

Disrupted Post-Transcriptional Regulation of Gene Expression as a Hallmark of Fatty Liver Progression


Abstract
1. Introduction
2. Results
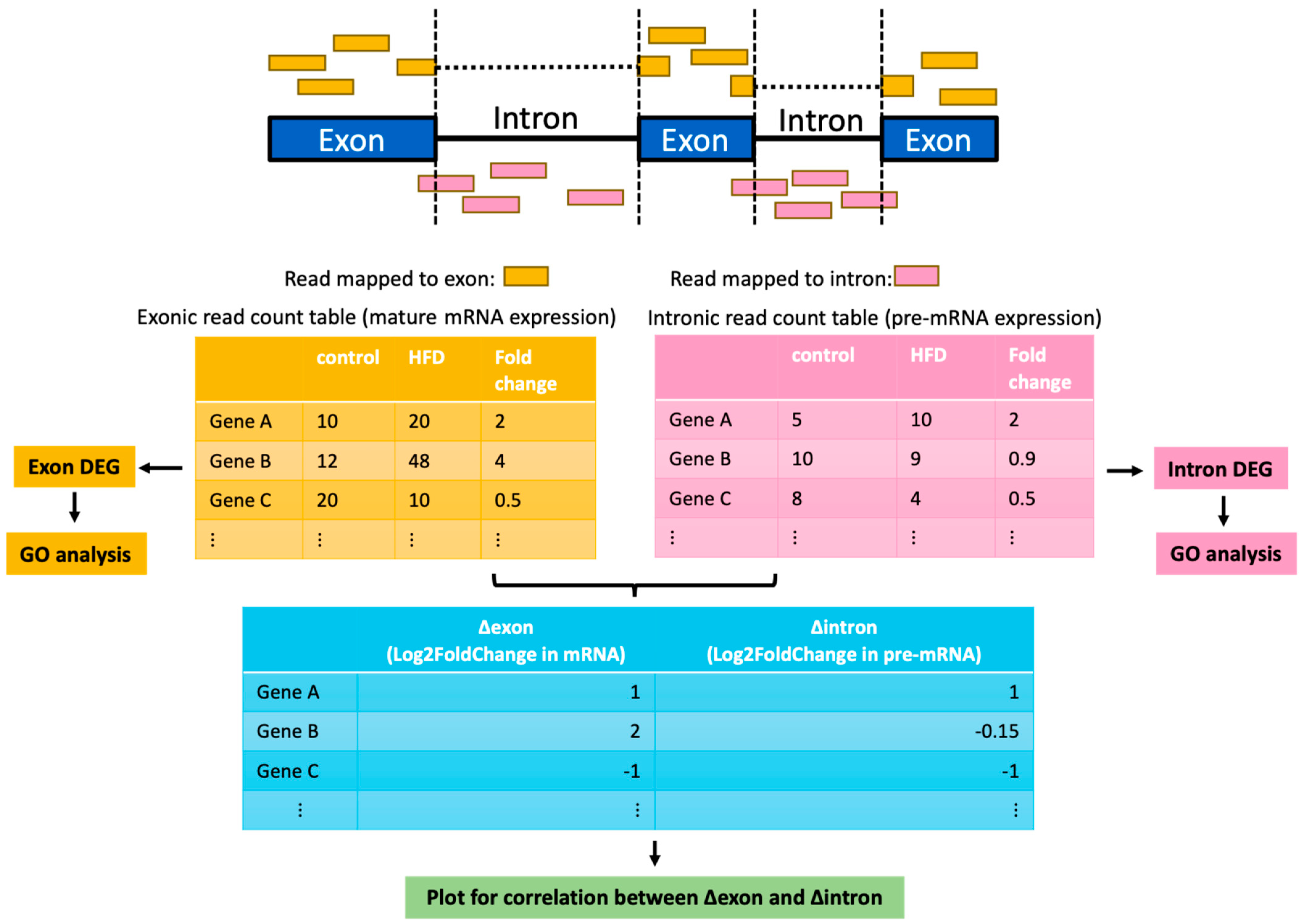
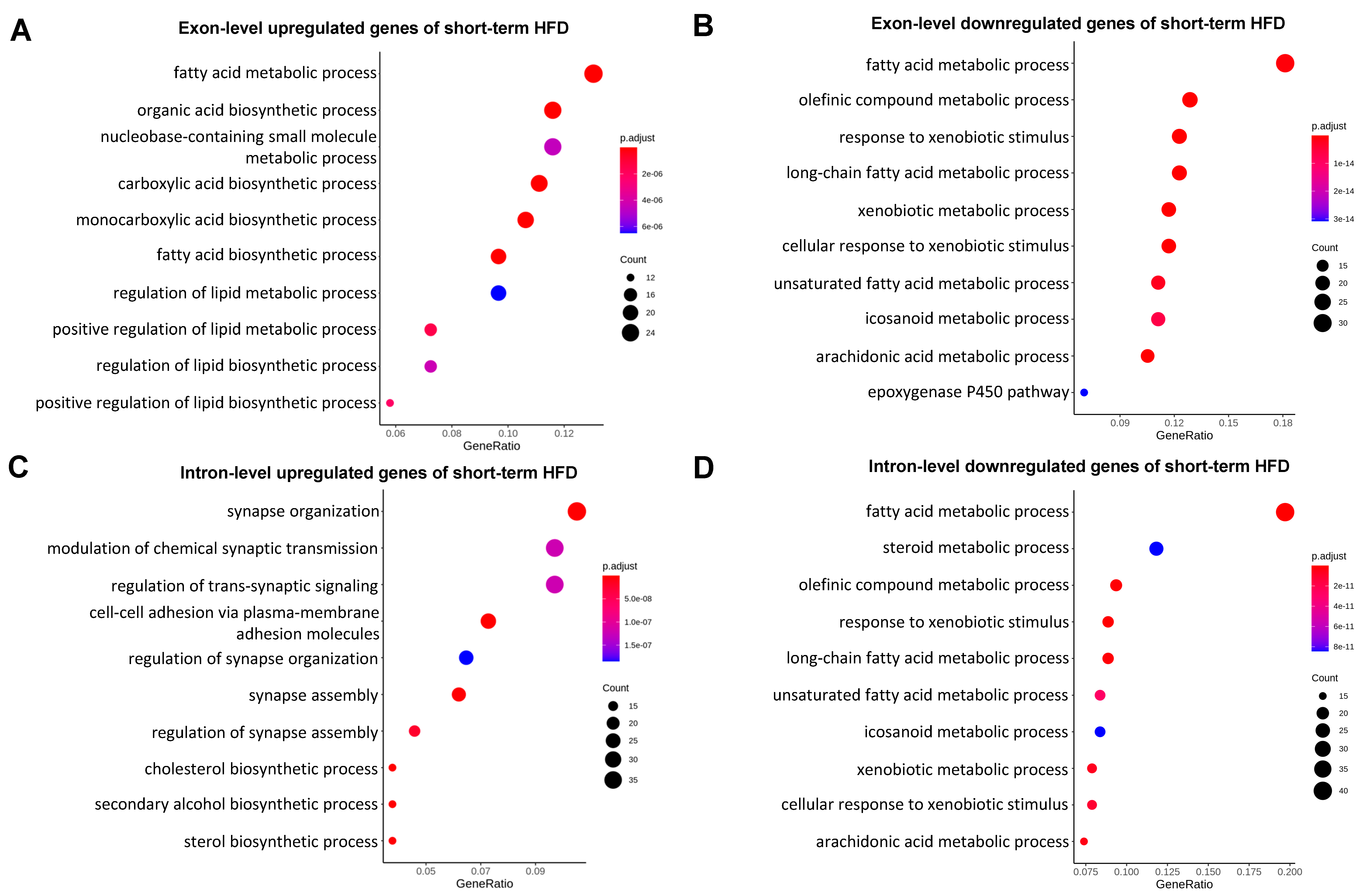
2.1. Broad Post-Transcriptional Gene Suppression at Early Stage of Fatty Liver
2.2. Genes Involved in Synapse Organization and Cell Adhesion Were Post-Transcriptionally Suppressed in Mild Fatty Liver
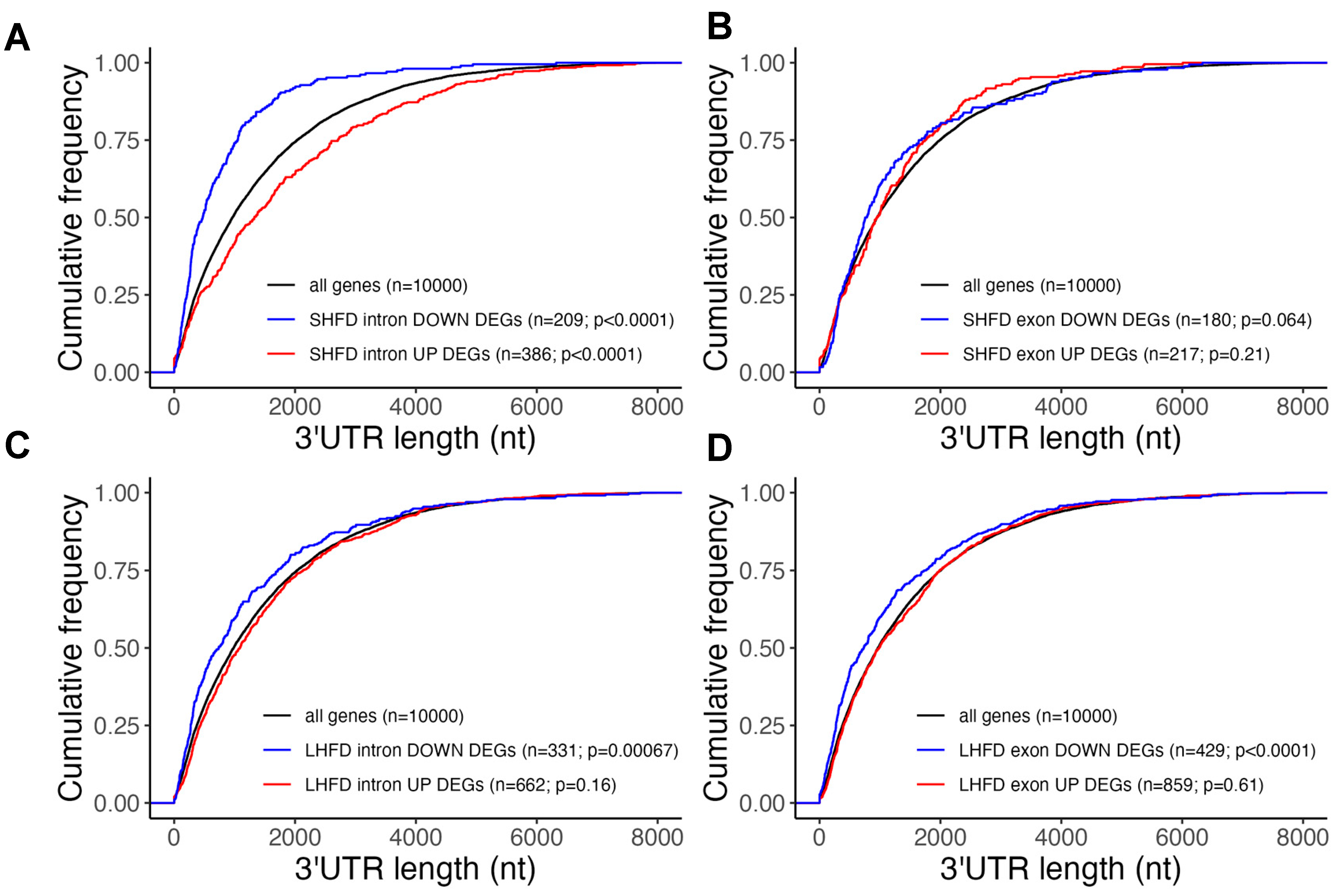
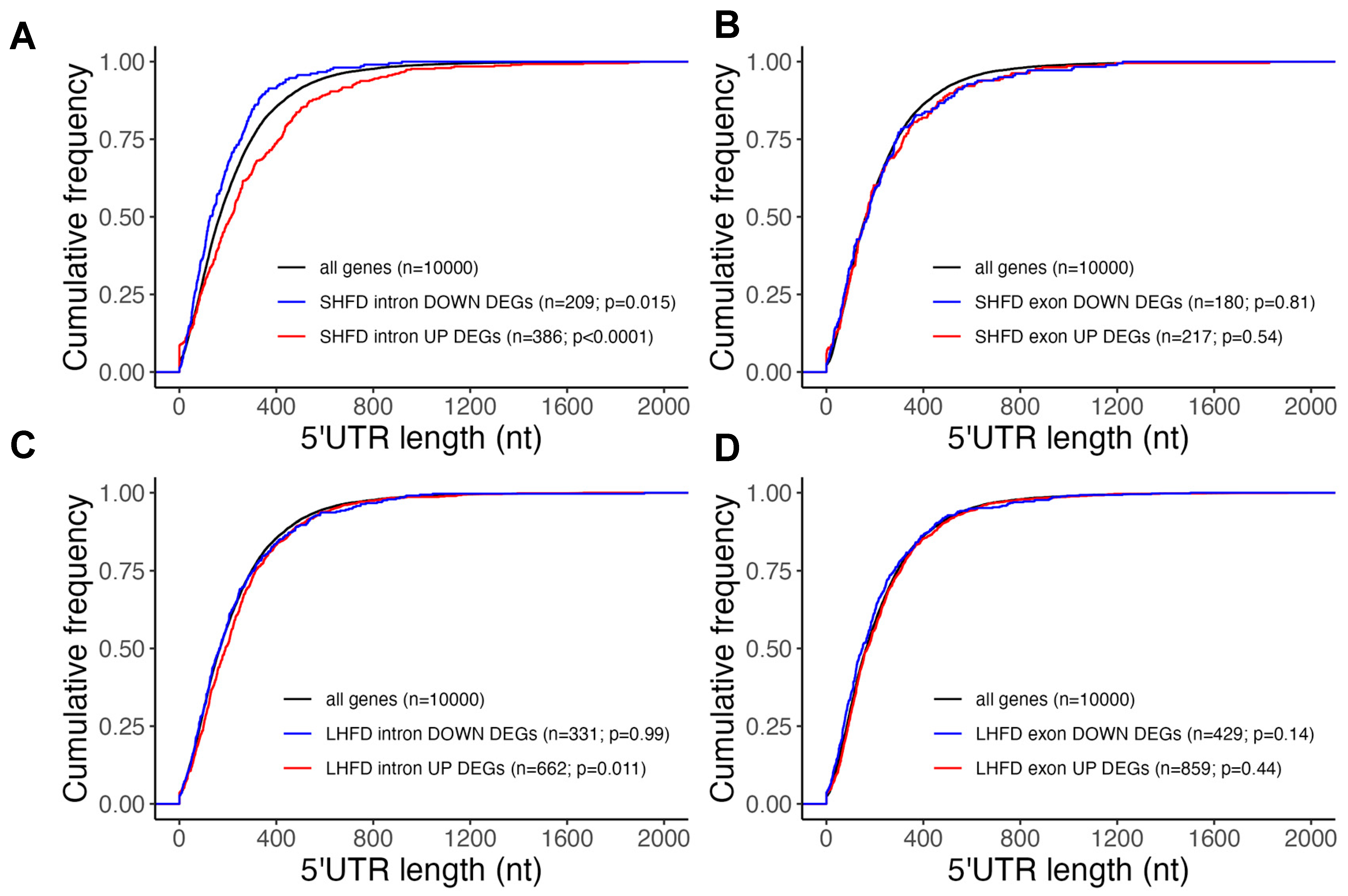
2.3. Post-Transcriptionally Suppressed Genes in Mild Fatty Liver Have Long UTRs
2.4. Post-Transcriptionally Suppressed Genes in Early Fatty Liver Are NMD Targets
3. Discussion
4. Materials and Methods
4.1. Reanalysis of Publicly Available RNA-Seq Data
4.2. UTR Feature Analysis
4.3. Mouse Study
4.4. RNA Extraction and qPCR
4.5. Protein Extraction and Western Blotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Hasty, A.H.; Osuga, J.; Tamura, Y.; Shionoiri, F.; Iizuka, Y.; Ohashi, K.; Harada, K.; et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J. Biol. Chem. 1999, 274, 35832–35839. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Corbett, A.H. Post-transcriptional regulation of gene expression and human disease. Curr. Opin. Cell Biol. 2018, 52, 96–104. [Google Scholar] [CrossRef]
- Takahashi, A.; Suzuki, T.; Soeda, S.; Takaoka, S.; Kobori, S.; Yamaguchi, T.; Mohamed, H.M.A.; Yanagiya, A.; Abe, T.; Shigeta, M.; et al. The CCR4-NOT complex maintains liver homeostasis through mRNA deadenylation. Life Sci. Alliance 2020, 3, e201900494. [Google Scholar] [CrossRef]
- Takaoka, S.; Yanagiya, A.; Mohamed, H.M.A.; Higa, R.; Abe, T.; Inoue, K.I.; Takahashi, A.; Stoney, P.; Yamamoto, T. Neuronal XRN1 is required for maintenance of whole-body metabolic homeostasis. iScience 2021, 24, 103151. [Google Scholar] [CrossRef]
- Zhang, Z.; Zong, C.; Jiang, M.; Hu, H.; Cheng, X.; Ni, J.; Yi, X.; Jiang, B.; Tian, F.; Chang, M.W.; et al. Hepatic HuR modulates lipid homeostasis in response to high-fat diet. Nat. Commun. 2020, 11, 3067. [Google Scholar] [CrossRef]
- Li, Y.; Xu, J.; Lu, Y.; Bian, H.; Yang, L.; Wu, H.; Zhang, X.; Zhang, B.; Xiong, M.; Chang, Y.; et al. DRAK2 aggravates nonalcoholic fatty liver disease progression through SRSF6-associated RNA alternative splicing. Cell Metab. 2021, 33, 2004–2020.e2009. [Google Scholar] [CrossRef]
- Arif, W.; Mathur, B.; Saikali, M.F.; Chembazhi, U.V.; Toohill, K.; Song, Y.J.; Hao, Q.; Karimi, S.; Blue, S.M.; Yee, B.A.; et al. Splicing factor SRSF1 deficiency in the liver triggers NASH-like pathology and cell death. Nat. Commun. 2023, 14, 551. [Google Scholar] [CrossRef]
- Kumar, D.; Das, M.; Sauceda, C.; Ellies, L.G.; Kuo, K.; Parwal, P.; Kaur, M.; Jih, L.; Bandyopadhyay, G.K.; Burton, D.; et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J. Clin. Investig. 2019, 129, 4477–4491. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Becskei, A. Impact of Methods on the Measurement of mRNA Turnover. Int. J. Mol. Sci. 2017, 18, 2723. [Google Scholar] [CrossRef] [PubMed]
- Faloona, G.R.; Stewart, B.N.; Fried, M. The effects of actinomycin D on the siosynthesis of plasma lipoproteins. Biochemistry 1968, 7, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Wai, H.; Douglas, A.G.L.; Baralle, D. RNA splicing analysis in genomic medicine. Int. J. Biochem. Cell Biol. 2019, 108, 61–71. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, S.; Sheng, Q.; Guo, M.; Lehmann, B.; Pietenpol, J.; Samuels, D.C.; Shyr, Y. RNAseq by Total RNA Library Identifies Additional RNAs Compared to Poly(A) RNA Library. BioMed Res. Int. 2015, 2015, 862130. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, A.Y.; Su, S.; Ng, A.P.; Holik, A.Z.; Asselin-Labat, M.L.; Ritchie, M.E.; Law, C.W. Covering all your bases: Incorporating intron signal from RNA-seq data. NAR Genom. Bioinform. 2020, 2, lqaa073. [Google Scholar] [CrossRef]
- Gaidatzis, D.; Burger, L.; Florescu, M.; Stadler, M.B. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat. Biotechnol. 2015, 33, 722–729. [Google Scholar] [CrossRef]
- Brummer, A.; Bergmann, S. Disentangling genetic effects on transcriptional and post-transcriptional gene regulation through integrating exon and intron expression QTLs. Nat. Commun. 2024, 15, 3786. [Google Scholar] [CrossRef]
- Manaig, Y.J.Y.; Marmol-Sanchez, E.; Castello, A.; Esteve-Codina, A.; Sandrini, S.; Savoini, G.; Agazzi, A.; Sanchez, A.; Folch, J.M. Exon-intron split analysis reveals posttranscriptional regulatory signals induced by high and low n-6/n-3 polyunsaturated fatty acid ratio diets in piglets. J. Anim. Sci. 2023, 101, skad271. [Google Scholar] [CrossRef]
- Siersbaek, M.; Varticovski, L.; Yang, S.; Baek, S.; Nielsen, R.; Mandrup, S.; Hager, G.L.; Chung, J.H.; Grontved, L. High fat diet-induced changes of mouse hepatic transcription and enhancer activity can be reversed by subsequent weight loss. Sci. Rep. 2017, 7, 40220. [Google Scholar] [CrossRef]
- Pradas-Juni, M.; Hansmeier, N.R.; Link, J.C.; Schmidt, E.; Larsen, B.D.; Klemm, P.; Meola, N.; Topel, H.; Loureiro, R.; Dhaouadi, I.; et al. A MAFG-lncRNA axis links systemic nutrient abundance to hepatic glucose metabolism. Nat. Commun. 2020, 11, 644. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xiong, C.; Jia, K.; Jin, J.; Li, Z.; Huang, Y.; Liu, Y.; Wang, L.; Luo, H.; Li, H.; et al. Hepatic transcriptome analysis from HFD-fed mice defines a long noncoding RNA regulating cellular cholesterol levels. J. Lipid Res. 2019, 60, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated regions of mRNAs. Genome Biol. 2002, 3, REVIEWS0004. [Google Scholar] [CrossRef]
- Hug, N.; Longman, D.; Caceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Peccarelli, M.; Kebaara, B.W. Regulation of natural mRNAs by the nonsense-mediated mRNA decay pathway. Eukaryot. Cell 2014, 13, 1126–1135. [Google Scholar] [CrossRef]
- Karousis, E.D.; Nasif, S.; Muhlemann, O. Nonsense-mediated mRNA decay: Novel mechanistic insights and biological impact. Wiley Interdiscip. Rev. RNA 2016, 7, 661–682. [Google Scholar] [CrossRef]
- Thoren, L.A.; Norgaard, G.A.; Weischenfeldt, J.; Waage, J.; Jakobsen, J.S.; Damgaard, I.; Bergstrom, F.C.; Blom, A.M.; Borup, R.; Bisgaard, H.C.; et al. UPF2 is a critical regulator of liver development, function and regeneration. PLoS ONE 2010, 5, e11650. [Google Scholar] [CrossRef]
- Lopez-Perrote, A.; Castano, R.; Melero, R.; Zamarro, T.; Kurosawa, H.; Ohnishi, T.; Uchiyama, A.; Aoyagi, K.; Buchwald, G.; Kataoka, N.; et al. Human nonsense-mediated mRNA decay factor UPF2 interacts directly with eRF3 and the SURF complex. Nucleic Acids Res. 2016, 44, 1909–1923. [Google Scholar] [CrossRef] [PubMed]
- Kiourtis, C.; Wilczynska, A.; Nixon, C.; Clark, W.; May, S.; Bird, T.G. Specificity and off-target effects of AAV8-TBG viral vectors for the manipulation of hepatocellular gene expression in mice. Biol. Open 2021, 10, bio058678. [Google Scholar] [CrossRef]
- Isken, O.; Kim, Y.K.; Hosoda, N.; Mayeur, G.L.; Hershey, J.W.; Maquat, L.E. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 2008, 133, 314–327. [Google Scholar] [CrossRef]
- Hansen, H.H.; HM, A.E.; Oro, D.; Evers, S.S.; Heeboll, S.; Eriksen, P.L.; Thomsen, K.L.; Bengtsson, A.; Veidal, S.S.; Feigh, M.; et al. Human translatability of the GAN diet-induced obese mouse model of non-alcoholic steatohepatitis. BMC Gastroenterol. 2020, 20, 210. [Google Scholar] [CrossRef] [PubMed]
- Basha, S.; Jin-Smith, B.; Sun, C.; Pi, L. The SLIT/ROBO Pathway in Liver Fibrosis and Cancer. Biomolecules 2023, 13, 785. [Google Scholar] [CrossRef]
- Xiao, Y.; Batmanov, K.; Hu, W.; Zhu, K.; Tom, A.Y.; Guan, D.; Jiang, C.; Cheng, L.; McCright, S.J.; Yang, E.C.; et al. Hepatocytes demarcated by EphB2 contribute to the progression of nonalcoholic steatohepatitis. Sci. Transl. Med. 2023, 15, eadc9653. [Google Scholar] [CrossRef] [PubMed]
- Paigen, B. Genetics of responsiveness to high-fat and high-cholesterol diets in the mouse. Am. J. Clin. Nutr. 1995, 62, 458S–462S. [Google Scholar] [CrossRef]
- Li, Z.; Vuong, J.K.; Zhang, M.; Stork, C.; Zheng, S. Inhibition of nonsense-mediated RNA decay by ER stress. RNA 2017, 23, 378–394. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yamashita, A.; Fujimura, M. Environmental stresses suppress nonsense-mediated mRNA decay (NMD) and affect cells by stabilizing NMD-targeted gene expression. Sci. Rep. 2019, 9, 1279. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Weischenfeldt, J.; Damgaard, I.; Bryder, D.; Theilgaard-Monch, K.; Thoren, L.A.; Nielsen, F.C.; Jacobsen, S.E.; Nerlov, C.; Porse, B.T. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 2008, 22, 1381–1396. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence |
|---|---|
| 18S-f | AGTCCCTGCCCTTTGTACACA |
| 18S-r | CGATCCGAGGGCCTCACTA |
| Ephb2-f | AGGCAAGCAACAAGGAAAGG |
| Ephb2-r | GCAGGGCCCAGAGTATATGT |
| Gabrb3-f | ATAAACCGGGTGGATGCTCA |
| Gabrb3-r | TGGGCATGCTCTGTTTCCTA |
| Gpc6-f | AAGCCAGATACCTGCCTGAG |
| Gpc6-r | TGTTGGTCATCACACGGAGA |
| Robo1-f | GCTGCCAAGCGGGTCTTTAT |
| Robo1-r | CTCCGAGGTAATTCCTAGCCA |
| Dlg2-f | GTGAGATTTGTAGCAGAAAGGGG |
| Dlg2-r | GGCGACTTGTAACCGCTTAATAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takaoka, S.; Jaso-Vera, M.E.; Ruan, X. Disrupted Post-Transcriptional Regulation of Gene Expression as a Hallmark of Fatty Liver Progression. Int. J. Mol. Sci. 2024, 25, 11054. https://doi.org/10.3390/ijms252011054
Takaoka S, Jaso-Vera ME, Ruan X. Disrupted Post-Transcriptional Regulation of Gene Expression as a Hallmark of Fatty Liver Progression. International Journal of Molecular Sciences. 2024; 25(20):11054. https://doi.org/10.3390/ijms252011054
Chicago/Turabian StyleTakaoka, Shohei, Marcos E. Jaso-Vera, and Xiangbo Ruan. 2024. "Disrupted Post-Transcriptional Regulation of Gene Expression as a Hallmark of Fatty Liver Progression" International Journal of Molecular Sciences 25, no. 20: 11054. https://doi.org/10.3390/ijms252011054
APA StyleTakaoka, S., Jaso-Vera, M. E., & Ruan, X. (2024). Disrupted Post-Transcriptional Regulation of Gene Expression as a Hallmark of Fatty Liver Progression. International Journal of Molecular Sciences, 25(20), 11054. https://doi.org/10.3390/ijms252011054