
Interactive Structural Analysis of KH3-4 Didomains of IGF2BPs with Preferred RNA Motif Having m6A Through Dynamics Simulation Studies



Abstract
1. Introduction
2. Results
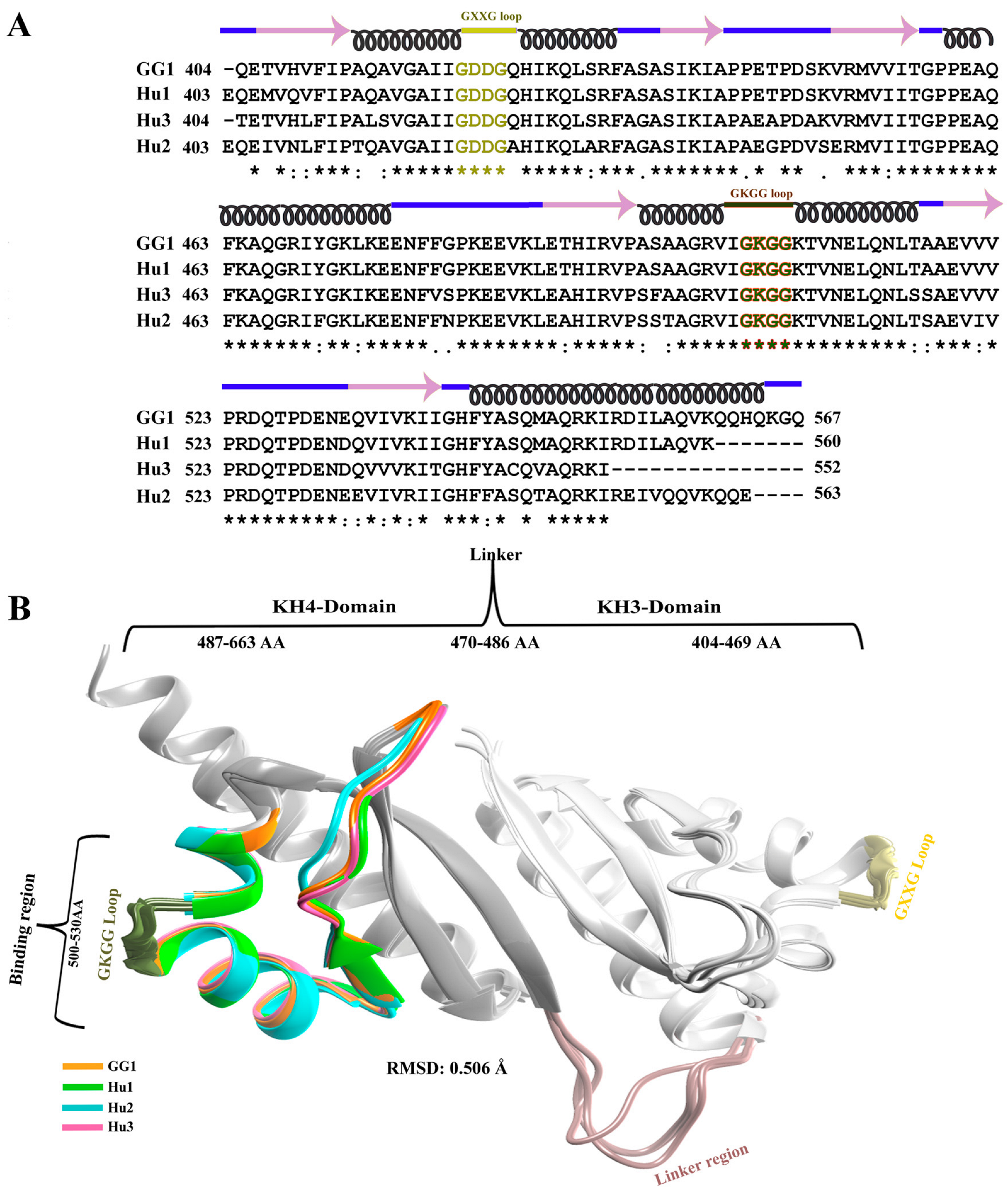
2.1. Modeling and Comparative Analysis of KH3-4 Domains of IGF2BPs
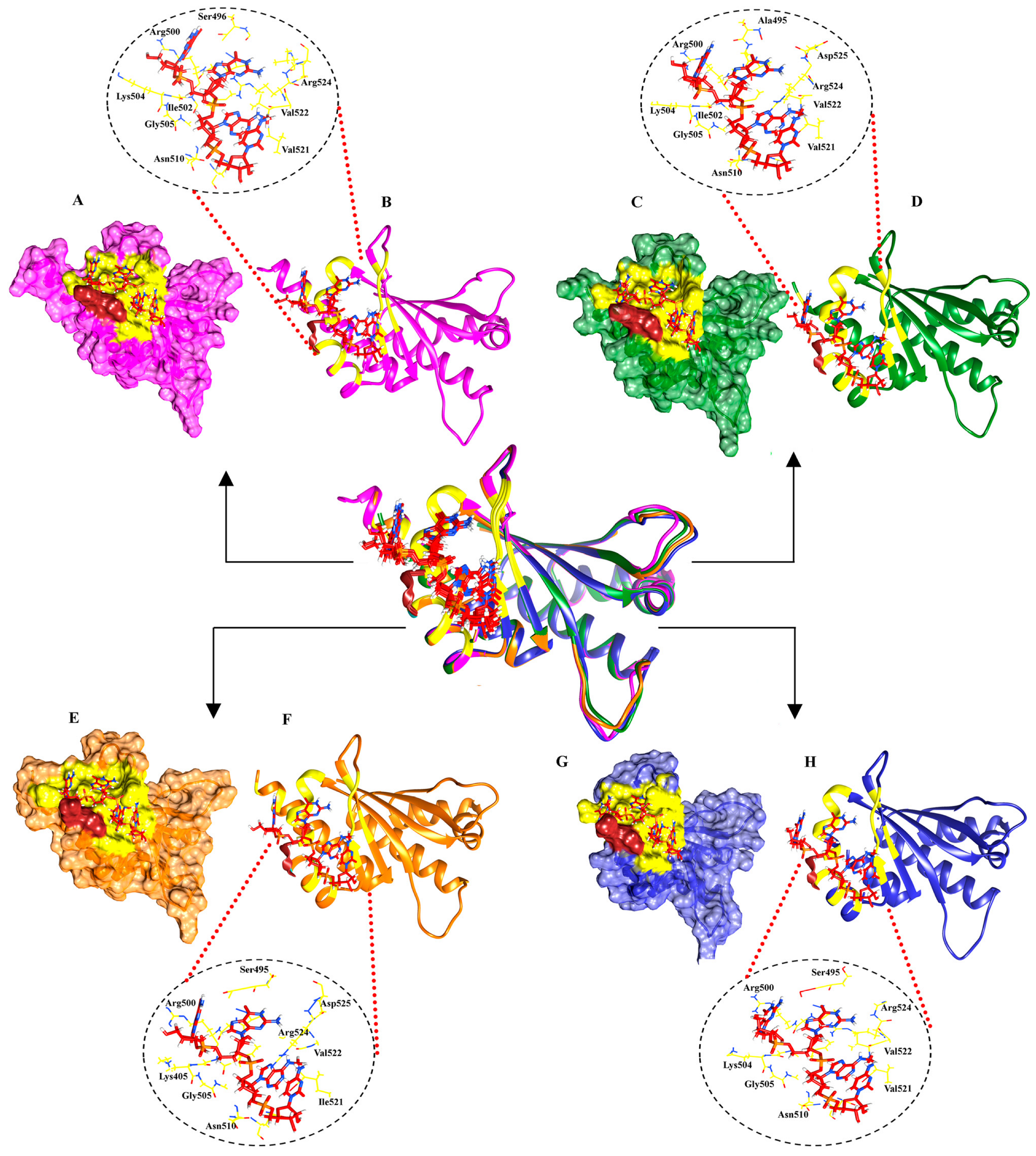
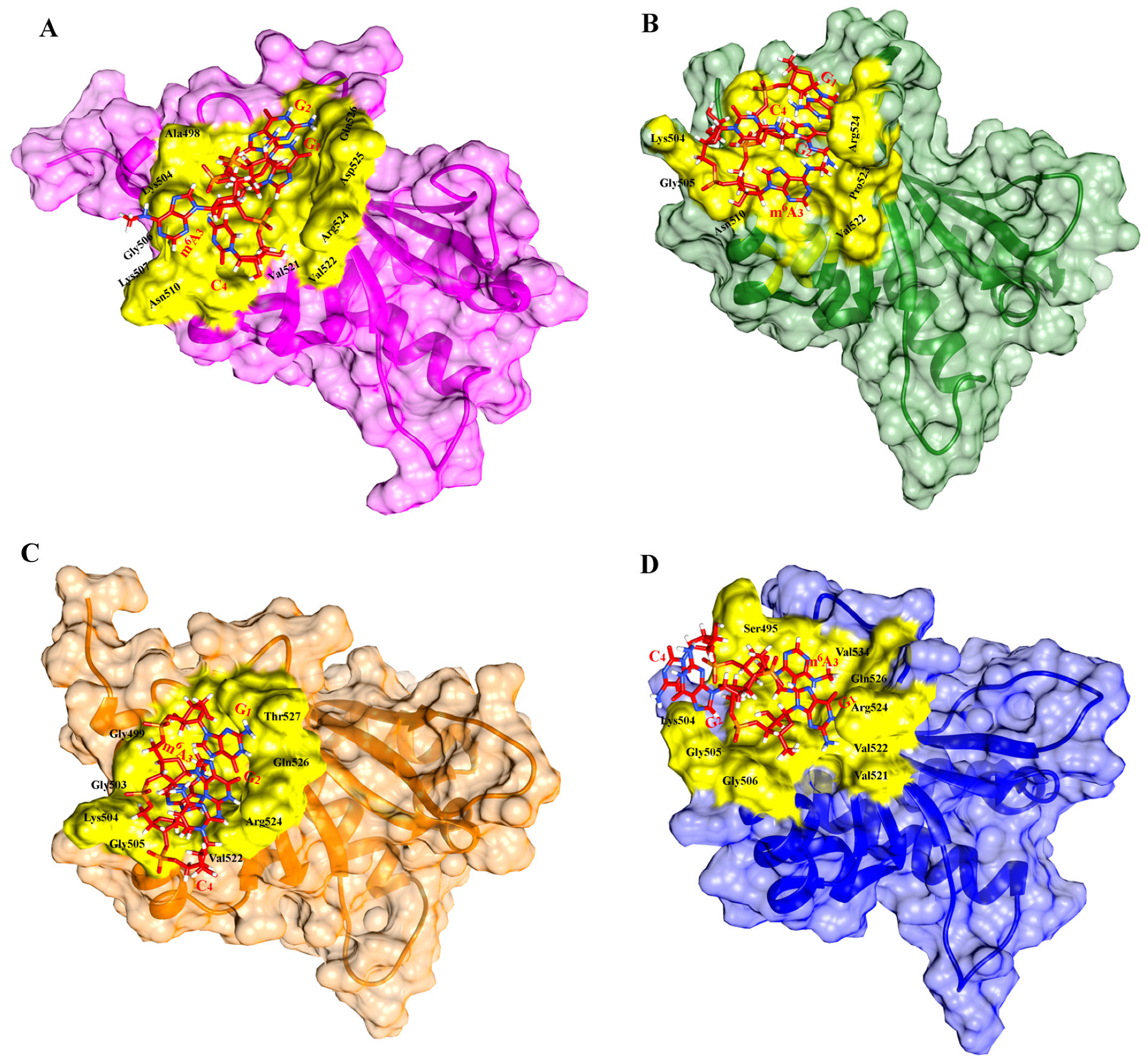
2.2. Molecular Docking Analysis
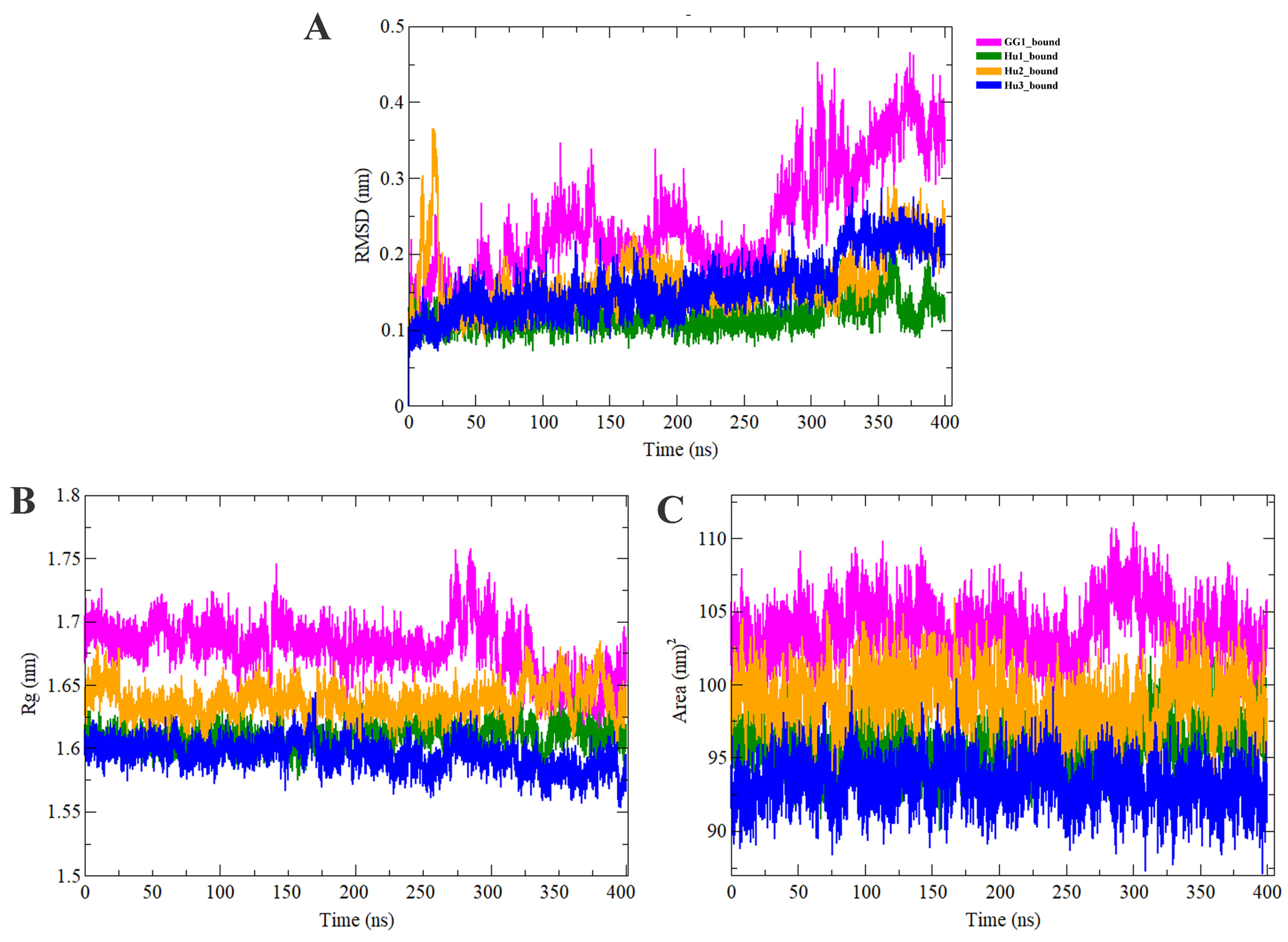
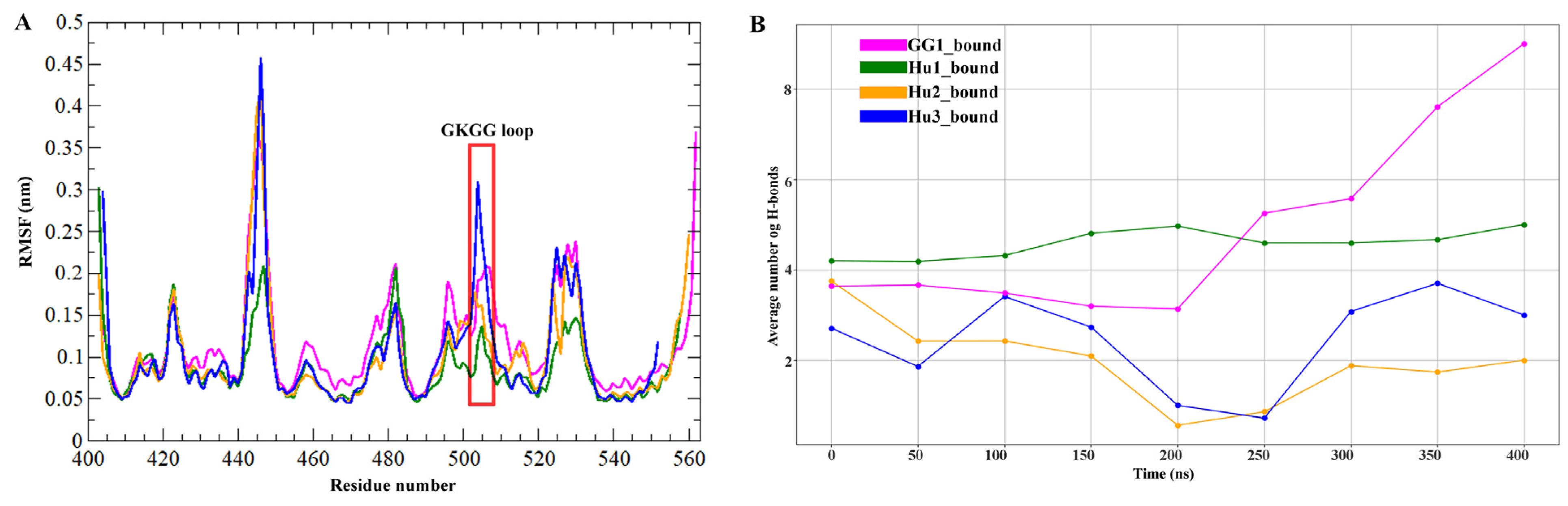
2.3. Molecular Dynamics Simulation Analysis
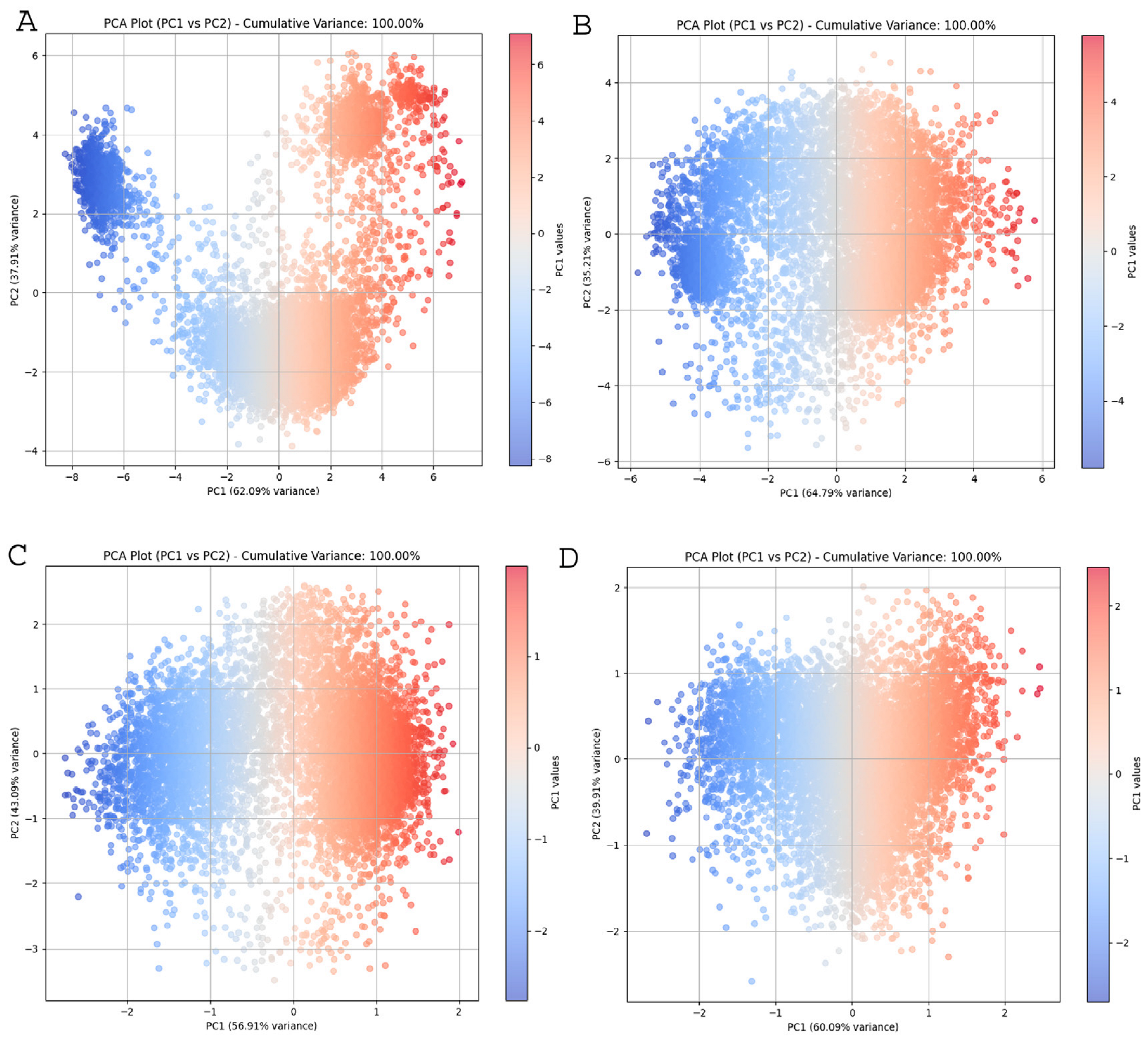
2.4. Principal Component Analysis
3. Discussion
4. Material and Methods
4.1. Data Set
4.2. Molecular Docking
4.3. Molecular Dynamics Simulation Analysis
4.4. Principal Component Analysis
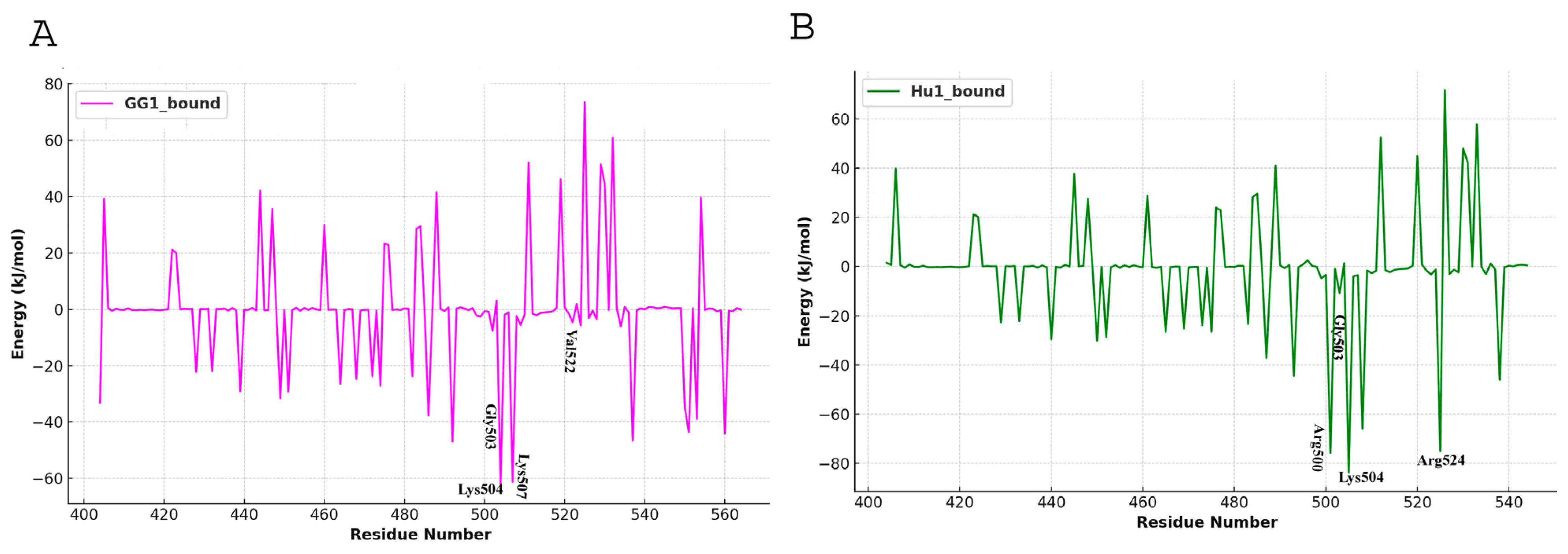
4.5. Binding Energy Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, S.; Lv, W.; Li, T.; Zhang, S.; Wang, H.; Li, X.; Wang, L.; Ma, D.; Zang, Y.; Shen, J.; et al. Dynamic Regulation and Functions of MRNA M6A Modification. Cancer Cell Int. 2022, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wu, R.; Ming, L. The Role of M6A RNA Methylation in Cancer. Biomed. Pharmacother. 2019, 112, 108613. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Salmon-Divon, M.; Amariglio, N.; Rechavi, G. Transcriptome-Wide Mapping of N 6-Methyladenosine by M6A-Seq Based on Immunocapturing and Massively Parallel Sequencing. Nat. Protoc. 2013, 8, 176–189. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of MRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.-S.; Hao, Y.-J.; Sun, B.-F.; Sun, H.-Y.; Li, A.; Ping, X.-L.; Lai, W.-Y.; et al. Nuclear M6A Reader YTHDC1 Regulates MRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N 6-Methyladenosine Marks Primary MicroRNAs for Processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The M6A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-Methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Wang, R.; Xiong, F.; Krakowiak, J.; Liao, Z.; Nguyen, P.T.; Moroz-Omori, E.V.; Shao, J.; Zhu, X.; Bolt, M.J.; et al. Enhancer RNA M6A Methylation Facilitates Transcriptional Condensate Formation and Gene Activation. Mol. Cell 2021, 81, 3368–3385. [Google Scholar] [CrossRef]
- Ben-Haim, M.S.; Moshitch-Moshkovitz, S.; Rechavi, G. FTO: Linking M6A Demethylation to Adipogenesis. Cell Res. 2015, 25, 3–4. [Google Scholar] [CrossRef]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-Methyladenosine and Its Role in Cancer. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Satterlee, J.S.; Basanta-Sanchez, M.; Blanco, S.; Li, J.B.; Meyer, K.; Pollock, J.; Sadri-Vakili, G.; Rybak-Wolf, A. Novel RNA Modifications in the Nervous System: Form and Function. J. Neurosci. 2014, 34, 15170–15177. [Google Scholar] [CrossRef] [PubMed]
- Ramesh-Kumar, D.; Guil, S. The IGF2BP Family of RNA Binding Proteins Links Epitranscriptomics to Cancer. Semin. Cancer Biol. 2022, 86, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Jaffrey, S.R. Rethinking M6A Readers, Writers, and Erasers. Annu. Rev. Cell Dev. Biol. 2017, 33, 319–342. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Y.; Sun, B.-F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.-J.; Ping, X.-L.; Chen, Y.-S.; Wang, W.-J.; et al. FTO-Dependent Demethylation of N6-Methyladenosine Regulates MRNA Splicing and Is Required for Adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N 6-Methyladenosine by IGF2BP Proteins Enhances MRNA Stability and Translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Bell, J.L.; Wächter, K.; Mühleck, B.; Pazaitis, N.; Köhn, M.; Lederer, M.; Hüttelmaier, S. Insulin-like Growth Factor 2 MRNA-Binding Proteins (IGF2BPs): Post-Transcriptional Drivers of Cancer Progression? Cell. Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef]
- Zhou, H.; Sun, Q.; Feng, M.; Gao, Z.; Jia, S.; Cao, L.; Yu, X.; Gao, S.; Wu, H.; Li, K. Regulatory Mechanisms and Therapeutic Implications of Insulin-like Growth Factor 2 MRNA-Binding Proteins, the Emerging Crucial M6A Regulators of Tumors. Theranostics 2023, 13, 4247. [Google Scholar] [CrossRef]
- Biswas, J.; Patel, V.L.; Bhaskar, V.; Chao, J.A.; Singer, R.H.; Eliscovich, C. The Structural Basis for RNA Selectivity by the IMP Family of RNA-Binding Proteins. Nat. Commun. 2019, 10, 4440. [Google Scholar] [CrossRef]
- Korn, S.M.; Ulshöfer, C.J.; Schneider, T.; Schlundt, A. Structures and Target RNA Preferences of the RNA-Binding Protein Family of IGF2BPs: An Overview. Structure 2021, 29, 787–803. [Google Scholar] [CrossRef]
- Valverde, R.; Edwards, L.; Regan, L. Structure and Function of KH Domains. FEBS J. 2008, 275, 2712–2726. [Google Scholar] [CrossRef] [PubMed]
- Grishin, N. V KH Domain: One Motif, Two Folds. Nucleic Acids Res. 2001, 29, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Cukier, C.D.; Ramos, A. Modular Protein-RNA Interactions Regulating MRNA Metabolism: A Role for NMR. Eur. Biophys. J. 2011, 40, 1317–1325. [Google Scholar] [CrossRef]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-Binding Proteins: Modular Design for Efficient Function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef]
- Nicastro, G.; Taylor, I.A.; Ramos, A. KH–RNA Interactions: Back in the Groove. Curr. Opin. Struct. Biol. 2015, 30, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Paziewska, A.; Wyrwicz, L.S.; Bujnicki, J.M.; Bomsztyk, K.; Ostrowski, J. Cooperative Binding of the HnRNP K Three KH Domains to MRNA Targets. FEBS Lett. 2004, 577, 134–140. [Google Scholar] [CrossRef]
- Wächter, K.; Köhn, M.; Stöhr, N.; Hüttelmaier, S. Subcellular Localization and RNP Formation of IGF2BPs (IGF2 MRNA-Binding Proteins) Is Modulated by Distinct RNA-Binding Domains. Biol. Chem. 2013, 394, 1077–1090. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 1–3. [Google Scholar] [CrossRef]
- Singh, A.; Copeland, M.M.; Kundrotas, P.J.; Vakser, I.A. GRAMM Web Server for Protein Docking. In Computational Drug Discovery and Design; Springer: Berlin/Heidelberg, Germany, 2023; pp. 101–112. [Google Scholar]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A Web Server for Protein–Protein and Protein–DNA/RNA Docking Based on a Hybrid Strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK Server for Integrated Protein–Protein Docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Duan, M.; Liu, H.; Xu, S.; Yang, Z.; Zhang, F.; Wang, G.; Wang, Y.; Zhao, S.; Jiang, X. IGF2BPs as Novel M6A Readers: Diverse Roles in Regulating Cancer Cell Biological Functions, Hypoxia Adaptation, Metabolism, and Immunosuppressive Tumor Microenvironment. Genes Dis. 2024, 11, 890–920. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Liu, P.Y.; Haase, J.; Bell, J.L.; Hüttelmaier, S.; Liu, T. The Critical Role of RNA M6A Methylation in Cancer. Cancer Res. 2019, 79, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shi, X.; Huang, T.; Zhao, X.; Chen, W.; Gu, N.; Zhang, R. Dynamic Landscape and Evolution of M6A Methylation in Human. Nucleic Acids Res. 2020, 48, 6251–6264. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Patel, H.; Chen, J.; Wang, J.; Chen, Z.-S.; Wang, H. Epigenetic Modification of M6A Regulator Proteins in Cancer. Mol. Cancer 2023, 22, 102. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Yuan, X.; Wang, X.; Liu, S. Crosstalk between M6A Modification and Non-Coding RNAs in HCC. Cell Signal 2024, 117, 111076. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; He, F.; Hou, Y.; Tu, G.; Li, Q.; Jin, T.; Zeng, H.; Qin, Y.; Wan, X.; Qiao, Y.; et al. A Novel Hypoxic Long Noncoding RNA KB-1980E6. 3 Maintains Breast Cancer Stem Cell Stemness via Interacting with IGF2BP1 to Facilitate c-Myc MRNA Stability. Oncogene 2021, 40, 1609–1627. [Google Scholar] [CrossRef]
- Chen, H.-M.; Lin, C.-C.; Chen, W.-S.; Jiang, J.-K.; Yang, S.-H.; Chang, S.-C.; Ho, C.-L.; Yang, C.-C.; Huang, S.-C.; Chao, Y.; et al. Insulin-like Growth Factor 2 MRNA-Binding Protein 1 (IGF2BP1) Is a Prognostic Biomarker and Associated with Chemotherapy Responsiveness in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 6940. [Google Scholar] [CrossRef]
- Müller, S.; Glaß, M.; Singh, A.K.; Haase, J.; Bley, N.; Fuchs, T.; Lederer, M.; Dahl, A.; Huang, H.; Chen, J.; et al. IGF2BP1 Promotes SRF-Dependent Transcription in Cancer in a M6A-and MiRNA-Dependent Manner. Nucleic Acids Res. 2019, 47, 375–390. [Google Scholar] [CrossRef]
- Liu, L.; Wu, Y.; Li, Q.; Liang, J.; He, Q.; Zhao, L.; Chen, J.; Cheng, M.; Huang, Z.; Ren, H.; et al. METTL3 Promotes Tumorigenesis and Metastasis through BMI1 M6A Methylation in Oral Squamous Cell Carcinoma. Mol. Ther. 2020, 28, 2177–2190. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, C.; Ding, Q.; Zhao, Y.; Wang, Z.; Chen, J.; Jiang, Z.; Zhang, Y.; Xu, G.; Zhang, J.; et al. METTL3-Mediated M6A Modification of HDGF MRNA Promotes Gastric Cancer Progression and Has Prognostic Significance. Gut 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, Y.; Mao, B.; You, Q.; Guo, X. Targeting Insulin-like Growth Factor 2 MRNA-Binding Proteins (IGF2BPs) for the Treatment of Cancer. Eur. J. Med. Chem. 2024, 268, 116241. [Google Scholar] [CrossRef]
- Nicastro, G.; Candel, A.M.; Uhl, M.; Oregioni, A.; Hollingworth, D.; Backofen, R.; Martin, S.R.; Ramos, A. Mechanism of β-Actin MRNA Recognition by ZBP1. Cell Rep. 2017, 18, 1187–1199. [Google Scholar] [CrossRef]
- Chao, J.A.; Patskovsky, Y.; Patel, V.; Levy, M.; Almo, S.C.; Singer, R.H. ZBP1 Recognition of β-Actin Zipcode Induces RNA Looping. Genes. Dev. 2010, 24, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, G.; Abis, G.; Klein, P.; Esteban-Serna, S.; Gallagher, C.; Chaves-Arquero, B.; Cai, Y.; Figueiredo, A.M.; Martin, S.R.; Patani, R.; et al. Direct M6A Recognition by IMP1 Underlays an Alternative Model of Target Selection for Non-Canonical Methyl-Readers. Nucleic Acids Res. 2023, 51, 8774–8786. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Jo, S.; MacKerell, A.D.; Klauda, J.B.; Im, W. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. Biophys. J. 2016, 110, 641a. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; MacKerell, A.D., Jr. Impact of 2’-Hydroxyl Sampling on the Conformational Properties of RNA: Update of the CHARMM All-Atom Additive Force Field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef] [PubMed]
- Gul, M.; Navid, A.; Fakhar, M.; Rashid, S. SHP-1 Tyrosine Phosphatase Binding to c-Src Kinase Phosphor-Dependent Conformations: A Comparative Structural Framework. PLoS ONE 2023, 18, e0278448. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Alexei, I. Implementation of the Particle Mesh Ewald Method on a GPU. Master’s Thesis, KTH Royal Institute of Technology, Stockholm, Sweden, 2016. [Google Scholar]
- McDonald, I.R. NpT-Ensemble Monte Carlo Calculations for Binary Liquid Mixtures. Mol. Phys. 1972, 23, 41–58. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Ausaf Ali, S.; Hassan, I.; Islam, A.; Ahmad, F. A Review of Methods Available to Estimate Solvent-Accessible Surface Areas of Soluble Proteins in the Folded and Unfolded States. Curr. Protein Pept. Sci. 2014, 15, 456–476. [Google Scholar]
- Lee, B.; Richards, F.M. The Interpretation of Protein Structures: Estimation of Static Accessibility. J. Mol. Biol. 1971, 55, 379–400, IN3–IN4. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, T.; Srinivasan, P. Exploring the Binding Properties of Agonists Interacting with Human TGR5 Using Structural Modeling, Molecular Docking and Dynamics Simulations. RSC Adv. 2015, 5, 14202–14213. [Google Scholar] [CrossRef]
- Ali, I.; Rasheed, M.A.; Cavalu, S.; Rahim, K.; Ijaz, S.; Yahya, G.; Goh, L.P.W.; Popoviciu, M.S. Identification of Natural Lead Compounds against Hemagglutinin-Esterase Surface Glycoprotein in Human Coronaviruses Investigated via MD Simulation, Principal Component Analysis, Cross-Correlation, H-Bond Plot and MMGBSA. Biomedicines 2023, 11, 793. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.N.N.; Ali, S.K.; Zhu, H. Structural Signature of the G719S-T790M Double Mutation in the EGFR Kinase Domain and Its Response to Inhibitors. Sci. Rep. 2014, 4, 5868. [Google Scholar]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Honig, B.; Nicholls, A. Classical Electrostatics in Biology and Chemistry. Science (1979) 1995, 268, 1144–1149. [Google Scholar] [CrossRef]
- Fakhar, M.; Gul, M.; Li, W. Interactive Structural Analysis of KH3-4 Didomains of IGF2BPs with Preferred RNA Motif Having m6A through Dynamics Simulation Studies. Zenodo 2024. [Data set]. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Evdw | Eelec | Gsol-polar | Gsol-non-polar | ΔGbinding |
|---|---|---|---|---|---|
| GG1_bound | −166.518 +/− 38.842 kJ/mol | −342.658 +/− 158.491 kJ/mol | 450.713 +/− 181.749 kJ/mol | −20.032 +/− 4.320 kJ/mol | −78.494 +/− 71.542 kJ/mol |
| Hu1_bound | −179.233 +/− 28.346 kJ/mol | −493.531 +/− 99.375 kJ/mol | 433.563 +/− 124.414 kJ/mol | −19.425 +/− 3.174 kJ/mol | −258.627 +/− 52.896 kJ/mol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakhar, M.; Gul, M.; Li, W. Interactive Structural Analysis of KH3-4 Didomains of IGF2BPs with Preferred RNA Motif Having m6A Through Dynamics Simulation Studies. Int. J. Mol. Sci. 2024, 25, 11118. https://doi.org/10.3390/ijms252011118
Fakhar M, Gul M, Li W. Interactive Structural Analysis of KH3-4 Didomains of IGF2BPs with Preferred RNA Motif Having m6A Through Dynamics Simulation Studies. International Journal of Molecular Sciences. 2024; 25(20):11118. https://doi.org/10.3390/ijms252011118
Chicago/Turabian StyleFakhar, Muhammad, Mehreen Gul, and Wenjin Li. 2024. "Interactive Structural Analysis of KH3-4 Didomains of IGF2BPs with Preferred RNA Motif Having m6A Through Dynamics Simulation Studies" International Journal of Molecular Sciences 25, no. 20: 11118. https://doi.org/10.3390/ijms252011118
APA StyleFakhar, M., Gul, M., & Li, W. (2024). Interactive Structural Analysis of KH3-4 Didomains of IGF2BPs with Preferred RNA Motif Having m6A Through Dynamics Simulation Studies. International Journal of Molecular Sciences, 25(20), 11118. https://doi.org/10.3390/ijms252011118