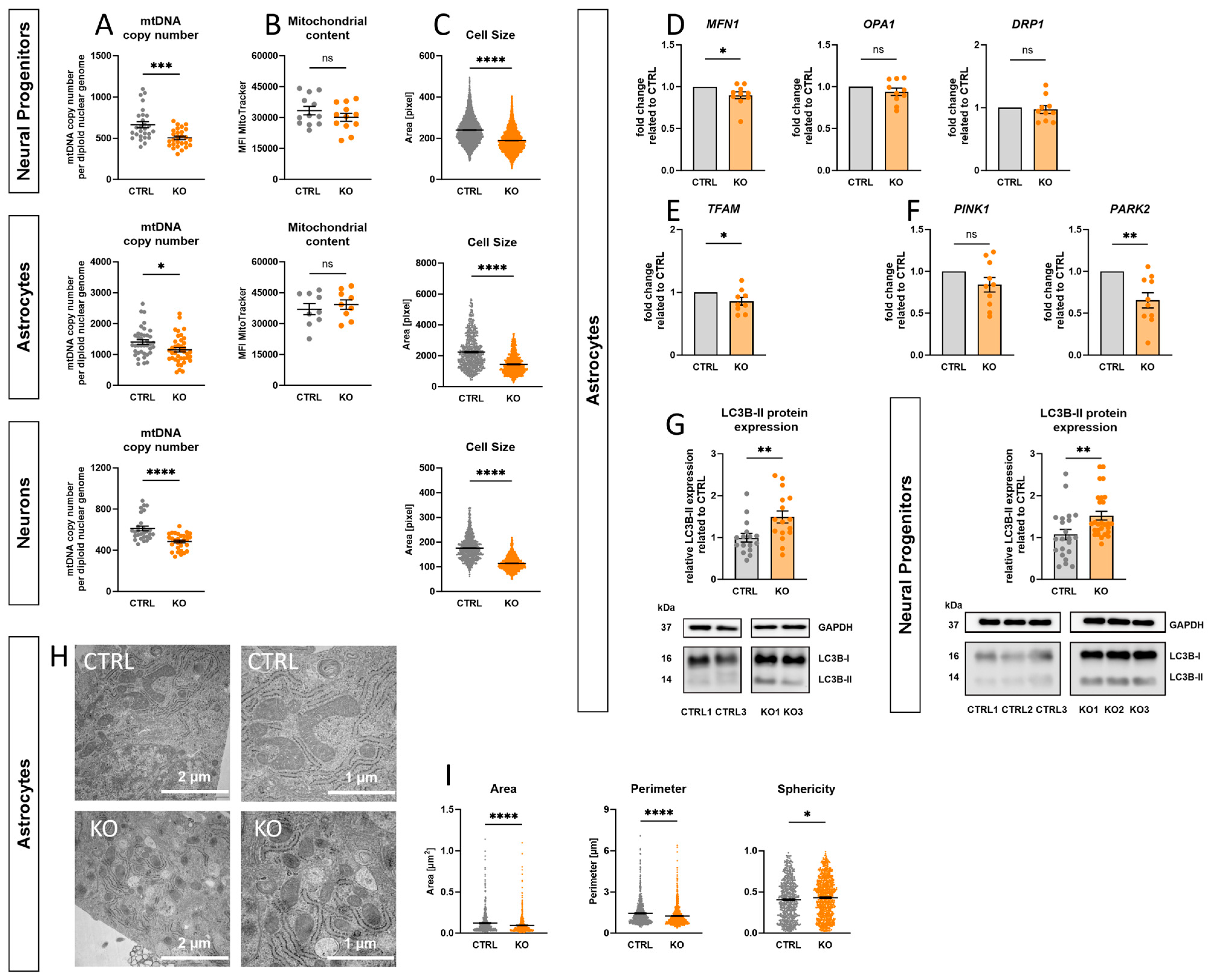
2.11. Effect of TSPO-Deficiency on mtDNA Copy Number, Mitochondrial Content, and Cell Size
2.12. Impact of TSPO on Mitochondrial Dynamics and Morphology
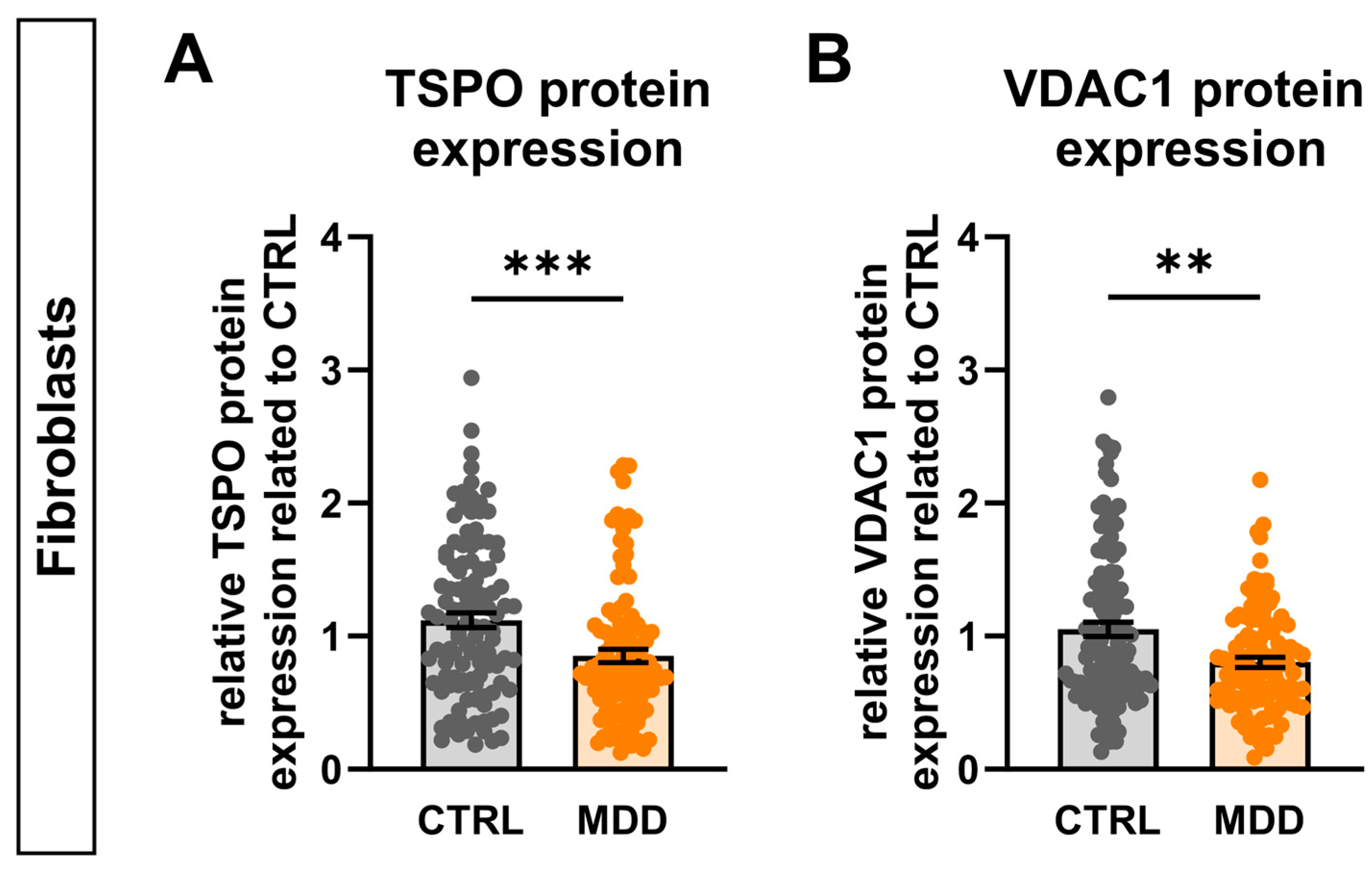
2.13. TSPO Expression in Major Depressive Disorder
3. Discussion
3.1. Role of TSPO in Neurosteroidogenesis
3.2. Impact of TSPO on Mitochondrial and Cellular Respiration
3.3. Mitochondrial Membrane Potential and Ca2+ and Redox Homeostasis
3.4. Involvement of TSPO in Mitochondrial Dynamics and Mitophagy
3.5. Possible Role for TSPO in Mitochondrial Dysfunction in Depression
4. Materials and Methods
4.1. Fibroblasts and Human-Induced Pluripotent Stem Cells
4.2. C20 Microglia and H295-R Cells
4.3. Generation of TSPO Knockout in iPSC Using CRISPR/Cas9 Genome Editing
4.4. Differentiation of iPSCs into Neural Progenitor Cells (NPCs), Neurons, and Astrocytes
4.5. Western Blotting
4.6. Immunofluorescence
4.7. Fluorescent Live-Cell Imaging
4.8. Quantitative Real-Time PCR and mtDNA Copy Number Analysis
4.9. Mitochondrial Respirometry
4.10. Total ATP Content Quantification
4.11. Scanning Electron Microscopy (SEM)
4.12. Pregnenolone Quantification
4.13. Flow Cytometry Analyses
4.14. Cytosolic and Mitochondrial Reactive Oxygen Species/Oxidative Stress
4.15. Mitochondrial Mass
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapere, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18 kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Wolf, L.; Bauer, A.; Melchner, D.; Hallof-Buestrich, H.; Stoertebecker, P.; Haen, E.; Kreutz, M.; Sarubin, N.; Milenkovic, V.M.; Wetzel, C.H.; et al. Enhancing neurosteroid synthesis—Relationship to the pharmacology of translocator protein (18 kDa) (TSPO) ligands and benzodiazepines. Pharmacopsychiatry 2015, 48, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocrinol. 2018, 30, e12500. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Fan, J.; Campioli, E.; Venugopal, S.; Midzak, A.; Daly, E.; Harlay, A.; Issop, L.; Libri, V.; Kalogiannopoulou, D.; et al. TSPO mutations in rats and a human polymorphism impair the rate of steroid synthesis. Biochem. J. 2017, 474, 3985–3999. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V. On the role of the translocator protein (18-kDa) TSPO in steroid hormone biosynthesis. Endocrinology 2014, 155, 15–20. [Google Scholar] [CrossRef]
- Liu, G.J.; Middleton, R.J.; Kam, W.W.; Chin, D.Y.; Hatty, C.R.; Chan, R.H.; Banati, R.B. Functional gains in energy and cell metabolism after TSPO gene insertion. Cell Cycle 2017, 16, 436–447. [Google Scholar] [CrossRef]
- Milenkovic, V.M.; Slim, D.; Bader, S.; Koch, V.; Heinl, E.S.; Alvarez-Carbonell, D.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. CRISPR-Cas9 Mediated TSPO Gene Knockout alters Respiration and Cellular Metabolism in Human Primary Microglia Cells. Int. J. Mol. Sci. 2019, 20, 3359. [Google Scholar] [CrossRef]
- Bader, S.; Wurfel, T.; Jahner, T.; Nothdurfter, C.; Rupprecht, R.; Milenkovic, V.M.; Wetzel, C.H. Impact of Translocator Protein 18 kDa (TSPO) Deficiency on Mitochondrial Function and the Inflammatory State of Human C20 Microglia Cells. Cells 2023, 12, 954. [Google Scholar] [CrossRef]
- Tu, L.N.; Zhao, A.H.; Hussein, M.; Stocco, D.M.; Selvaraj, V. Translocator Protein (TSPO) Affects Mitochondrial Fatty Acid Oxidation in Steroidogenic Cells. Endocrinology 2016, 157, 1110–1121. [Google Scholar] [CrossRef]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279–2296. [Google Scholar] [CrossRef]
- Gatliff, J.; East, D.A.; Singh, A.; Alvarez, M.S.; Frison, M.; Matic, I.; Ferraina, C.; Sampson, N.; Turkheimer, F.; Campanella, M. A role for TSPO in mitochondrial Ca2+ homeostasis and redox stress signaling. Cell Death Dis. 2017, 8, e2896. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Lecanu, L. Translocator protein (18 kDa) TSPO: An emerging therapeutic target in neurotrauma. Exp. Neurol. 2009, 219, 53–57. [Google Scholar] [CrossRef]
- Bader, S.; Wolf, L.; Milenkovic, V.M.; Gruber, M.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. Differential effects of TSPO ligands on mitochondrial function in mouse microglia cells. Psychoneuroendocrinology 2019, 106, 65–76. [Google Scholar] [CrossRef]
- Rupprecht, R.; Wetzel, C.H.; Dorostkar, M.; Herms, J.; Albert, N.L.; Schwarzbach, J.; Schumacher, M.; Neumann, I.D. Translocator protein (18kDa) TSPO: A new diagnostic or therapeutic target for stress-related disorders? Mol. Psychiatry 2022, 27, 2918–2926. [Google Scholar] [CrossRef] [PubMed]
- Ammer, L.M.; Vollmann-Zwerenz, A.; Ruf, V.; Wetzel, C.H.; Riemenschneider, M.J.; Albert, N.L.; Beckhove, P.; Hau, P. The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma. Cancers 2020, 12, 2973. [Google Scholar] [CrossRef]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Barron, A.M.; Higuchi, M.; Hattori, S.; Kito, S.; Suhara, T.; Ji, B. Regulation of Anxiety and Depression by Mitochondrial Translocator Protein-Mediated Steroidogenesis: The Role of Neurons. Mol. Neurobiol. 2021, 58, 550–563. [Google Scholar] [CrossRef]
- Zorumski, C.F.; Paul, S.M.; Covey, D.F.; Mennerick, S. Neurosteroids as novel antidepressants and anxiolytics: GABA-A receptors and beyond. Neurobiol. Stress 2019, 11, 100196. [Google Scholar] [CrossRef]
- Gunduz-Bruce, H.; Silber, C.; Kaul, I.; Rothschild, A.J.; Riesenberg, R.; Sankoh, A.J.; Li, H.; Lasser, R.; Zorumski, C.F.; Rubinow, D.R.; et al. Trial of SAGE-217 in Patients with Major Depressive Disorder. N. Engl. J. Med. 2019, 381, 903–911. [Google Scholar] [CrossRef]
- Nutma, E.; Fancy, N.; Weinert, M.; Tsartsalis, S.; Marzin, M.C.; Muirhead, R.C.J.; Falk, I.; Breur, M.; de Bruin, J.; Hollaus, D.; et al. Translocator protein is a marker of activated microglia in rodent models but not human neurodegenerative diseases. Nat. Commun. 2023, 14, 5247. [Google Scholar] [CrossRef] [PubMed]
- Weidner, L.; Lorenz, J.; Quach, S.; Braun, F.K.; Rothhammer-Hampl, T.; Ammer, L.M.; Vollmann-Zwerenz, A.; Bartos, L.M.; Dekorsy, F.J.; Holzgreve, A.; et al. Translocator protein (18kDA) (TSPO) marks mesenchymal glioblastoma cell populations characterized by elevated numbers of tumor-associated macrophages. Acta Neuropathol. Commun. 2023, 11, 147. [Google Scholar] [CrossRef] [PubMed]
- Batarseh, A.; Papadopoulos, V. Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol. Cell. Endocrinol. 2010, 327, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rone, M.B.; Midzak, A.S.; Issop, L.; Rammouz, G.; Jagannathan, S.; Fan, J.; Ye, X.; Blonder, J.; Veenstra, T.; Papadopoulos, V. Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol. Endocrinol. 2012, 26, 1868–1882. [Google Scholar] [CrossRef]
- Hiser, C.; Montgomery, B.L.; Ferguson-Miller, S. TSPO protein binding partners in bacteria, animals, and plants. J. Bioenerg. Biomembr. 2021, 53, 463–487. [Google Scholar] [CrossRef]
- Cardon, I.; Grobecker, S.; Kucukoktay, S.; Bader, S.; Jahner, T.; Nothdurfter, C.; Koschitzki, K.; Berneburg, M.; Weber, B.H.F.; Stohr, H.; et al. Mitochondrial and Cellular Function in Fibroblasts, Induced Neurons, and Astrocytes Derived from Case Study Patients: Insights into Major Depression as a Mitochondria-Associated Disease. Int. J. Mol. Sci. 2024, 25, 963. [Google Scholar] [CrossRef]
- Triebelhorn, J.; Cardon, I.; Kuffner, K.; Bader, S.; Jahner, T.; Meindl, K.; Rothhammer-Hampl, T.; Riemenschneider, M.J.; Drexler, K.; Berneburg, M.; et al. Induced neural progenitor cells and iPS-neurons from major depressive disorder patients show altered bioenergetics and electrophysiological properties. Mol. Psychiatry 2024, 29, 1217–1227. [Google Scholar] [CrossRef]
- Kuffner, K.; Triebelhorn, J.; Meindl, K.; Benner, C.; Manook, A.; Sudria-Lopez, D.; Siebert, R.; Nothdurfter, C.; Baghai, T.C.; Drexler, K.; et al. Major Depressive Disorder is Associated with Impaired Mitochondrial Function in Skin Fibroblasts. Cells 2020, 9, 884. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, C.T.; Chen, J.; Pankratz, M.T.; Xi, J.; Li, J.; Yang, Y.; Lavaute, T.M.; Li, X.J.; Ayala, M.; et al. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 2010, 7, 90–100. [Google Scholar] [CrossRef]
- Tcw, J.; Wang, M.; Pimenova, A.A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef]
- Yan, Y.; Shin, S.; Jha, B.S.; Liu, Q.; Sheng, J.; Li, F.; Zhan, M.; Davis, J.; Bharti, K.; Zeng, X.; et al. Efficient and rapid derivation of primitive neural stem cells and generation of brain subtype neurons from human pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Aguado, F.; Espinosa-Parrilla, J.F.; Carmona, M.A.; Soriano, E. Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J. Neurosci. 2002, 22, 9430–9444. [Google Scholar] [CrossRef]
- Barbar, L.; Jain, T.; Zimmer, M.; Kruglikov, I.; Sadick, J.S.; Wang, M.; Kalpana, K.; Rose, I.V.L.; Burstein, S.R.; Rusielewicz, T.; et al. CD49f Is a Novel Marker of Functional and Reactive Human iPSC-Derived Astrocytes. Neuron 2020, 107, 436–453.E12. [Google Scholar] [CrossRef]
- Scemes, E.; Giaume, C. Astrocyte calcium waves: What they are and what they do. Glia 2006, 54, 716–725. [Google Scholar] [CrossRef]
- Volterra, A.; Liaudet, N.; Savtchouk, I. Astrocyte Ca(2)(+) signalling: An unexpected complexity. Nat. Rev. Neurosci. 2014, 15, 327–335. [Google Scholar] [CrossRef]
- Uddin, L.Q. Bring the Noise: Reconceptualizing Spontaneous Neural Activity. Trends Cogn. Sci. 2020, 24, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Takeuchi, A.; Hashizume, M.; Kitamura, K.; Kano, M. A highly sensitive fluorescent indicator dye for calcium imaging of neural activity in vitro and in vivo. Eur. J. Neurosci. 2014, 39, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Artiga, D.J.; Abiria, S.A.; Clapham, D.E. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. USA 2016, 113, E1872–E1880. [Google Scholar] [CrossRef]
- Karabatsiakis, A.; Bock, C.; Salinas-Manrique, J.; Kolassa, S.; Calzia, E.; Dietrich, D.E.; Kolassa, I.T. Mitochondrial respiration in peripheral blood mononuclear cells correlates with depressive subsymptoms and severity of major depression. Transl. Psychiatry 2014, 4, e397. [Google Scholar] [CrossRef]
- Manji, H.; Kato, T.; Di Prospero, N.A.; Ness, S.; Beal, M.F.; Krams, M.; Chen, G. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 293–307. [Google Scholar] [CrossRef]
- Garbett, K.A.; Vereczkei, A.; Kalman, S.; Wang, L.; Korade, Z.; Shelton, R.C.; Mirnics, K. Fibroblasts from patients with major depressive disorder show distinct transcriptional response to metabolic stressors. Transl. Psychiatry 2015, 5, e523. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Da Pozzo, E.; Martini, C. Translocator protein and steroidogenesis. Biochem. J. 2018, 475, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Hauet, T.; Yao, Z.X.; Bose, H.S.; Wall, C.T.; Han, Z.; Li, W.; Hales, D.B.; Miller, W.L.; Culty, M.; Papadopoulos, V. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol. Endocrinol. 2005, 19, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Hershkovitz, E.; Weizman, R.; Spanier, I.; Leschiner, S.; Lahav, M.; Weisinger, G.; Gavish, M. Effects of peripheral-type benzodiazepine receptor antisense knockout on MA-10 Leydig cell proliferation and steroidogenesis. J. Biol. Chem. 1998, 273, 5478–5483. [Google Scholar] [CrossRef]
- Mukhin, A.G.; Papadopoulos, V.; Costa, E.; Krueger, K.E. Mitochondrial benzodiazepine receptors regulate steroid biosynthesis. Proc. Natl. Acad. Sci. USA 1989, 86, 9813–9816. [Google Scholar] [CrossRef]
- Gardella, K.A.; Muro, I.; Fang, G.; Sarkar, K.; Mendez, O.; Wright, C.W. Aryl hydrocarbon receptor nuclear translocator (ARNT) isoforms control lymphoid cancer cell proliferation through differentially regulating tumor suppressor p53 activity. Oncotarget 2016, 7, 10710–10722. [Google Scholar] [CrossRef]
- Banati, R.B.; Middleton, R.J.; Chan, R.; Hatty, C.R.; Kam, W.W.; Quin, C.; Graeber, M.B.; Parmar, A.; Zahra, D.; Callaghan, P.; et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014, 5, 5452. [Google Scholar] [CrossRef]
- Tu, L.N.; Morohaku, K.; Manna, P.R.; Pelton, S.H.; Butler, W.R.; Stocco, D.M.; Selvaraj, V. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J. Biol. Chem. 2014, 289, 27444–27454. [Google Scholar] [CrossRef]
- Morohaku, K.; Pelton, S.H.; Daugherty, D.J.; Butler, W.R.; Deng, W.; Selvaraj, V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology 2014, 155, 89–97. [Google Scholar] [CrossRef]
- Selvaraj, V.; Tu, L.N. Current status and future perspectives: TSPO in steroid neuroendocrinology. J. Endocrinol. 2016, 231, R1–R30. [Google Scholar] [CrossRef]
- Angeloni, E.; Germelli, L.; Marchetti, L.; Da Pozzo, E.; Tremolanti, C.; Wetzel, C.H.; Baglini, E.; Taliani, S.; Da Settimo, F.; Martini, C.; et al. The human microglial surveillant phenotype is preserved by de novo neurosteroidogenesis through the control of cholesterol homeostasis: Crucial role of 18 kDa Translocator Protein. Biochim. Biophys. Acta Mol. Basis. Dis. 2023, 1869, 166751. [Google Scholar] [CrossRef] [PubMed]
- Leverve, X.M. Mitochondrial function and substrate availability. Crit. Care Med. 2007, 35, S454–S460. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, Interactions, and Associated Functions in Health and Disease States. Int. J. Mol. Sci. 2019, 20, 3348. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Barichello, T.; Fries, G.R.; Kennon, E.A.; Andrews, T.; Nix, B.R.; Zunta-Soares, G.; Valvassori, S.S.; Soares, J.C.; Quevedo, J. TSPO upregulation in bipolar disorder and concomitant downregulation of mitophagic proteins and NLRP3 inflammasome activation. Neuropsychopharmacology 2019, 44, 1291–1299. [Google Scholar] [CrossRef]
- Schmidt, C.A.; Fisher-Wellman, K.H.; Neufer, P.D. From OCR and ECAR to energy: Perspectives on the design and interpretation of bioenergetics studies. J. Biol. Chem. 2021, 297, 101140. [Google Scholar] [CrossRef]
- Yao, R.; Pan, R.; Shang, C.; Li, X.; Cheng, J.; Xu, J.; Li, Y. Translocator Protein 18 kDa (TSPO) Deficiency Inhibits Microglial Activation and Impairs Mitochondrial Function. Front. Pharmacol. 2020, 11, 986. [Google Scholar] [CrossRef]
- Fairley, L.H.; Lai, K.O.; Wong, J.H.; Chong, W.J.; Vincent, A.S.; D’Agostino, G.; Wu, X.; Naik, R.R.; Jayaraman, A.; Langley, S.R.; et al. Mitochondrial control of microglial phagocytosis by the translocator protein and hexokinase 2 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2209177120. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Behera, S.; Alam, M.F.; Syed, G.H. Endoplasmic reticulum & mitochondrial calcium homeostasis: The interplay with viruses. Mitochondrion 2021, 58, 227–242. [Google Scholar]
- Fu, Y.; Wang, D.; Wang, H.; Cai, M.; Li, C.; Zhang, X.; Chen, H.; Hu, Y.; Zhang, X.; Ying, M.; et al. TSPO deficiency induces mitochondrial dysfunction, leading to hypoxia, angiogenesis, and a growth-promoting metabolic shift toward glycolysis in glioblastoma. Neuro Oncol. 2020, 22, 240–252. [Google Scholar] [CrossRef]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 2012, 1819, 921–929. [Google Scholar] [CrossRef]
- Jeng, J.Y.; Yeh, T.S.; Lee, J.W.; Lin, S.H.; Fong, T.H.; Hsieh, R.H. Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J. Cell. Biochem. 2008, 103, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Man, D.; Lu, J.; Jiang, Y.; Ding, B.; Su, R.; Tong, R.; Chen, J.; Yang, B.; Zheng, S.; et al. Mitochondrial TSPO Promotes Hepatocellular Carcinoma Progression through Ferroptosis Inhibition and Immune Evasion. Adv. Sci. 2023, 10, e2206669. [Google Scholar] [CrossRef] [PubMed]
- Frison, M.; Faccenda, D.; Abeti, R.; Rigon, M.; Strobbe, D.; England-Rendon, B.S.; Cash, D.; Barnes, K.; Sadeghian, M.; Sajic, M.; et al. The translocator protein (TSPO) is prodromal to mitophagy loss in neurotoxicity. Mol. Psychiatry 2021, 26, 2721–2739. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Bjorklund, M. Cellular Allometry of Mitochondrial Functionality Establishes the Optimal Cell Size. Dev. Cell 2016, 39, 370–382. [Google Scholar] [CrossRef]
- Mourier, A.; Motori, E.; Brandt, T.; Lagouge, M.; Atanassov, I.; Galinier, A.; Rappl, G.; Brodesser, S.; Hultenby, K.; Dieterich, C.; et al. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J. Cell. Biol. 2015, 208, 429–442. [Google Scholar] [CrossRef]
- Gardner, A.; Boles, R.G. Beyond the serotonin hypothesis: Mitochondria, inflammation and neurodegeneration in major depression and affective spectrum disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 730–743. [Google Scholar] [CrossRef]
- Klinedinst, N.J.; Regenold, W.T. A mitochondrial bioenergetic basis of depression. J. Bioenerg. Biomembr. 2015, 47, 155–171. [Google Scholar] [CrossRef]
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Dobeln, U.; Hagenfeldt, L.; Hallstrom, T. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 2003, 76, 55–68. [Google Scholar] [CrossRef]
- Hroudova, J.; Fisar, Z.; Kitzlerova, E.; Zverova, M.; Raboch, J. Mitochondrial respiration in blood platelets of depressive patients. Mitochondrion 2013, 13, 795–800. [Google Scholar] [CrossRef]
- Sjovall, F.; Ehinger, J.K.; Marelsson, S.E.; Morota, S.; Frostner, E.A.; Uchino, H.; Lundgren, J.; Arnbjornsson, E.; Hansson, M.J.; Fellman, V.; et al. Mitochondrial respiration in human viable platelets—Methodology and influence of gender, age and storage. Mitochondrion 2013, 13, 7–14. [Google Scholar] [CrossRef]
- Richards, E.M.; Zanotti-Fregonara, P.; Fujita, M.; Newman, L.; Farmer, C.; Ballard, E.D.; Machado-Vieira, R.; Yuan, P.; Niciu, M.J.; Lyoo, C.H.; et al. PET radioligand binding to translocator protein (TSPO) is increased in unmedicated depressed subjects. EJNMMI Res. 2018, 8, 57. [Google Scholar] [CrossRef]
- Setiawan, E.; Attwells, S.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Xu, C.; Sharma, S.; Kish, S.; Houle, S.; et al. Association of translocator protein total distribution volume with duration of untreated major depressive disorder: A cross-sectional study. Lancet Psychiatry 2018, 5, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Kanes, S.; Colquhoun, H.; Gunduz-Bruce, H.; Raines, S.; Arnold, R.; Schacterle, A.; Doherty, J.; Epperson, C.N.; Deligiannidis, K.M.; Riesenberg, R.; et al. Brexanolone (SAGE-547 injection) in post-partum depression: A randomised controlled trial. Lancet 2017, 390, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Buck, D.J.; McCracken, K.; Cox, G.W.; Das, S. Interleukin-1β-induced inflammatory signaling in C20 human microglial cells. Neuroimmunol. Neuroinflamm. 2018, 5, 50. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Arnaud, L.; Benech, P.; Greetham, L.; Stephan, D.; Jimenez, A.; Jullien, N.; Garcia-Gonzalez, L.; Tsvetkov, P.O.; Devred, F.; Sancho-Martinez, I.; et al. APOE4 drives inflammation in human astrocytes via TAGLN3 repression and NF-κB activation. Cell Rep. 2022, 40, 111200. [Google Scholar] [CrossRef]
- Brandl, C.; Zimmermann, S.J.; Milenkovic, V.M.; Rosendahl, S.M.; Grassmann, F.; Milenkovic, A.; Hehr, U.; Federlin, M.; Wetzel, C.H.; Helbig, H.; et al. In-depth characterisation of Retinal Pigment Epithelium (RPE) cells derived from human induced pluripotent stem cells (hiPSC). Neuromolecular. Med. 2014, 16, 551–564. [Google Scholar] [CrossRef]
- Germelli, L.; Da Pozzo, E.; Giacomelli, C.; Tremolanti, C.; Marchetti, L.; Wetzel, C.H.; Barresi, E.; Taliani, S.; Da Settimo, F.; Martini, C.; et al. De novo Neurosteroidogenesis in Human Microglia: Involvement of the 18 kDa Translocator Protein. Int. J. Mol. Sci. 2021, 22, 3115. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bader, S.; Jahner, T.; Dörfelt, A.; Melchner, D.; Cardon, I.; Siegmund, H.I.; Brochhausen, C.; Rupprecht, R.; Milenkovic, V.M.; Wetzel, C.H. A Comprehensive Functional Investigation of the Human Translocator Protein 18 kDa (TSPO) in a Novel Human Neuronal Cell Knockout Model. Int. J. Mol. Sci. 2024, 25, 12882. https://doi.org/10.3390/ijms252312882
Bader S, Jahner T, Dörfelt A, Melchner D, Cardon I, Siegmund HI, Brochhausen C, Rupprecht R, Milenkovic VM, Wetzel CH. A Comprehensive Functional Investigation of the Human Translocator Protein 18 kDa (TSPO) in a Novel Human Neuronal Cell Knockout Model. International Journal of Molecular Sciences. 2024; 25(23):12882. https://doi.org/10.3390/ijms252312882
Chicago/Turabian StyleBader, Stefanie, Tatjana Jahner, Anett Dörfelt, Doris Melchner, Iseline Cardon, Heiko I. Siegmund, Christoph Brochhausen, Rainer Rupprecht, Vladimir M. Milenkovic, and Christian H. Wetzel. 2024. "A Comprehensive Functional Investigation of the Human Translocator Protein 18 kDa (TSPO) in a Novel Human Neuronal Cell Knockout Model" International Journal of Molecular Sciences 25, no. 23: 12882. https://doi.org/10.3390/ijms252312882
APA StyleBader, S., Jahner, T., Dörfelt, A., Melchner, D., Cardon, I., Siegmund, H. I., Brochhausen, C., Rupprecht, R., Milenkovic, V. M., & Wetzel, C. H. (2024). A Comprehensive Functional Investigation of the Human Translocator Protein 18 kDa (TSPO) in a Novel Human Neuronal Cell Knockout Model. International Journal of Molecular Sciences, 25(23), 12882. https://doi.org/10.3390/ijms252312882