
Investigating p.Ala1035Val in NPC1: New Cellular Models for Niemann–Pick Type C Disease

 , , ,
, , ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Niemann–Pick Type C Disease
1.2. Pathogenesis
1.3. Study Overview
2. Results
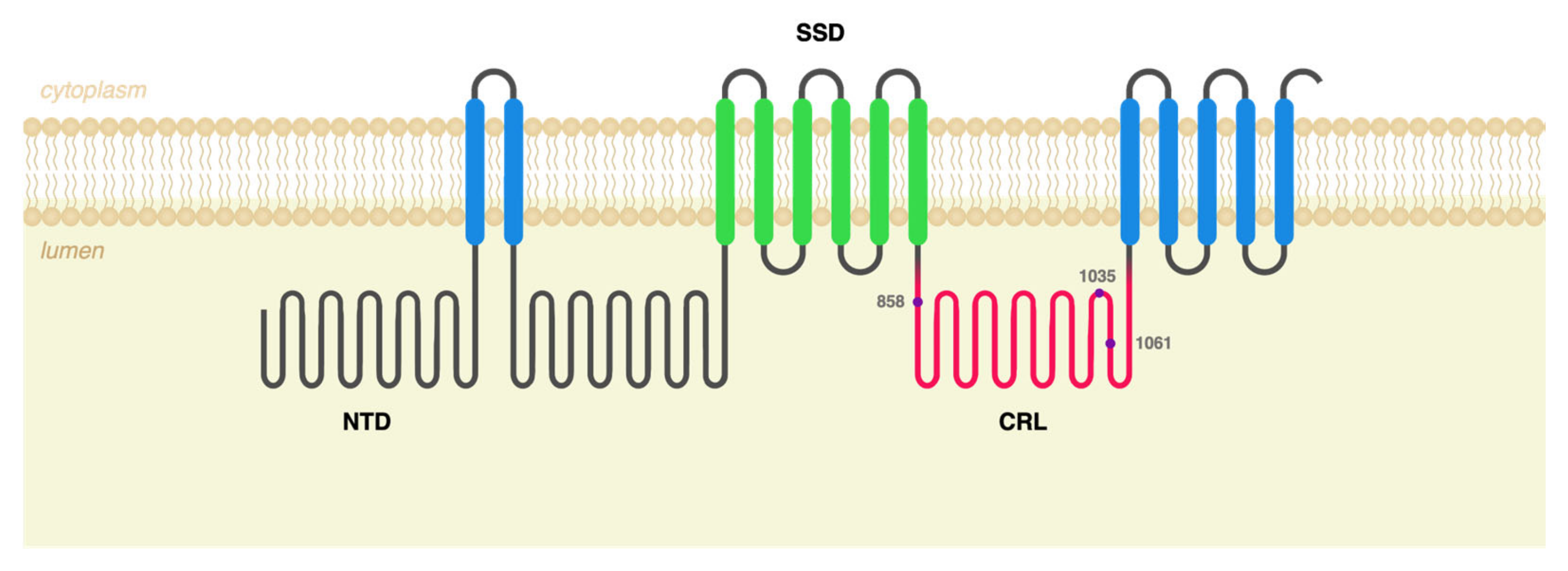
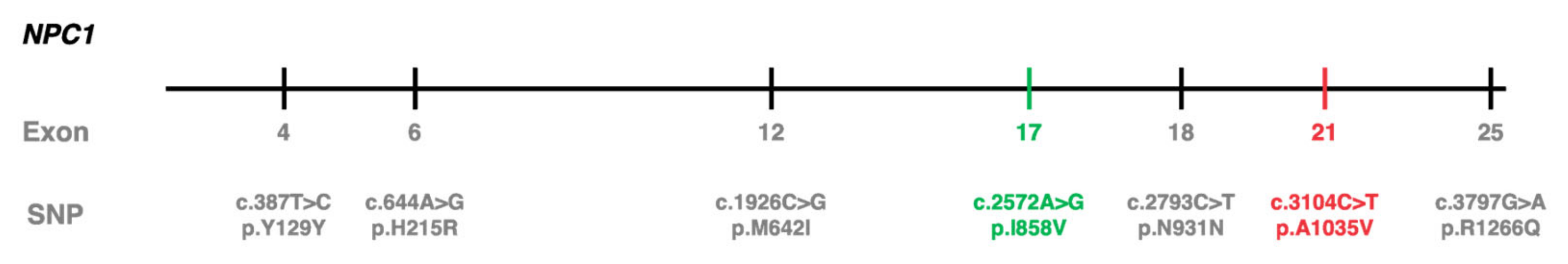
2.1. c.3104C>T (p.Ala1035Val) Was Identified in cis with Other Polymorphisms in 10 Portuguese NPC Patients
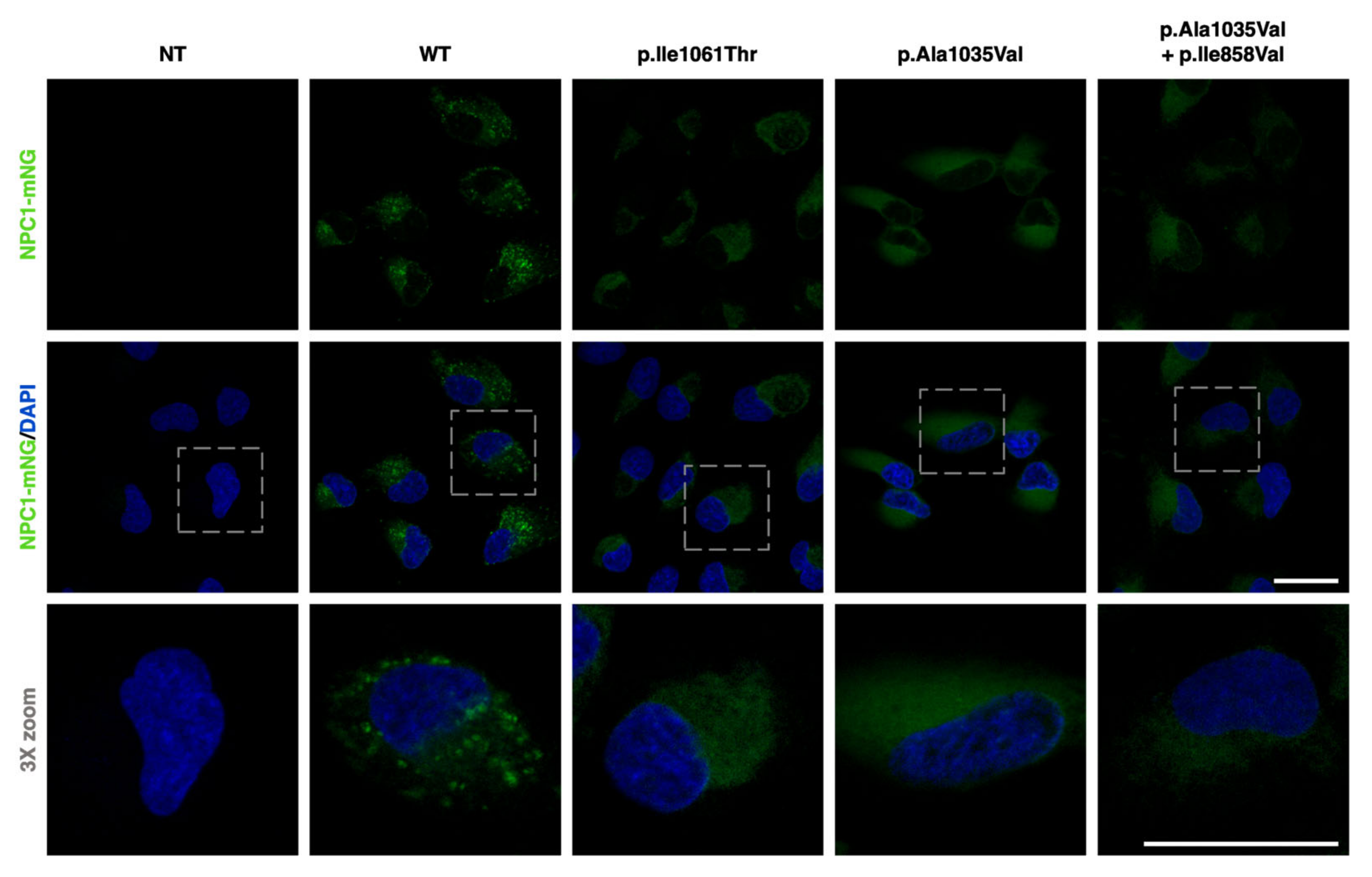
2.2. NPC1−/− ARPE-19 Cells Retrovirally Transduced with Either WT or Mutant NPC1 Forms Mimic the Expected Cellular Phenotype
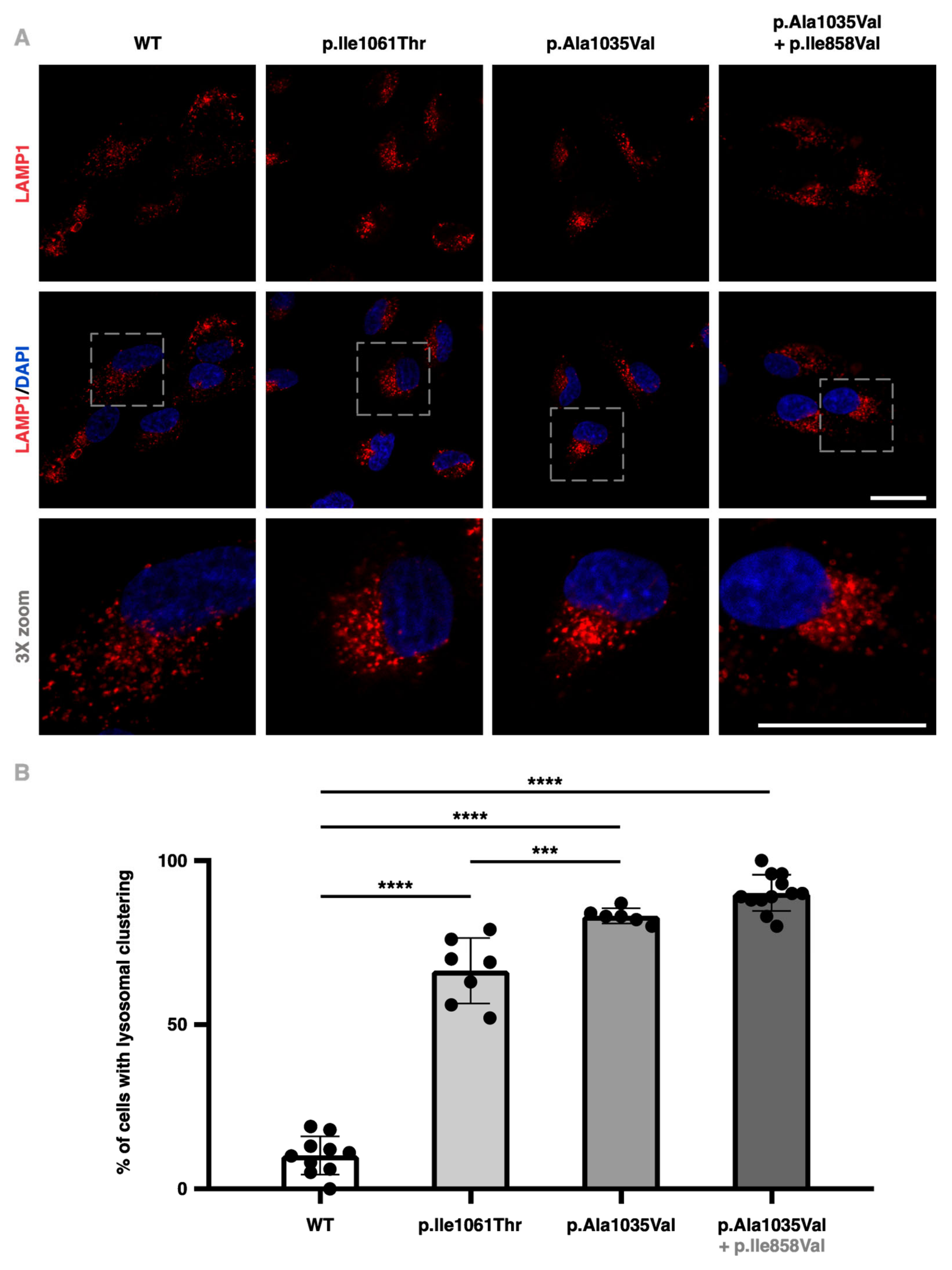
2.3. NPC1−/− ARPE-19 Cells Retrovirally Transduced with the p.Ala1035Val Variant of NPC1 Display Higher Perinuclear Lysosome Clustering
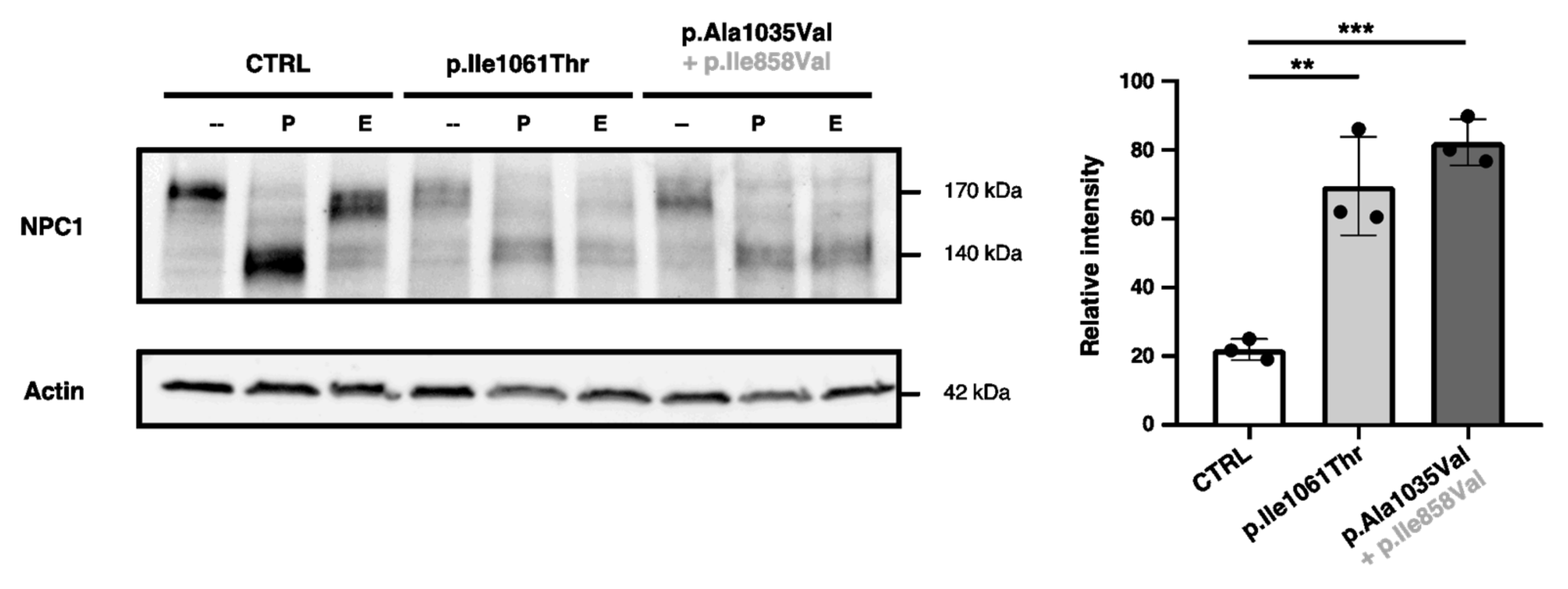
2.4. p.Ala1035Val-NPC1 and p.Il11061Thr-NPC1 Are More Endo-H-Sensitive than WT-NPC1
2.5. NPC1−/− ARPE-19 Cells Retrovirally Transduced with the p.Ala1035Val + p.Ile858Val Variant Combination of NPC1 Display Attenuated LAMP1 Colocalization
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Plasmids
4.3. Antibodies
4.4. Site-Directed Mutagenesis
4.5. Generation of Stably Transduced NPC1−/− ARPE-19 Cells
4.6. Immunofluorescence Assays
4.7. Enzymatic Deglycosylation and Western Blot
4.8. Lysosome Clustering Scoring
4.9. Colocalization Quantification
4.10. Image Processing
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lloyd-Evans, E.; Platt, F.M. Lipids on Trial: The Search for the Offending Metabolite in Niemann-Pick Type C Disease. Traffic 2010, 11, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Mengel, E.; Klünemann, H.-H.; Lourenço, C.M.; Hendriksz, C.J.; Sedel, F.; Walterfang, M.; Kolb, S.A. Niemann-Pick Disease Type C Symptomatology: An Expert-Based Clinical Description. Orphanet J. Rare Dis. 2013, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Geberhiwot, T.; Moro, A.; Dardis, A.; Ramaswami, U.; Sirrs, S.; Marfa, M.P.; Vanier, M.T.; Walterfang, M.; Bolton, S.; Dawson, C.; et al. Consensus Clinical Management Guidelines for Niemann-Pick Disease Type C. Orphanet J. Rare Dis. 2018, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Raas-Rothschild, A.; Reish, O.; Regev, M.; Meiner, V.; Bargal, R.; Sury, V.; Meir, K.; Nadjari, M.; Hermann, G.; et al. The Clinical Spectrum of Fetal Niemann-Pick Type C. Am. J. Med. Genet. A 2009, 149A, 446–450. [Google Scholar] [CrossRef]
- Kelly, D.A.; Portmann, B.; Mowat, A.P.; Sherlock, S.; Lake, B.D. Niemann-Pick Disease Type C: Diagnosis and Outcome in Children, with Particular Reference to Liver Disease. J. Pediatr. 1993, 123, 242–247. [Google Scholar] [CrossRef]
- Bolton, S.C.; Soran, V.; Marfa, M.P.; Imrie, J.; Gissen, P.; Jahnova, H.; Sharma, R.; Jones, S.; Santra, S.; Crushell, E.; et al. Clinical Disease Characteristics of Patients with Niemann-Pick Disease Type C: Findings from the International Niemann-Pick Disease Registry (INPDR). Orphanet J. Rare Dis. 2022, 17, 51. [Google Scholar] [CrossRef]
- Klarner, B.; Klünemann, H.H.; Lürding, R.; Aslanidis, C.; Rupprecht, R. Neuropsychological Profile of Adult Patients with Niemann-Pick C1 (NPC1) Mutations. J. Inherit. Metab. Dis. 2007, 30, 60–67. [Google Scholar] [CrossRef]
- Millat, G.; Baïlo, N.; Molinero, S.; Rodriguez, C.; Chikh, K.; Vanier, M.T. Niemann-Pick C Disease: Use of Denaturing High Performance Liquid Chromatography for the Detection of NPC1 and NPC2 Genetic Variations and Impact on Management of Patients and Families. Mol. Genet. Metab. 2005, 86, 220–232. [Google Scholar] [CrossRef]
- Carstea, E.D.; Polymeropoulos, M.H.; Parker, C.C.; Detera-Wadleigh, S.D.; O’Neill, R.R.; Patterson, M.C.; Goldin, E.; Xiao, H.; Straub, R.E.; Vanier, M.T. Linkage of Niemann-Pick Disease Type C to Human Chromosome 18. Proc. Natl. Acad. Sci. USA 1993, 90, 2002–2004. [Google Scholar] [CrossRef]
- Brogden, G.; Shammas, H.; Walters, F.; Maalouf, K.; Das, A.M.; Naim, H.Y.; Rizk, S. Different Trafficking Phenotypes of Niemann-Pick C1 Gene Mutations Correlate with Various Alterations in Lipid Storage, Membrane Composition and Miglustat Amenability. Int. J. Mol. Sci. 2020, 21, 2101. [Google Scholar] [CrossRef]
- Millat, G.; Marçais, C.; Rafi, M.A.; Yamamoto, T.; Morris, J.A.; Pentchev, P.G.; Ohno, K.; Wenger, D.A.; Vanier, M.T. Niemann-Pick C1 Disease: The I1061T Substitution Is a Frequent Mutant Allele in Patients of Western European Descent and Correlates with a Classic Juvenile Phenotype. Am. J. Hum. Genet. 1999, 65, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, I.; Marcão, A.; Amaral, O.; Sá Miranda, M.C.; Vanier, M.T.; Millat, G. Niemann-Pick Type C Disease: NPC1 Mutations Associated with Severe and Mild Cellular Cholesterol Trafficking Alterations. Hum. Genet. 2001, 109, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Guatibonza Moreno, P.; Pardo, L.M.; Pereira, C.; Schroeder, S.; Vagiri, D.; Almeida, L.S.; Juaristi, C.; Hosny, H.; Loh, C.C.Y.; Leubauer, A.; et al. At a Glance: The Largest Niemann-Pick Type C1 Cohort with 602 Patients Diagnosed over 15 Years. Eur. J. Hum. Genet. 2023, 31, 1108–1116. [Google Scholar] [CrossRef]
- Fernandez-Valero, E.M.; Ballart, A.; Iturriaga, C.; Lluch, M.; Macias, J.; Vanier, M.T.; Pineda, M.; Coll, M.J. Identification of 25 New Mutations in 40 Unrelated Spanish Niemann-Pick Type C Patients: Genotype-Phenotype Correlations. Clin. Genet. 2005, 68, 245–254. [Google Scholar] [CrossRef]
- Bychkov, I.; Filatova, A.; Perelman, G.; Proshlyakova, T.; Korotkova, D.; Klyushnikov, S.; Karpova, M.; Tabakov, V.; Baydakova, G.; Ilyushkina, A.; et al. Additive Effect of Frequent Polymorphism and Rare Synonymous Variant Alters Splicing in Twin Patients with Niemann-Pick Disease Type C. Eur. J. Hum. Genet. 2021, 30, 133–136. [Google Scholar] [CrossRef]
- Schultz, M.L.; Schache, K.J.; Azaria, R.D.; Kuiper, E.Q.; Erwood, S.; Ivakine, E.A.; Farhat, N.Y.; Porter, F.D.; Pathmasiri, K.C.; Cologna, S.M.; et al. Species-Specific Differences in NPC1 Protein Trafficking Govern Therapeutic Response in Niemann-Pick Type C Disease. JCI Insight 2022, 7, e160308. [Google Scholar] [CrossRef]
- Schultz, M.L.; Krus, K.L.; Lieberman, A.P. Lysosome and Endoplasmic Reticulum Quality Control Pathways in Niemann-Pick Type C Disease. Brain Res. 2016, 1649, 181–188. [Google Scholar] [CrossRef]
- Höglinger, D.; Burgoyne, T.; Sanchez-Heras, E.; Hartwig, P.; Colaco, A.; Newton, J.; Futter, C.E.; Spiegel, S.; Platt, F.M.; Eden, E.R. NPC1 Regulates ER Contacts with Endocytic Organelles to Mediate Cholesterol Egress. Nat. Commun. 2019, 10, 4276. [Google Scholar] [CrossRef]
- Daniele, T.; Hackmann, Y.; Ritter, A.T.; Wenham, M.; Booth, S.; Bossi, G.; Schintler, M.; Auer-Grumbach, M.; Griffiths, G.M. A Role for Rab7 in the Movement of Secretory Granules in Cytotoxic T Lymphocytes. Traffic 2011, 12, 902–911. [Google Scholar] [CrossRef]
- Encarnação, M.; Espada, L.; Escrevente, C.; Mateus, D.; Ramalho, J.; Michelet, X.; Santarino, I.; Hsu, V.W.; Brenner, M.B.; Barral, D.C.; et al. A Rab3a-Dependent Complex Essential for Lysosome Positioning and Plasma Membrane Repair. J. Cell Biol. 2016, 213, 631–640. [Google Scholar] [CrossRef]
- Barral, D.C.; Staiano, L.; Guimas Almeida, C.; Cutler, D.F.; Eden, E.R.; Futter, C.E.; Galione, A.; Marques, A.R.A.; Medina, D.L.; Napolitano, G.; et al. Current Methods to Analyze Lysosome Morphology, Positioning, Motility and Function. Traffic 2022, 23, 238–269. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.M.; Cassady, J.; Cohen, M.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired Autophagy in the Lipid Storage Disorder Niemann–Pick Type C1 Disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar] [CrossRef]
- Bauer, P.; Knoblich, R.; Bauer, C.; Finckh, U.; Hufen, A.; Kropp, J.; Braun, S.; Kustermann-Kuhn, B.; Schmidt, D.; Harzer, K.; et al. NPC1: Complete Genomic Sequence, Mutation Analysis, and Characterization of Haplotypes. Hum. Mutat. 2002, 19, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Shumate, J.L.; Caldwell, J.G.; Elghobashi-Meinhardt, N.; Lu, A.; Zhang, L.; Olsson, N.E.; Elias, J.E.; Pfeffer, S.R. Inter-Domain Dynamics Drive Cholesterol Transport by NPC1 and NPC1L1 Proteins. eLife 2020, 9, e57089. [Google Scholar] [CrossRef] [PubMed]
- Encarnação, M.; Coutinho, M.F.; Cho, S.M.; Cardoso, M.T.; Ribeiro, I.; Chaves, P.; Santos, J.I.; Quelhas, D.; Lacerda, L.; Leão Teles, E.; et al. NPC1 Silent Variant Induces Skipping of Exon 11 (p.V562V) and Unfolded Protein Response Was Found in a Specific Niemann-Pick Type C Patient. Mol. Genet. Genom. Med. 2020, 8, e1451. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Santos, J.I.; Moreira, L.; Duarte, A.J.; Gaspar, P.; Rocha, H.; Encarnação, M.; Ribeiro, D.; Barbosa Almeida, M.; Gonçalves, M.; et al. Modeling Lysosomal Storage Disorders in an Innovative Way: Establishment and Characterization of Stem Cell Lines from Human Exfoliated Deciduous Teeth of Mucopolysaccharidosis Type II Patients. Int. J. Mol. Sci. 2024, 25, 3546. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
David, H.; Monfregola, J.; Ribeiro, I.; Cardoso, M.T.; Sandiares, A.C.; Moreira, L.; Coutinho, M.F.; Quelhas, D.; Ballabio, A.; Alves, S.; et al. Investigating p.Ala1035Val in NPC1: New Cellular Models for Niemann–Pick Type C Disease. Int. J. Mol. Sci. 2024, 25, 12186. https://doi.org/10.3390/ijms252212186
David H, Monfregola J, Ribeiro I, Cardoso MT, Sandiares AC, Moreira L, Coutinho MF, Quelhas D, Ballabio A, Alves S, et al. Investigating p.Ala1035Val in NPC1: New Cellular Models for Niemann–Pick Type C Disease. International Journal of Molecular Sciences. 2024; 25(22):12186. https://doi.org/10.3390/ijms252212186
Chicago/Turabian StyleDavid, Hugo, Jlenia Monfregola, Isaura Ribeiro, Maria Teresa Cardoso, Ana Catarina Sandiares, Luciana Moreira, Maria Francisca Coutinho, Dulce Quelhas, Andrea Ballabio, Sandra Alves, and et al. 2024. "Investigating p.Ala1035Val in NPC1: New Cellular Models for Niemann–Pick Type C Disease" International Journal of Molecular Sciences 25, no. 22: 12186. https://doi.org/10.3390/ijms252212186
APA StyleDavid, H., Monfregola, J., Ribeiro, I., Cardoso, M. T., Sandiares, A. C., Moreira, L., Coutinho, M. F., Quelhas, D., Ballabio, A., Alves, S., & Encarnação, M. (2024). Investigating p.Ala1035Val in NPC1: New Cellular Models for Niemann–Pick Type C Disease. International Journal of Molecular Sciences, 25(22), 12186. https://doi.org/10.3390/ijms252212186