
How Do Gepotidacin and Zoliflodacin Stabilize DNA Cleavage Complexes with Bacterial Type IIA Topoisomerases? 1. Experimental Definition of Metal Binding Sites

 ,
,  , , ,
, , ,
Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. Ref. | PDB Code + Res in Å | Inhibitor Type | DNA Sequence Central Six Base-Pairs 1 | Inhibitor Pockets Occupied 2 | Catal. Tyr 123 3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1′ | 2D | 2A | 3 | 3′ | |||||
| 1 [11] | 2xcs-v2-BA-x.pdb* 2.1 Å | NBTI | 5′ GGGCCC 3′ | - | - | x | x | - | - | Phe |
| 2 [27] | 4bul-BA-x.pdb 2.6 Å | NBTI | 5′ TGTACC 3′ 3′ ACATGG 5′ | - | - | x | x | - | - | Phe |
| 3 [29] | 4plb 2.69 Å | NBTI | 5′ GGGCCC 3′ | - | - | x | x | - | - | Phe |
| 4 [28] | 5bs3 2.65 Å | NBTI | 5′ GGGCCC 3′ | - | - | x | x | - | - | Phe |
| 5 [9] | 5cdm-v2-BA-x.pdb* 2.5 Å | SPT | 5′ C-YGGCCG 3′ | x | x | - | - | - | - | TyrP |
| 6 [9] | 5cdp-BA-x.pdb 2.45 Å | Etop. | 5′ G-PGTACC 3′ | x | - | - | - | - | - | Phe |
| 7 [9] | 5cdr-v2-BA-x.pdb* 2.65 Å | - | 5′ G-PGTACC 3′ | - | - | - | - | - | - | Phe |
| 8 [26] | 5iwi-v2-BA-x.pdb* 1.98 Å | NBTI | 5′ GGTAC A 3′ 3′ CCATG-T 5′ | - | - | x | x | - | - | Phe |
| 9 [26] | 5cdp-BA-x.pdb 2.5 Å | NBTI | 5′ GGTTCA 3′ 3′ CCAAGT 5′ | - | - | x | x | - | - | Phe |
| 10 [39] | 5npp-BA-x.pdb 2.22 Å | NBTI + thiop. | 5′ G-PGTACC 3′ | - | - | x | x | x | x | Tyr |
| 11 [30] | 6fm4 2.7 Å | NBTI | 5′ GGGCCC 3′ | - | - | x | x | - | - | Phe |
| 12 [33] | 6fqs-BA-x.pdb 3.11 Å | IPY | 5′ C-YGGCCG 3′ | x | x | - | - | - | - | TyrP |
| 13 [6] | 6qtk-BA-x.pdb 2.31 Å | NBTI | 5′ G-PGTACC 3′ | - | - | x | x | - | - | Phe |
| 14 [40] | 6qx1-BA-x.pdb 2.65 Å | benzoi’ | 5′ G-PGTACC 3′ | - | - | - | - | x | x | Phe |
| 15 [41] | 6z1a 2.3 Å | NBTI | 5′ G-PGTACC 3′ | - | - | x | x | - | - | Phe |
| 16 [31] | 7fvs 2.16 Å | NBTI | 5′ GGGCCC 3′ | - | - | x | x | - | - | Phe |
| 17 [31] | 7fvt 2.08 Å | NBTI | 5′ GGGCCC 3′ | - | - | x | x | - | - | Phe |
| 18 [32] | 7mvs 2.60 Å | NBTI | 5′ GGGCCC 3′ | - | - | x | x | - | - | Phe |
| 19 [8] | 8bp2-BA-x.pdb 2.80 Å | SPT | 5′ C-YGGCCG 3′ | x | x | - | - | - | - | TyrP |
| 20 t.p. | 9fz6-BA-x.pdb 2.58 Å | - | 5′ G-PGTACC 3′ | - | - | - | - | - | - | Phe |
2. Results
2.1. High-Resolution Structures Have Clearly Defined Metals at Either the 3′(A)-Site or Y(B)-Site
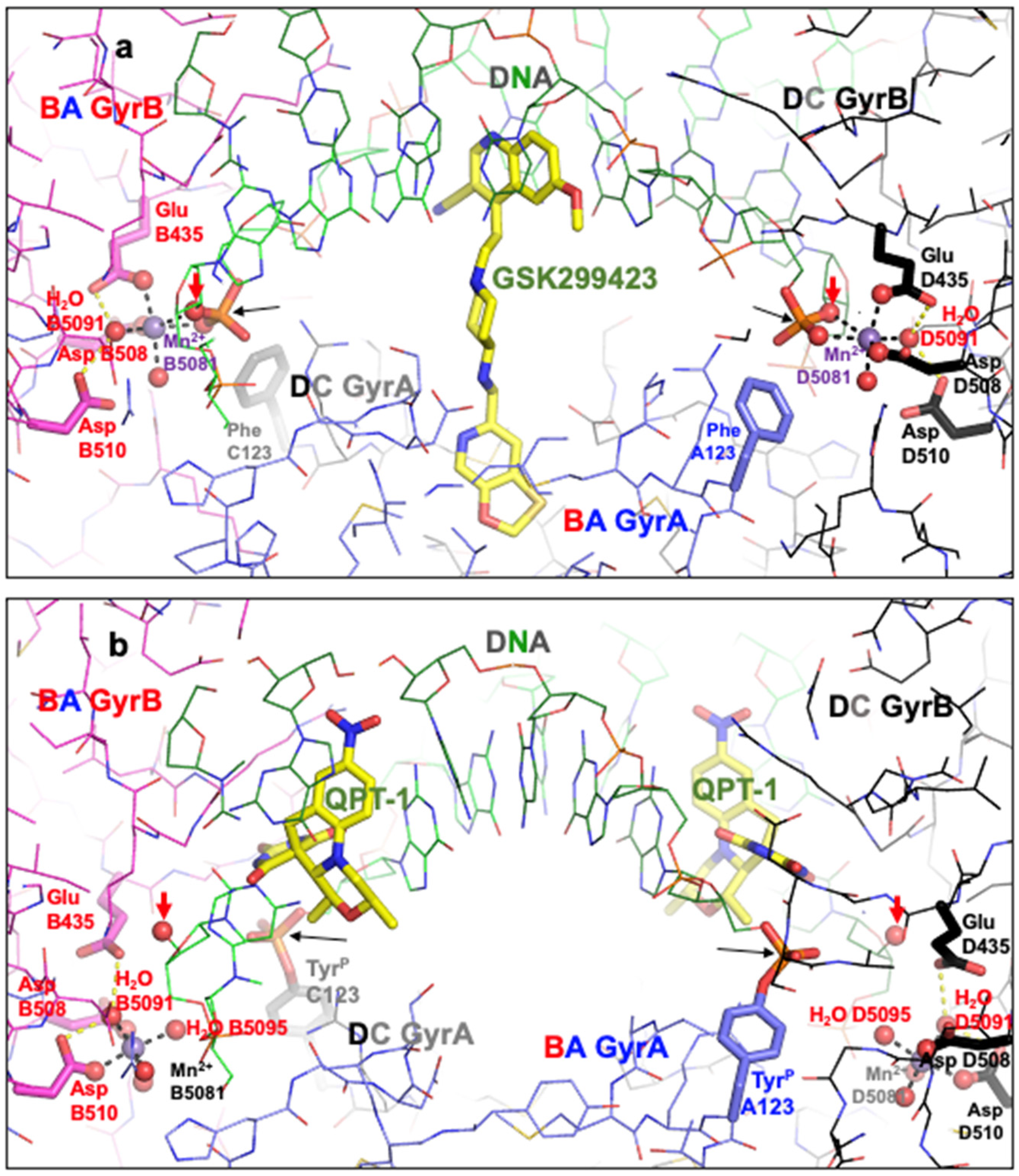

2.2. A Comparison of the Original 3l4k Structure Interpreted as Having the Same Metal Ion Coordination Geometry Independent of the Active Sites with Re-Refined Coordinates
2.3. Re-Refined P61 S. aureus Structures Suggest a Manganese Ion and BisTris Mediate an Important Crystal Contact
2.4. An Anomalous Dataset Confirmed Catalytic Metal and Crystal Contact Manganese Ions, but Unexpectedly Showed Additional Manganese Density near Serine 84
| Mn Site | Occupancy | Peak Height (rmsd) |
|---|---|---|
| B/5081 | 1.0 | 15.5 |
| D/5081 | 1.0 | 15.1 |
| E/1999 | 0.6 | 7.0 |
| F/1999 | 0.5 | 4.3 |
| C/901 | 0.2 | 4.0 |

3. Discussion
4. Materials and Methods
4.1. The Standard BA-x Nomenclature for S. aureus DNA Gyrase Crystal Structures and the Y(B), 3′(A) Nomenclature for Catalytic Metals
4.2. Re-Refining Fully and Partially Occupied Metal Binding Sites with Chemically Reasonable Geometry
4.3. Protein Purification and Crystallization of a DNA Gyrase—DNA Complex
4.4. Data Collection, Structure Determination and Refinement
4.5. The Generation of a Movie of Geoptidacin Binding
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tran, P.T.; Antonelli, P.J.; Hincapie-Castillo, J.M.; Winterstein, A.G. Association of US Food and Drug Administration removal of indications for use of oral quinolones with prescribing trends. JAMA Intern. Med. 2021, 181, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C.; Jacoby, G.A. Topoisomerase inhibitors: Fluoroquinolone mechanisms of action and resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025320. [Google Scholar] [CrossRef] [PubMed]
- Wohlkonig, A.; Chan, P.F.; Fosberry, A.P.; Homes, P.; Huang, J.; Kranz, M.; Leydon, V.R.; Miles, T.J.; Pearson, N.D.; Perera, R.L.; et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol 2010, 17, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Jacobsson, S.; Golparian, D.; Oxelbark, J.; Kong, F.Y.; Da Costa, R.M.A.; Franceschi, F.; Brown, D.; Louie, A.; Drusano, G.; Unemo, M. Pharmacodynamics of zoliflodacin plus doxycycline combination therapy against Neisseria gonorrhoeae in a gonococcal hollow-fiber infection model. Front. Pharmacol. 2023, 14, 1291885. [Google Scholar] [CrossRef]
- Hooton, T.M.; Perry, C.R.; Janmohamed, S.; Sheets, A.; Dennison, J.; Millns, H.; Jarvis, E.; Scangarella-Oman, N.E.; Huang, C. 2832. Gepotidacin Efficacy in E. coli Drug-Resistant Phenotypes: A Pooled Analysis of the EAGLE-2 and EAGLE-3 Randomized Controlled Trials in Uncomplicated Urinary Tract Infection. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2023. [Google Scholar] [CrossRef]
- Gibson, E.G.; Bax, B.; Chan, P.F.; Osheroff, N. Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase. ACS Infect. Dis. 2019, 5, 570–581. [Google Scholar] [CrossRef]
- Miller, A.A.; Bundy, G.L.; Mott, J.E.; Skepner, J.E.; Boyle, T.P.; Harris, D.W.; Hromockyj, A.E.; Marotti, K.R.; Zurenko, G.E.; Munzner, J.B.; et al. Discovery and characterization of QPT-1, the progenitor of a new class of bacterial topoisomerase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 2806–2812. [Google Scholar] [CrossRef]
- Morgan, H.; Lipka-Lloyd, M.; Warren, A.J.; Hughes, N.; Holmes, J.; Burton, N.P.; Mahenthiralingam, E.; Bax, B.D. A 2.8 Å Structure of Zoliflodacin in a DNA Cleavage Complex with Staphylococcus aureus DNA Gyrase. Int. J. Mol. Sci. 2023, 24, 1634. [Google Scholar] [CrossRef]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef]
- Blower, T.R.; Williamson, B.H.; Kerns, R.J.; Berger, J.M. Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2016, 113, 1706–1713. [Google Scholar] [CrossRef]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef]
- Dadgostar, P. Antimicrobial resistance: Implications and costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef]
- Founou, R.C.; Founou, L.L.; Essack, S.Y. Clinical and economic impact of antibiotic resistance in developing countries: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0189621. [Google Scholar] [CrossRef] [PubMed]
- Llor, C.; Bjerrum, L. Antimicrobial resistance: Risk associated with antibiotic overuse and initiatives to reduce the problem. Ther. Adv. Drug Saf. 2014, 5, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control (ECDC). Antimicrobial Resistance Surveillance in Europe 2023—2021 Data; European Centre for Disease Prevention and Control: Solna, Sweden, 2023; p. 186. [Google Scholar]
- Allocati, N.; Masulli, M.; Alexeyev, M.F.; Di Ilio, C. Escherichia coli in Europe: An overview. Int. J. Environ. Res. Public Health 2013, 10, 6235–6254. [Google Scholar] [CrossRef]
- Manaia, C.M.; Aga, D.S.; Cytryn, E.; Gaze, W.H.; Graham, D.W.; Guo, J.; Leonard, A.F.; Li, L.; Murray, A.K.; Nunes, O.C. The Complex interplay between antibiotic resistance and pharmaceutical and personal care products in the environment. Environ. Toxicol. Chem. 2024, 43, 637–652. [Google Scholar] [CrossRef]
- Wagenlehner, F.; Perry, C.R.; Hooton, T.M.; Scangarella-Oman, N.E.; Millns, H.; Powell, M.; Jarvis, E.; Dennison, J.; Sheets, A.; Butler, D. Oral gepotidacin versus nitrofurantoin in patients with uncomplicated urinary tract infection (EAGLE-2 and EAGLE-3): Two randomised, controlled, double-blind, double-dummy, phase 3, non-inferiority trials. Lancet 2024, 403, 741–755. [Google Scholar] [CrossRef]
- Taylor, S.N.; Marrazzo, J.; Batteiger, B.E.; Hook, E.W., 3rd; Sena, A.C.; Long, J.; Wierzbicki, M.R.; Kwak, H.; Johnson, S.M.; Lawrence, K.; et al. Single-Dose Zoliflodacin (ETX0914) for Treatment of Urogenital Gonorrhea. N. Engl. J. Med. 2018, 379, 1835–1845. [Google Scholar] [CrossRef]
- Allan-Blitz, L.-T.; Fifer, H.; Klausner, J.D. Managing treatment failure in Neisseria gonorrhoeae infection: Current guidelines and future directions. Lancet Infect. Dis. 2024, 24, e532–e538. [Google Scholar] [CrossRef]
- Oviatt, A.A.; Gibson, E.G.; Huang, J.; Mattern, K.; Neuman, K.C.; Chan, P.F.; Osheroff, N. Interactions between Gepotidacin and Escherichia coli Gyrase and Topoisomerase IV: Genetic and Biochemical Evidence for Well-Balanced Dual-Targeting. ACS Infect. Dis. 2024, 10, 1137–1151. [Google Scholar] [CrossRef]
- Schoeffler, A.J.; Berger, J.M. DNA topoisomerases: Harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 2008, 41, 41–101. [Google Scholar] [CrossRef] [PubMed]
- Bates, A.D.; Maxwell, A. DNA Topology; Oxford University Press: New York, NY, USA, 2005. [Google Scholar]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Miles, T.J.; Hennessy, A.J.; Bax, B.; Brooks, G.; Brown, B.S.; Brown, P.; Cailleau, N.; Chen, D.; Dabbs, S.; Davies, D.T. Novel tricyclics (eg, GSK945237) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2016, 26, 2464–2469. [Google Scholar] [CrossRef]
- Miles, T.J.; Hennessy, A.J.; Bax, B.; Brooks, G.; Brown, B.S.; Brown, P.; Cailleau, N.; Chen, D.; Dabbs, S.; Davies, D.T.; et al. Novel hydroxyl tricyclics (e.g., GSK966587) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2013, 23, 5437–5441. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Meinke, P.T.; Olsen, D.B.; Lagrutta, A.; Wei, C.; Liao, Y.; et al. Structure activity relationship of pyridoxazinone substituted RHS analogs of oxabicyclooctane-linked 1,5-naphthyridinyl novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents (Part-6). Bioorg. Med. Chem. Lett. 2015, 25, 3636–3643. [Google Scholar] [CrossRef]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Meinke, P.T.; Olsen, D.; Lagrutta, A.; Bradley, P.; Lu, J.; et al. Oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad spectrum antibacterial agents. ACS Med. Chem. Lett. 2014, 5, 609–614. [Google Scholar] [CrossRef]
- Magarò, G.; Prati, F.; Garofalo, B.; Corso, G.; Furlotti, G.; Apicella, C.; Mangano, G.; D’Atanasio, N.; Robinson, D.; Di Giorgio, F.P. Virtual Screening Approach and Investigation of Structure—Activity Relationships to Discover Novel Bacterial Topoisomerase Inhibitors Targeting Gram-Positive and Gram-Negative Pathogens. J. Med. Chem. 2019, 62, 7445–7472. [Google Scholar] [CrossRef]
- Cumming, J.G.; Kreis, L.; Kühne, H.; Wermuth, R.; Vercruysse, M.; Kramer, C.; Rudolph, M.G.; Xu, Z. Discovery of a Series of Indane-Containing NBTIs with Activity against Multidrug-Resistant Gram-Negative Pathogens. ACS Med. Chem. Lett. 2023, 14, 993–998. [Google Scholar] [CrossRef]
- Lu, Y.; Vibhute, S.; Li, L.; Okumu, A.; Ratigan, S.C.; Nolan, S.; Papa, J.L.; Mann, C.A.; English, A.; Chen, A. Optimization of TopoIV potency, ADMET properties, and hERG inhibition of 5-amino-1, 3-dioxane-linked novel bacterial topoisomerase inhibitors: Identification of a lead with in vivo efficacy against MRSA. J. Med. Chem. 2021, 64, 15214–15249. [Google Scholar] [CrossRef]
- Germe, T.; Voros, J.; Jeannot, F.; Taillier, T.; Stavenger, R.A.; Bacque, E.; Maxwell, A.; Bax, B.D. A new class of antibacterials, the imidazopyrazinones, reveal structural transitions involved in DNA gyrase poisoning and mechanisms of resistance. Nucleic Acids Res. 2018, 46, 4114–4128. [Google Scholar] [CrossRef]
- Bax, B.; Chung, C.W.; Edge, C. Getting the chemistry right: Protonation, tautomers and the importance of H atoms in biological chemistry. Acta Crystallogr. D Struct. Biol. 2017, 73, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, D.C.; Amoah, E.; Rogers, C.; Pearson, A.R. Using photocaging for fast time-resolved structural biology studies. Acta Crystallogr. Sect. D Struct. Biol. 2021, 77, 1218–1232. [Google Scholar] [CrossRef] [PubMed]
- Carey, P.R. Raman crystallography and other biochemical applications of Raman microscopy. Annu. Rev. Phys. Chem. 2006, 57, 527–554. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.R.; Mehrabi, P. Serial synchrotron crystallography for time-resolved structural biology. Curr. Opin. Struct. Biol. 2020, 65, 168–174. [Google Scholar] [CrossRef]
- Bax, B.D. Dr Benjamin Bax’s Website; ‘Research’ Tab. Available online: https://profiles.cardiff.ac.uk/staff/baxb (accessed on 10 October 2024).
- Chan, P.F.; Germe, T.; Bax, B.D.; Huang, J.; Thalji, R.K.; Bacque, E.; Checchia, A.; Chen, D.; Cui, H.; Ding, X.; et al. Thiophene antibacterials that allosterically stabilize DNA-cleavage complexes with DNA gyrase. Proc. Natl. Acad. Sci. USA 2017, 114, E4492–E4500. [Google Scholar] [CrossRef]
- Thalji, R.K.; Raha, K.; Andreotti, D.; Checchia, A.; Cui, H.; Meneghelli, G.; Profeta, R.; Tonelli, F.; Tommasi, S.; Bakshi, T.; et al. Structure-guided design of antibacterials that allosterically inhibit DNA gyrase. Bioorg. Med. Chem. Lett. 2019, 29, 1407–1412. [Google Scholar] [CrossRef]
- Kolaric, A.; Germe, T.; Hrast, M.; Stevenson, C.E.M.; Lawson, D.M.; Burton, N.P.; Voros, J.; Maxwell, A.; Minovski, N.; Anderluh, M. Potent DNA gyrase inhibitors bind asymmetrically to their target using symmetrical bifurcated halogen bonds. Nat. Commun. 2021, 12, 150. [Google Scholar] [CrossRef]
- Bax, B.D.; Murshudov, G.; Maxwell, A.; Germe, T. DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J. Mol. Biol. 2019, 431, 3427–3449. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.H.; Burgin, A.B.; Deweese, J.E.; Osheroff, N.; Berger, J.M. A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature 2010, 465, 641–644. [Google Scholar] [CrossRef]
- Bock, C.W.; Katz, A.K.; Markham, G.D.; Glusker, J.P. Manganese as a Replacement for Magnesium and Zinc: Functional Comparison of the Divalent Ions. J. Am. Chem. Soc. 1999, 121, 7360–7372. [Google Scholar] [CrossRef]
- Katz, A.K.; Glusker, J.P.; Beebe, S.A.; Bock, C.W. Calcium ion coordination: A comparison with that of beryllium, magnesium, and zinc. J. Am. Chem. Soc. 1996, 118, 5752–5763. [Google Scholar] [CrossRef]
- Wlodawer, A.; Dauter, Z.; Porebski, P.J.; Minor, W.; Stanfield, R.; Jaskolski, M.; Pozharski, E.; Weichenberger, C.X.; Rupp, B. Detect, correct, retract: How to manage incorrect structural models. FEBS J. 2018, 285, 444–466. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Wojdyr, M.; Long, F.; Nicholls, R.A.; Murshudov, G.N. GEMMI and Servalcat restrain REFMAC5. Acta Crystallogr. Sect. D Struct. Biol. 2023, 79, 368–373. [Google Scholar] [CrossRef]
- El Omari, K.; Forsyth, I.; Duman, R.; Orr, C.M.; Mykhaylyk, V.; Mancini, E.J.; Wagner, A. Utilizing anomalous signals for element identification in macromolecular crystallography. Biol. Crystallogr. 2024, 80, 713–721. [Google Scholar] [CrossRef]
- Masmaliyeva, R.C.; Babai, K.H.; Murshudov, G.N. Local and global analysis of macromolecular atomic displacement parameters. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.A.; Morgan, H.; Warren, A.J.; Ward, S.E.; Long, F.; Murshudov, G.N.; Sutormin, D.; Bax, B.D. How do gepotidacin and zolifodacin stabilize DNA-cleavage complexes with bacterial type IIA topoisomerases? 2. A Single Moving Metal Mechanism. bioRxiv 2024. [Google Scholar] [CrossRef]
- Bucher, D.; Stouten, P.; Triballeau, N. Shedding light on important waters for drug design: Simulations versus grid-based methods. J. Chem. Inf. Model. 2018, 58, 692–699. [Google Scholar] [CrossRef]
- Alvarez-Garcia, D.; Barril, X. Molecular simulations with solvent competition quantify water displaceability and provide accurate interaction maps of protein binding sites. J. Med. Chem. 2014, 57, 8530–8539. [Google Scholar] [CrossRef]
- Deweese, J.E.; Osheroff, N. The use of divalent metal ions by type II topoisomerases. Metallomics 2010, 2, 450–459. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.5.0.4; Schrödinger, LLC: New York, NY, USA, 2013.
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef]
- Deweese, J.E.; Guengerich, F.P.; Burgin, A.B.; Osheroff, N. Metal ion interactions in the DNA cleavage/ligation active site of human topoisomerase IIα. Biochemistry 2009, 48, 8940–8947. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Burgin, A.B.; Osheroff, N. Human topoisomerase IIalpha uses a two-metal-ion mechanism for DNA cleavage. Nucleic Acids Res 2008, 36, 4883–4893. [Google Scholar] [CrossRef]
- Thorn, A.; Sheldrick, G.M. ANODE: Anomalous and heavy-atom density calculation. J. Appl. Crystallogr. 2011, 44, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Kolaric, A.; Minovski, N. Novel bacterial topoisomerase inhibitors: Challenges and perspectives in reducing hERG toxicity. Future Med. Chem. 2018, 10, 2241–2244. [Google Scholar] [CrossRef]
- Morgan, T.K., Jr.; Sullivan, M.E. 2 An Overview of Class III Electrophysiological Agents: A New Generation of Antiarrhythmic Therapy. Prog. Med. Chem. 1992, 29, 65–108. [Google Scholar]
- Jing, Y.; Easter, A.; Peters, D.; Kim, N.; Enyedy, I.J. In silico prediction of hERG inhibition. Future Med. Chem. 2015, 7, 571–586. [Google Scholar] [CrossRef]
- Douangamath, A.; Powell, A.; Fearon, D.; Collins, P.M.; Talon, R.; Krojer, T.; Skyner, R.; Brandao-Neto, J.; Dunnett, L.; Dias, A. Achieving efficient fragment screening at XChem facility at diamond light source. JoVE (J. Vis. Exp.) 2021, e62414. [Google Scholar] [CrossRef]
- Hartman, P.; Perdok, W. On the relations between structure and morphology of crystals: I. Acta Crystallogr. 1955, 8, 49–52. [Google Scholar] [CrossRef]
- Frey, M.; Genovesio-Taverne, J.-C.; Fontecilla-Camps, J.C. Application of the periodic bond chain (PBC) theory to the analysis of the molecular packing in protein crystals. J. Cryst. Growth 1988, 90, 245–258. [Google Scholar] [CrossRef]
- Agirre, J.; Atanasova, M.; Bagdonas, H.; Ballard, C.B.; Baslé, A.; Beilsten-Edmands, J.; Borges, R.J.; Brown, D.G.; Burgos-Mármol, J.J.; Berrisford, J.M. The CCP4 suite: Integrative software for macromolecular crystallography. Acta Crystallogr. Sect. D Struct. Biol. 2023, 79, 449–461. [Google Scholar] [CrossRef]
- Srikannathasan, V.; Wohlkonig, A.; Shillings, A.; Singh, O.; Chan, P.F.; Huang, J.; Gwynn, M.N.; Fosberry, A.P.; Homes, P.; Hibbs, M. Crystallization and initial crystallographic analysis of covalent DNA-cleavage complexes of Staphyloccocus aureus DNA gyrase with QPT-1, moxifloxacin and etoposide. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Duman, R.; Henderson, K.; Mykhaylyk, V. In-vacuum long-wavelength macromolecular crystallography. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.D.; Gurusaran, M.; Satheesh, S.N.; Radha, P.; Pavithra, S.; Thulaa Tharshan, K.P.S.; Helliwell, J.R.; Sekar, K. Online_DPI: A web server to calculate the diffraction precision index for a protein structure. J. Appl. Crystallogr. 2015, 48, 939–942. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Koulouris, C.R.; Gardiner, S.E.; Harris, T.K.; Elvers, K.T.; Mark Roe, S.; Gillespie, J.A.; Ward, S.E.; Grubisha, O.; Nicholls, R.A.; Atack, J.R. Tyrosine 121 moves revealing a ligandable pocket that couples catalysis to ATP-binding in serine racemase. Commun. Biol. 2022, 5, 346. [Google Scholar] [CrossRef]



| PDB | Compound | Resol. (Å) | Crystallization Buffer pH | Mn C901 Occupancy (Bfactor) | His C390 Side-Chain Bfactor |
|---|---|---|---|---|---|
| 2xcs-v2-BA-x.pdb | GSK299423 | 2.10 | 6.5 | 0.65 (23.3) | 23.7 |
| 5iwi-v2-BA-x.pdb * | GSK945237 | 1.98 | 6.2 | 0.30 (35.8) | 35.5 |
| 5cdm-v2-BA-x.pdb | QPT-1 | 2.50 | 6.2 | 0.40 (55.2) | 52.1 ** |
| 5cdr-v2-BA-x.pdb | - | 2.65 | 6.2 | 0.30 (37.5) | 45.8 |
| Mechanism Number and Name | Problems with Mechanism (Current Estimate of Mechanism Being Broadly Correct). Reference. |
|---|---|
(i) a two-metal mechanism from 2010 | No chemically sensible structure has yet been deposited in support of this mechanism. The X-ray data are consistent with a single moving metal mechanism (see Figure 3). The topo1A lysine is also consistent with the single moving metal mechanism in [50]. Some variation on this two-metal mechanism cannot be entirely excluded: (Low to very low) Schmidt et al. (2010) [43]. |
(ii) Single-metal mechanism from 2019 | The mechanism proposed does not detail any sensible chemistry. The mechanism seems to propose that the metal starts at the A-site and cleaves the DNA before moving to the B-site. Some variation on this mechanism cannot be excluded: (Low to very low) Bax et al. (2019) [42]. |
(iii) a single moving metal mechanism described in [50] | Only heavy atom (non-hydrogen atom) positions defined by experimental data. Mechanism suggests the red hydrogen atom from water 5093 will protonate the 3′ oxygen on the scissile phosphate to cleave the DNA, after which the metaphosphate-like intermediate will be accepted by the catalytic tyrosinate. (High to very high) (Nicholls et al., 2024) [50]. |
| (iv) Some unthought-of mechanism | (Low?). No reference. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, H.; Nicholls, R.A.; Warren, A.J.; Ward, S.E.; Evans, G.; Long, F.; Murshudov, G.N.; Duman, R.; Bax, B.D. How Do Gepotidacin and Zoliflodacin Stabilize DNA Cleavage Complexes with Bacterial Type IIA Topoisomerases? 1. Experimental Definition of Metal Binding Sites. Int. J. Mol. Sci. 2024, 25, 11688. https://doi.org/10.3390/ijms252111688
Morgan H, Nicholls RA, Warren AJ, Ward SE, Evans G, Long F, Murshudov GN, Duman R, Bax BD. How Do Gepotidacin and Zoliflodacin Stabilize DNA Cleavage Complexes with Bacterial Type IIA Topoisomerases? 1. Experimental Definition of Metal Binding Sites. International Journal of Molecular Sciences. 2024; 25(21):11688. https://doi.org/10.3390/ijms252111688
Chicago/Turabian StyleMorgan, Harry, Robert A. Nicholls, Anna J. Warren, Simon E. Ward, Gwyndaf Evans, Fei Long, Garib N. Murshudov, Ramona Duman, and Benjamin D. Bax. 2024. "How Do Gepotidacin and Zoliflodacin Stabilize DNA Cleavage Complexes with Bacterial Type IIA Topoisomerases? 1. Experimental Definition of Metal Binding Sites" International Journal of Molecular Sciences 25, no. 21: 11688. https://doi.org/10.3390/ijms252111688
APA StyleMorgan, H., Nicholls, R. A., Warren, A. J., Ward, S. E., Evans, G., Long, F., Murshudov, G. N., Duman, R., & Bax, B. D. (2024). How Do Gepotidacin and Zoliflodacin Stabilize DNA Cleavage Complexes with Bacterial Type IIA Topoisomerases? 1. Experimental Definition of Metal Binding Sites. International Journal of Molecular Sciences, 25(21), 11688. https://doi.org/10.3390/ijms252111688