
Antimicrobial Peptide with a Bent Helix Motif Identified in Parasitic Flatworm Mesocestoides corti

 ,
,  ,
,  , , , , , and
, , , , , and
Abstract

1. Introduction
2. Results
2.1. Peptide Sequences
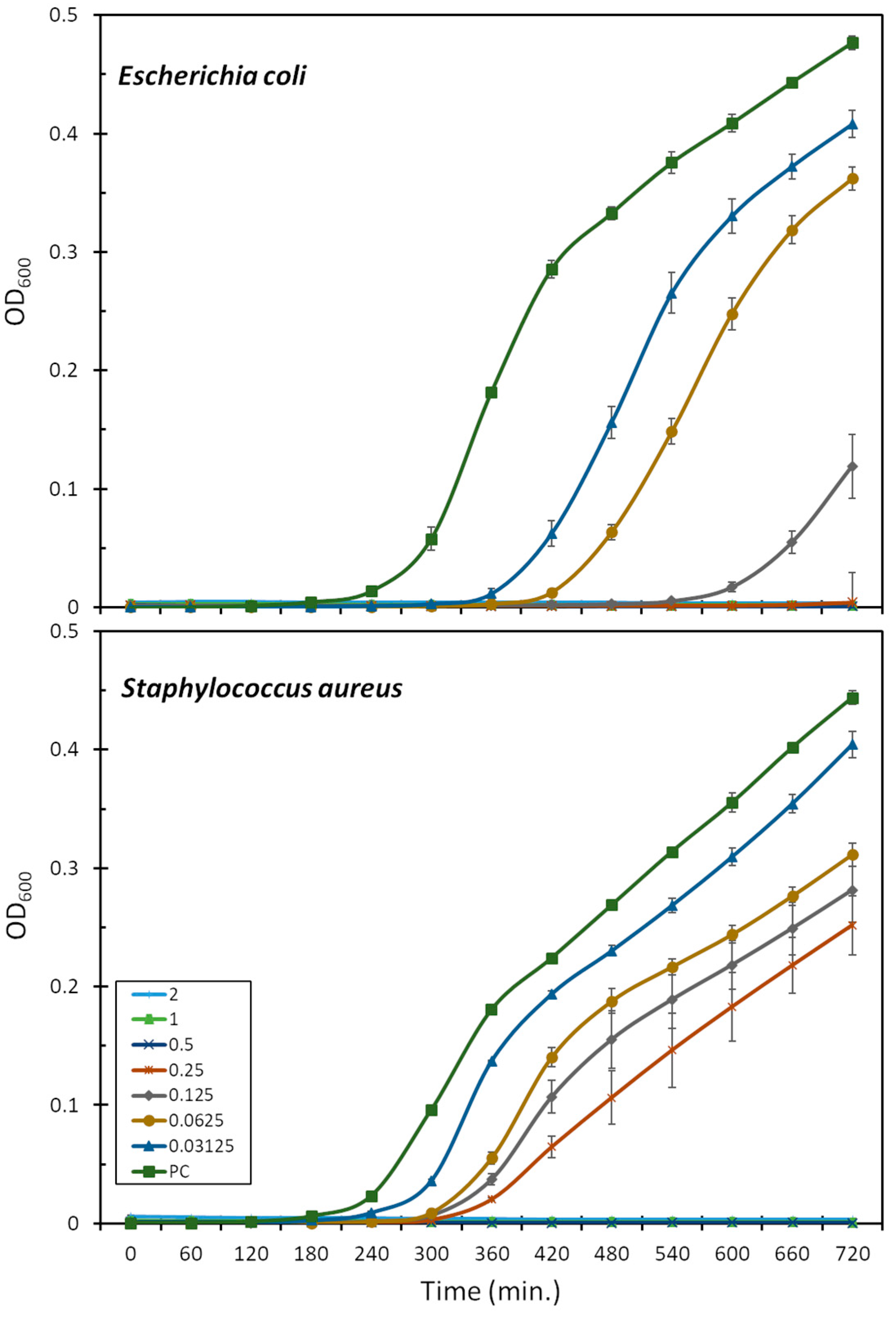
2.2. Antibacterial Activity
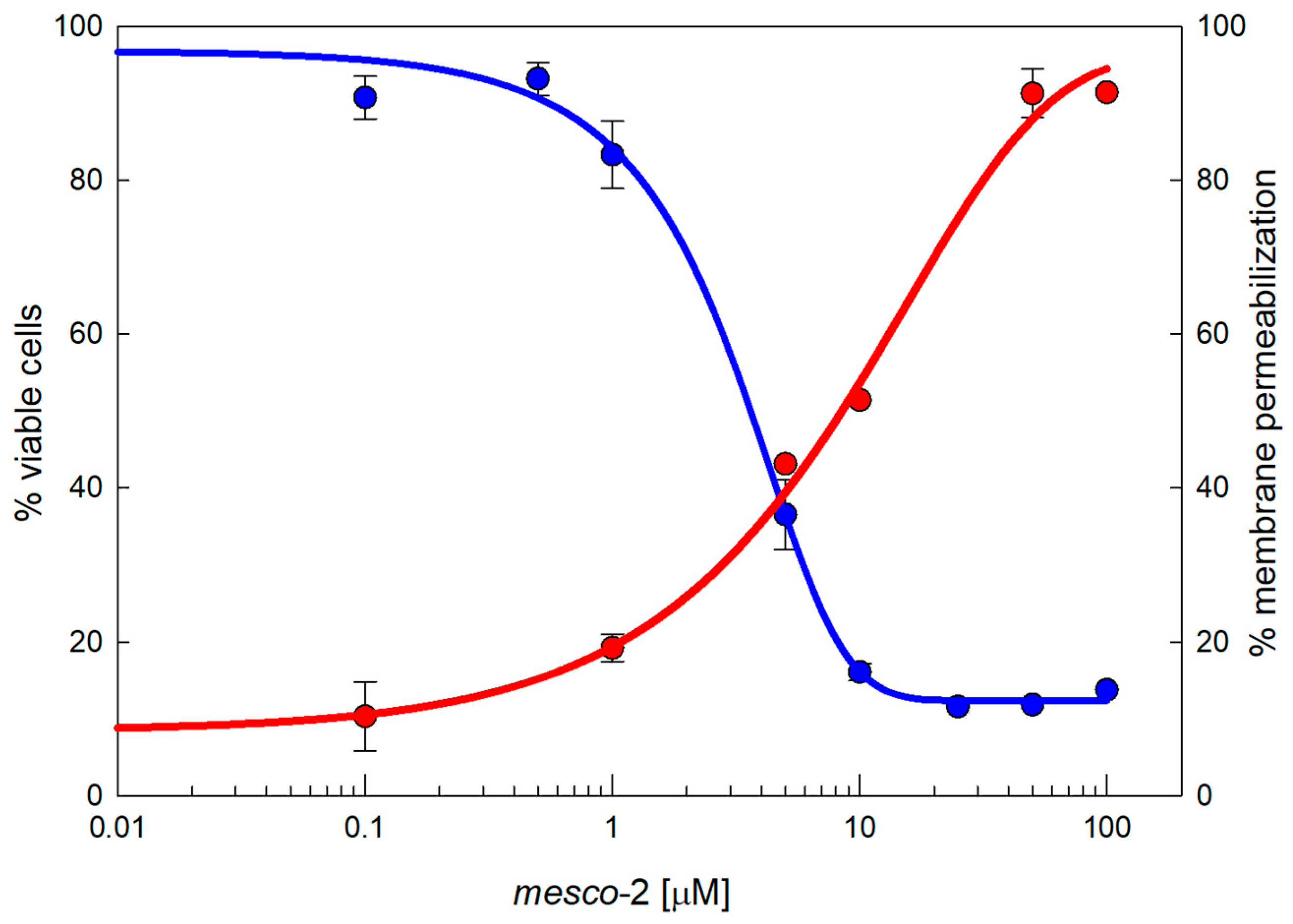
2.3. Toxicity
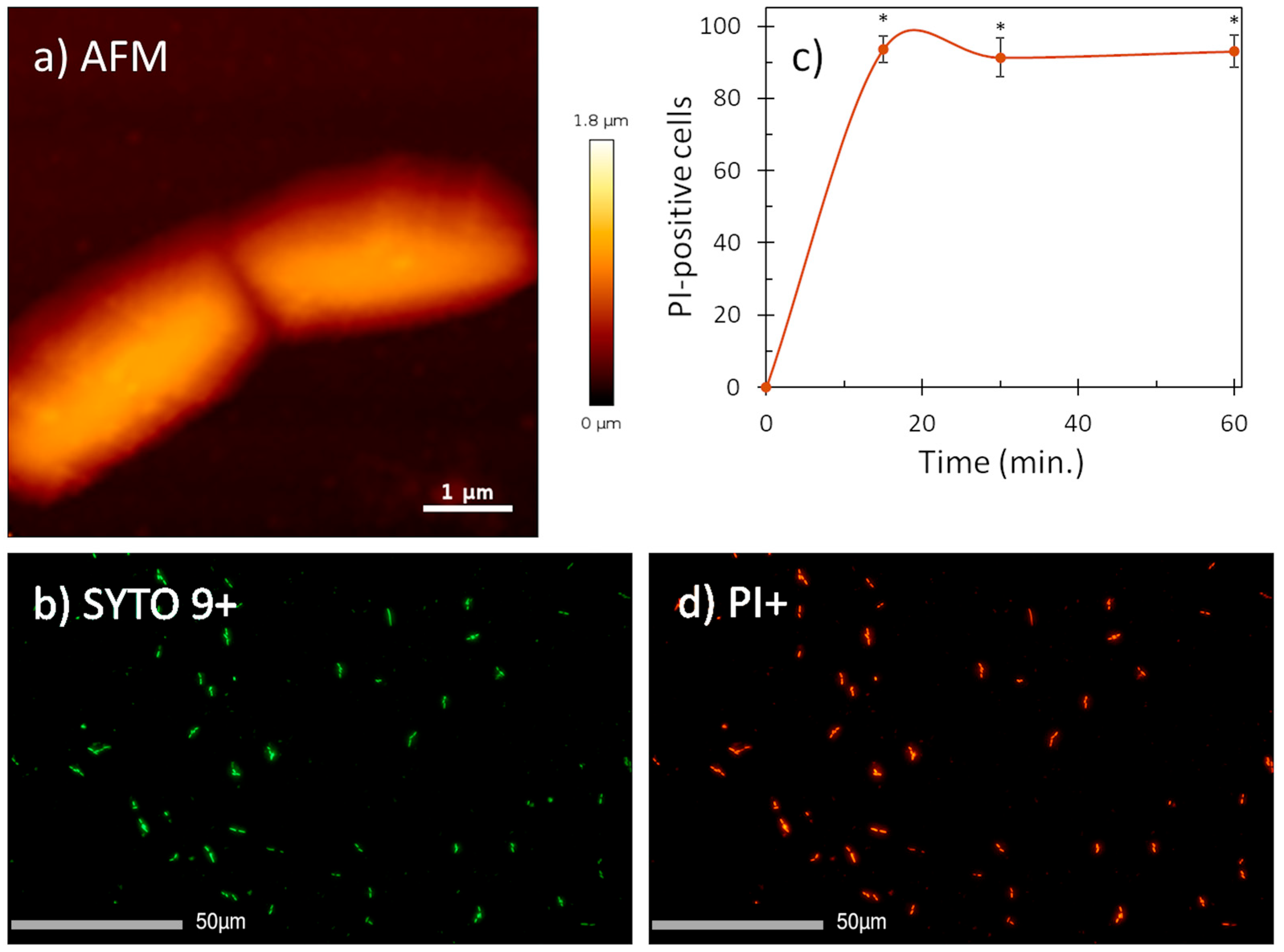
2.4. Peptide–Membrane Interaction
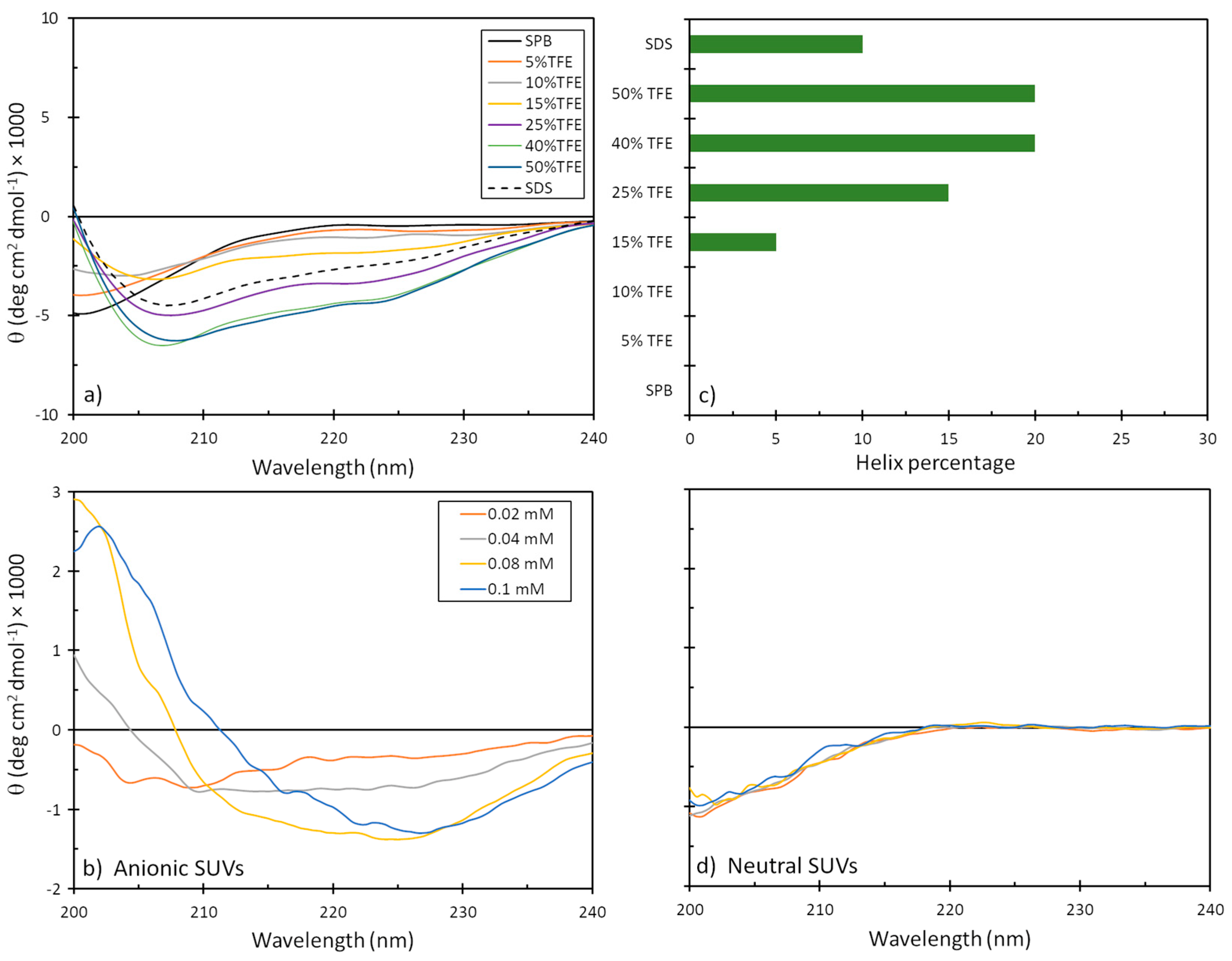
2.5. Secondary Structure
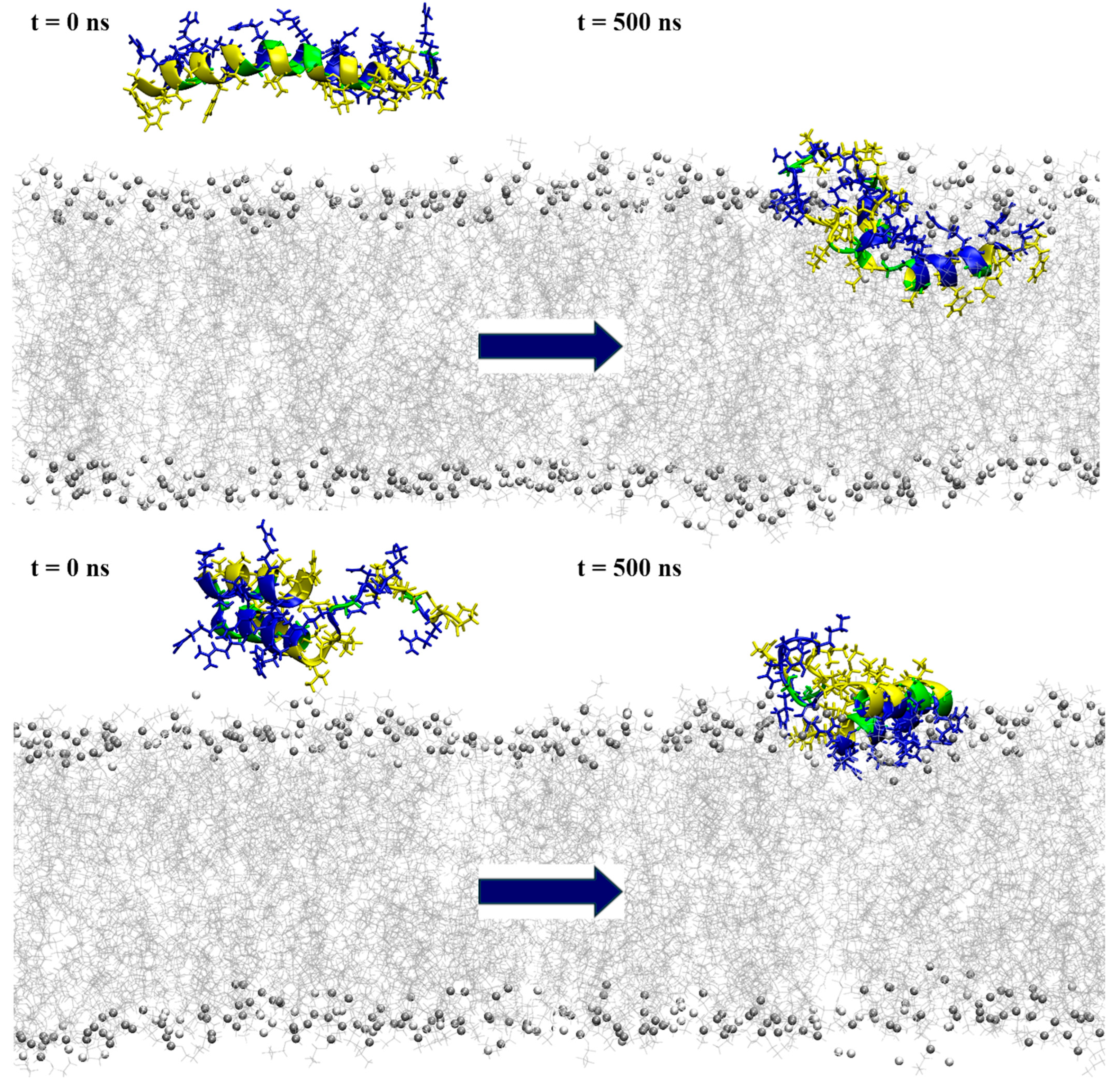
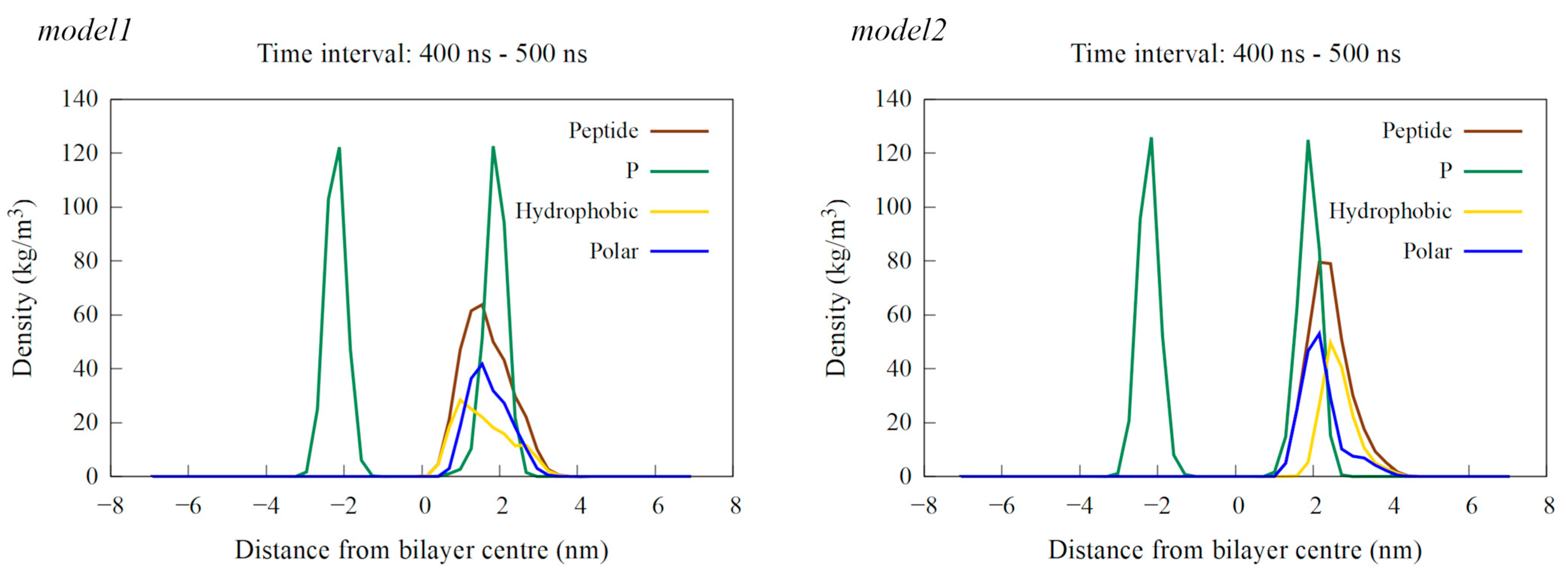
2.6. Structural Assessment via Molecular Modeling
3. Discussion
4. Materials and Methods
4.1. Peptide Identification
4.2. Peptide Synthesis
4.3. Bacterial Strains and Antibacterial Assays
4.4. Membrane Integrity Assay
4.5. Cytotoxicity Assays
4.6. Atomic Force Microscopy and Fluorescence Imaging
4.7. Liposome Preparation
4.8. Circular Dichroism
4.9. Structure Prediction and Molecular Dynamics Simulation Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 27 July 2023).
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef] [PubMed]
- Benfield, A.H.; Henriques, S.T. Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front. Med. Technol. 2020, 2, 610997. [Google Scholar] [CrossRef]
- Rončević, T.; Puizina, J.; Tossi, A. Antimicrobial Peptides as Anti-Infective Agents in Pre-Post-Antibiotic Era? Int. J. Mol. Sci. 2019, 20, 5713. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing Antimicrobial Peptides: Form Follows Function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Liang, W.; Diana, J. Mining the Bacterial Genome to Discover New Antimicrobial Molecules. EMBO Mol. Med. 2022, 14, e15409. [Google Scholar] [CrossRef]
- Gawde, U.; Chakraborty, S.; Waghu, F.H.; Barai, R.S.; Khanderkar, A.; Indraguru, R.; Shirsat, T.; Idicula-Thomas, S. CAMPR4: A Database of Natural and Synthetic Antimicrobial Peptides. Nucleic Acids Res. 2023, 51, D377–D383. [Google Scholar] [CrossRef]
- Littlewood, D.T.J. Marine Parasites and the Tree of Life. In Marine Parasitology; Rohde, K., Ed.; CABI Publishing: Wallingford, UK, 2005; pp. 6–10. ISBN 978-0-643-09025-5. [Google Scholar]
- Mladineo, I.; Rončević, T.; Gerdol, M.; Tossi, A. Helminthic Host Defense Peptides: Using the Parasite to Defend the Host. Trends Parasitol. 2023, 39, 345–357. [Google Scholar] [CrossRef]
- Rončević, T.; Gerdol, M.; Mardirossian, M.; Maleš, M.; Cvjetan, S.; Benincasa, M.; Maravić, A.; Gajski, G.; Krce, L.; Aviani, I.; et al. Anisaxins, Helical Antimicrobial Peptides from Marine Parasites, Kill Resistant Bacteria by Lipid Extraction and Membrane Disruption. Acta Biomater. 2022, 146, 131–144. [Google Scholar] [CrossRef] [PubMed]
- CDC—DPDx—Mesocestoidiasis. Available online: https://www.cdc.gov/dpdx/mesocestoidiasis/index.html (accessed on 30 September 2024).
- Kozić, M.; Vukičević, D.; Simunić, J.; Rončević, T.; Antcheva, N.; Tossi, A.; Juretić, D. Predicting the Minimal Inhibitory Concentration for Antimicrobial Peptides with Rana-Box Domain. J. Chem. Inf. Model. 2015, 55, 2275–2287. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L.; Giangaspero, A. New Consensus Hydrophobicity Scale Extended to Non-Proteinogenic Amino Acids. Peptides 2002, 27, 416. [Google Scholar]
- Shai, Y. Mode of Action of Membrane Active Antimicrobial Peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef]
- Sato, H.; Feix, J.B. Peptide–Membrane Interactions and Mechanisms of Membrane Destruction by Amphipathic α-Helical Antimicrobial Peptides. Biochim. Biophys. Acta BBA Biomembr. 2006, 1758, 1245–1256. [Google Scholar] [CrossRef]
- Rončević, T.; Vukičević, D.; Ilić, N.; Krce, L.; Gajski, G.; Tonkić, M.; Goić-Barišić, I.; Zoranić, L.; Sonavane, Y.; Benincasa, M.; et al. Antibacterial Activity Affected by the Conformational Flexibility in Glycine–Lysine Based α-Helical Antimicrobial Peptides. J. Med. Chem. 2018, 61, 2924–2936. [Google Scholar] [CrossRef]
- Rončević, T.; Krce, L.; Gerdol, M.; Pacor, S.; Benincasa, M.; Guida, F.; Aviani, I.; Čikeš-Čulić, V.; Pallavicini, A.; Maravić, A.; et al. Membrane-Active Antimicrobial Peptide Identified in Rana arvalis by Targeted DNA Sequencing. Biochim. Biophys. Acta BBA Biomembr. 2019, 1861, 651–659. [Google Scholar] [CrossRef]
- Lee, M.-T.; Sun, T.-L.; Hung, W.-C.; Huang, H.W. Process of Inducing Pores in Membranes by Melittin. Proc. Natl. Acad. Sci. USA 2013, 110, 14243–14248. [Google Scholar] [CrossRef]
- Wagschal, K.; Tripet, B.; Hodges, R.S. De Novo Design of a Model Peptide Sequence to Examine the Effects of Single Amino Acid Substitutions in the Hydrophobic Core on Both Stability and Oligomerization State of Coiled-Coils. J. Mol. Biol. 1999, 285, 785–803. [Google Scholar] [CrossRef]
- Wang, G.; Mishra, B.; Epand, R.F.; Epand, R.M. High-Quality 3D Structures Shine Light on Antibacterial, Anti-Biofilm and Antiviral Activities of Human Cathelicidin LL-37 and Its Fragments. Biochim. Biophys. Acta 2014, 1838, 2160–2172. [Google Scholar] [CrossRef]
- Xhindoli, D.; Pacor, S.; Guida, F.; Antcheva, N.; Tossi, A. Native Oligomerization Determines the Mode of Action and Biological Activities of Human Cathelicidin LL-37. Biochem. J. 2014, 457, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Tuerkova, A.; Kabelka, I.; Králová, T.; Sukeník, L.; Pokorná, Š.; Hof, M.; Vácha, R. Effect of Helical Kink in Antimicrobial Peptides on Membrane Pore Formation. eLife 2020, 9, e47946. [Google Scholar] [CrossRef] [PubMed]
- Bossemeyer, D. The Glycine-Rich Sequence of Protein Kinases: A Multifunctional Element. Trends Biochem. Sci. 1994, 19, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, A.; Hotta, Y. Glycine-Rich Proteins: A Class of Novel Proteins. Appl. Biochem. Biotechnol. 2005, 120, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Takeshima, K.; Park, C.B.; Kim, S.C.; Matsuzaki, K. Interactions of the Novel Antimicrobial Peptide Buforin 2 with Lipid Bilayers: Proline as a Translocation Promoting Factor. Biochemistry 2000, 39, 8648–8654. [Google Scholar] [CrossRef]
- Takeshima, K.; Chikushi, A.; Lee, K.-K.; Yonehara, S.; Matsuzaki, K. Translocation of Analogues of the Antimicrobial Peptides Magainin and Buforin across Human Cell Membranes*. J. Biol. Chem. 2003, 278, 1310–1315. [Google Scholar] [CrossRef]
- Elmore, D.E. Insights into Buforin II Membrane Translocation from Molecular Dynamics Simulations. Peptides 2012, 38, 357–362. [Google Scholar] [CrossRef]
- Harris, T.W.; Chen, N.; Cunningham, F.; Tello-Ruiz, M.; Antoshechkin, I.; Bastiani, C.; Bieri, T.; Blasiar, D.; Bradnam, K.; Chan, J.; et al. WormBase: A Multi-Species Resource for Nematode Biology and Genomics. Nucleic Acids Res. 2004, 32, D411–D417. [Google Scholar] [CrossRef]
- Leoni, G.; De Poli, A.; Mardirossian, M.; Gambato, S.; Florian, F.; Venier, P.; Wilson, D.N.; Tossi, A.; Pallavicini, A.; Gerdol, M. Myticalins: A Novel Multigenic Family of Linear, Cationic Antimicrobial Peptides from Marine Mussels (Mytilus spp.). Mar. Drugs 2017, 15, 261. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating Signal Peptides from Transmembrane Regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A Hidden Markov Model for Predicting Transmembrane Helices in Protein Sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar]
- Basika, T.; Paludo, G.P.; Araujo, F.M.; Salim, A.C.; Pais, F.; Maldonado, L.; Macchiaroli, N.; Camargo de Lima, J.; Rosenzvit, M.; Oliveira, G.C.; et al. Transcriptomic Profile of Two Developmental Stages of the Cestode Parasite Mesocestoides corti. Mol. Biochem. Parasitol. 2019, 229, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Costábile, A.; Domínguez, M.F.; Guarnaschelli, I.; Preza, M.; Koziol, U.; Castillo, E.; Tort, J.F. Purification and Transcriptomic Characterization of Proliferative Cells of Mesocestoides corti Selectively Affected by Irradiation. Front. Parasitol. 2024, 3, 1362199. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA Abundance Using RNA-seq Data: RPKM Measure Is Inconsistent among Samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Kuipers, B.J.H.; Gruppen, H. Prediction of Molar Extinction Coefficients of Proteins and Peptides Using UV Absorption of the Constituent Amino Acids at 214 nm To Enable Quantitative Reverse Phase High-Performance Liquid Chromatography−Mass Spectrometry Analysis. J. Agric. Food Chem. 2007, 55, 5445–5451. [Google Scholar] [CrossRef]
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 14.0. 2024. Available online: http://www.eucast.org (accessed on 22 July 2024).
- Rončević, T.; Vukičević, D.; Krce, L.; Benincasa, M.; Aviani, I.; Maravić, A.; Tossi, A. Selection and Redesign for High Selectivity of Membrane-Active Antimicrobial Peptides from a Dedicated Sequence/Function Database. Biochim. Biophys. Acta BBA Biomembr. 2019, 1861, 827–834. [Google Scholar] [CrossRef]
- Stacchini, A.; Aragno, M.; Vallario, A.; Alfarano, A.; Circosta, P.; Gottardi, D.; Faldella, A.; Rege-Cambrin, G.; Thunberg, U.; Nilsson, K. MEC1 and MEC2: Two New Cell Lines Derived from B-Chronic Lymphocytic Leukaemia in Prolymphocytoid Transformation. Leuk. Res. 1999, 23, 127–136. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Yang, J.T.; Chau, K.H. Determination of the Helix and β Form of Proteins in Aqueous Solution by Circular Dichroism. Biochemistry 1974, 13, 3350–3359. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing Structure Coverage for over 214 Million Protein Sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane BUILDER toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded And intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Murzyn, K.; Róg, T.; Pasenkiewicz-Gierula, M. Phosphatidylethanolamine-Phosphatidylglycerol Bilayer as a Model of the Inner Bacterial Membrane. Biophys. J. 2005, 88, 1091–1103. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Im, W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Gnuplot. Available online: http://www.gnuplot.info/ (accessed on 27 September 2024).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. ACPred: A Computational Tool for the Prediction and Analysis of Anticancer. Molecules 2019, 24, 1973. [Google Scholar] [CrossRef]
- Sangaraju, V.K.; Pham, N.T.; Wei, L.; Yu, X.; Manavalan, B. mACPpred 2.0: Stacked Deep Learning for Anticancer Peptide Prediction with Integrated Spatial and Probabilistic Feature Representations. J. Mol. Biol. 2024, 436, 68687. [Google Scholar] [CrossRef]
- Khatun, M.S.; Hasan, M.; Kurata, H. PreAIP: Computational Prediction of Anti-inflammatory Peptides by Integrating Multiple Complementary Features. Front. Genet. 2019, 10, 129. [Google Scholar] [CrossRef]
- Timmons, P.B.; Timmons, P.B.; Hewage, C.M.; Hewage, C.M. ENNAVIA is a novel method which employs neural networks for antiviral and anti-coronavirus activity prediction for therapeutic peptides. Briefings Bioinform. 2021, 22, bbab258. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. Meta-iAVP: A Sequence-Based Meta-Predictor for Improving the Prediction of Antiviral Peptides Using Effective Feature Representation. Int. J. Mol. Sci. 2019, 20, 5743. [Google Scholar] [CrossRef] [PubMed]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, srep42362. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Patra, M.C. MLCPP 2.0: An Updated Cell-penetrating Peptides and Their Uptake Efficiency Predictor. J. Mol. Biol. 2022, 434, 167604. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sharma, A.K.; Shastri, V.; Madhu, M.K.; Sharma, V.K. Prediction of anti-inflammatory proteins/peptides: An insilico approach. J. Transl. Med. 2017, 15, 1–11. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P. Peptide toxicity prediction. Methods Mol. Biol. 2015, 1268, 143–157. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Charge | H 1 | H rel 2 |
|---|---|---|---|---|
| mesco-1 | WRRLRRRISGGLRRIFRKPRRICFPYCPTGPRYPGPRPY | +13 | −0.475 | 0.071 |
| mesco-2 | FFRRIGRAFSRVGRGIGRGFRQLGRLMPRGNYKICLGRCP | +11 | −0.232 | 0.106 |
| mesco-3 | FLRRIGRAFSRVGRGIGRGFRQLGRLMPRGNYRICLGRCPR | +12 | −0.316 | 0.095 |
| Bacterial Strain | Mesco-2 | |
|---|---|---|
| MIC | MBC | |
| Escherichia coli ATCC 25922 | 0.5 | 0.5 |
| Acinetobacter baumannii ATCC 19606 | 1–2 | 1–2 |
| Klebsiella pneumoniae ATCC 13883 | 0.5 | 0.5 |
| Pseudomonas aeruginosa ATCC 27853 | 2 | 2 |
| Staphylococcus aureus ATCC 29213 | 1 | 1 |
| Staphylococcus aureus ATCC 25923 | 2 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rončević, T.; Gerdol, M.; Pacor, S.; Cvitanović, A.; Begić, A.; Weber, I.; Krce, L.; Caporale, A.; Mardirossian, M.; Tossi, A.; et al. Antimicrobial Peptide with a Bent Helix Motif Identified in Parasitic Flatworm Mesocestoides corti. Int. J. Mol. Sci. 2024, 25, 11690. https://doi.org/10.3390/ijms252111690
Rončević T, Gerdol M, Pacor S, Cvitanović A, Begić A, Weber I, Krce L, Caporale A, Mardirossian M, Tossi A, et al. Antimicrobial Peptide with a Bent Helix Motif Identified in Parasitic Flatworm Mesocestoides corti. International Journal of Molecular Sciences. 2024; 25(21):11690. https://doi.org/10.3390/ijms252111690
Chicago/Turabian StyleRončević, Tomislav, Marco Gerdol, Sabrina Pacor, Ana Cvitanović, Anamarija Begić, Ivana Weber, Lucija Krce, Andrea Caporale, Mario Mardirossian, Alessandro Tossi, and et al. 2024. "Antimicrobial Peptide with a Bent Helix Motif Identified in Parasitic Flatworm Mesocestoides corti" International Journal of Molecular Sciences 25, no. 21: 11690. https://doi.org/10.3390/ijms252111690
APA StyleRončević, T., Gerdol, M., Pacor, S., Cvitanović, A., Begić, A., Weber, I., Krce, L., Caporale, A., Mardirossian, M., Tossi, A., & Zoranić, L. (2024). Antimicrobial Peptide with a Bent Helix Motif Identified in Parasitic Flatworm Mesocestoides corti. International Journal of Molecular Sciences, 25(21), 11690. https://doi.org/10.3390/ijms252111690