
The Inhibition of Vessel Co-Option as an Emerging Strategy for Cancer Therapy
 , , , ,
, , , ,
Abstract
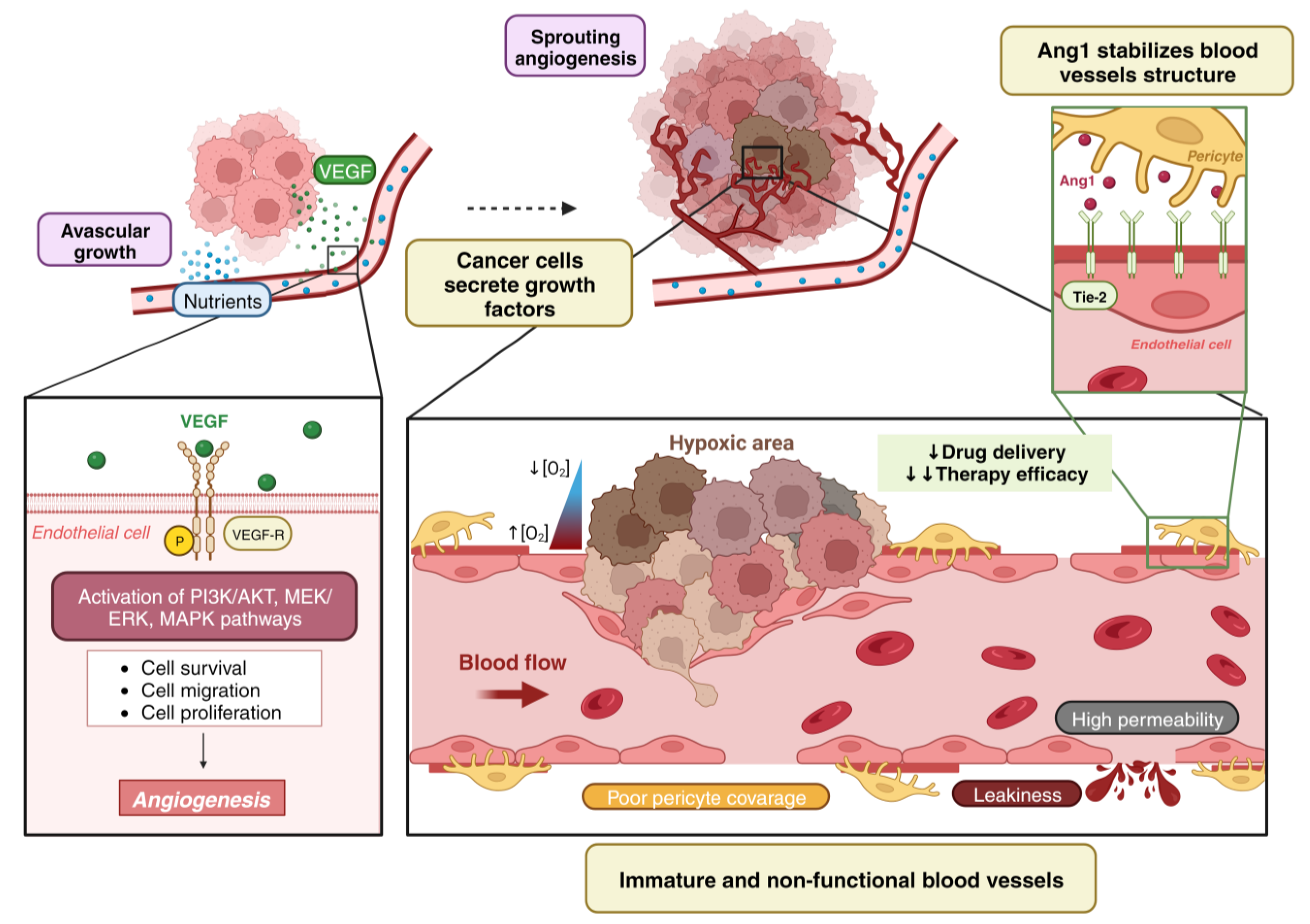
1. Tumor Vascularization
2. Angiogenesis vs. Vessel Co-Option
- (i)
- (ii)
- Tumors can modify their growth pattern depending on their stage or the different features of the neoplasia, the origin of the primary tumor and the location of the pre-metastatic niche [19].
- (iii)
- (iv)
- Transdifferentiation from the angiogenic to non-angiogenic phenotype has been described as a response to therapeutic selective pressure, being proposed as a major resistance to anti-angiogenic therapy (AAT) mechanism both intrinsic, from the beginning of treatment, and acquired, observed after an initial response and being able to return to the angiogenic process when therapy is interrupted [14,17].
- (v)
- VCO has been related with the metastatic cascade (mobility, cell adhesion and invasiveness), plasticity, latency and tumor cell survival, being associated with epithelial-to-mesenchymal (EMT) transition and paracrine interaction between cancer cells and pericytes or tumor cells from co-opted vessels [21,22].
3. VCO as a Resistance Mechanism to Anti-Angiogenic Therapy
3.1. Histological Features of VCO vs. Angiogenic Tumors
3.2. VCO Induced Resistance in Lung Cancer and Lung Metastases
3.3. VCO Induced Resistance in Brain Tumors and Brain Metastases
3.4. VCO Induced Resistance in Hepatocarcinoma and Liver Metastases
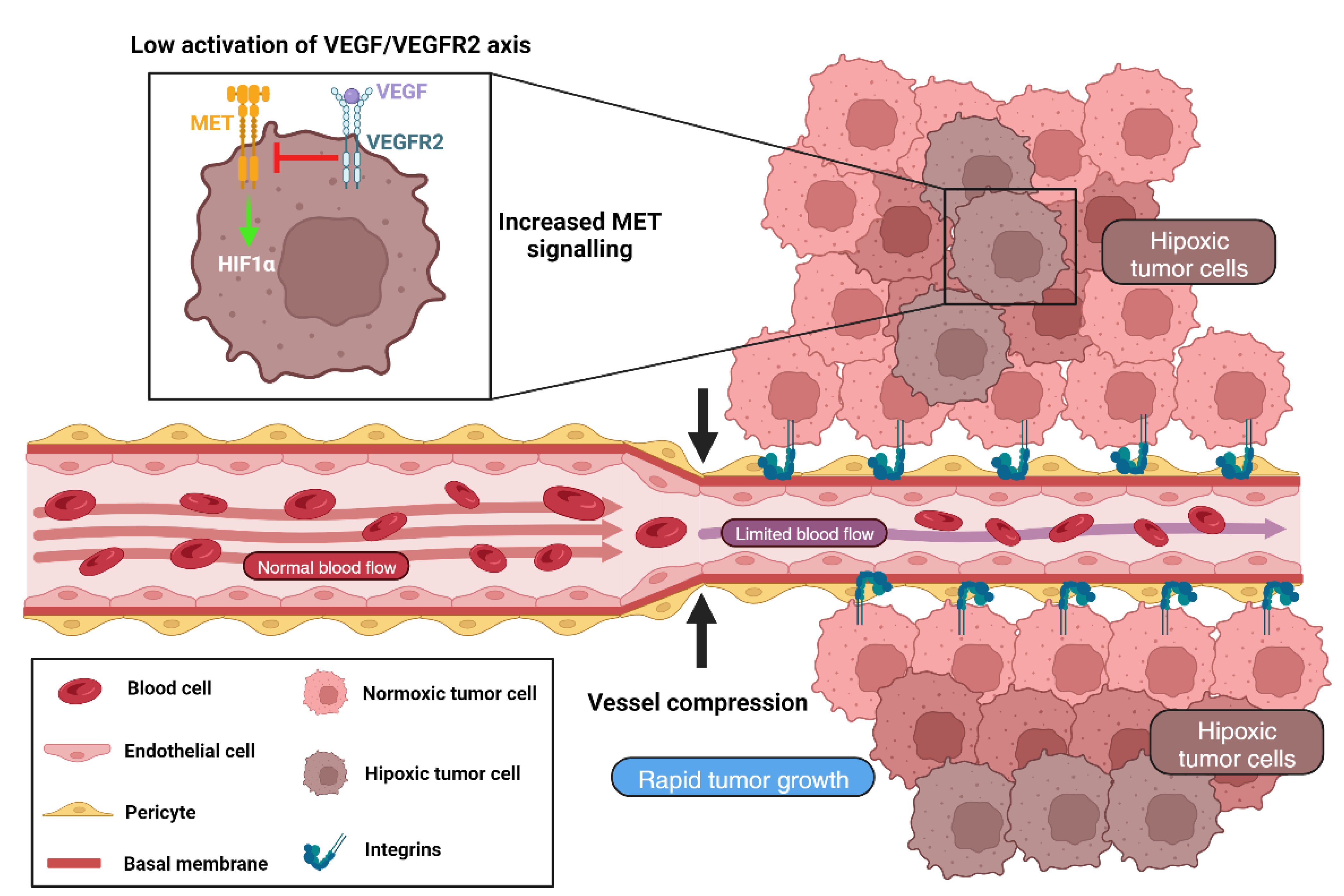
4. Cellular and Molecular Mechanisms Involved in VCO
4.1. Different Blood Vessels
Targeting Tumor Vasculature to Inhibit VCO
4.2. Cancer Cell Adhesion
Targeting Cancer Cell Adhesion to Inhibit VCO
4.3. Cancer Cell Motility
Inhibition of Cancer Cell Migration for VCO Inhibition
4.4. Epithelial-to-Mesenchymal Transition (EMT)
Inhibition of EMT as an Strategy for Vessel Co-Option Inhibition
4.5. Extracellular Matrix and VCO
Inhibition of Extracellular Matrix as Strategy for VCO Inhibition
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Z.L.; Chen, H.H.; Zheng, L.L.; Sun, L.P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Donnem, T.; Reynolds, A.R.; Kuczynski, E.A.; Gatter, K.; Vermeulen, P.B.; Kerbel, R.S.; Harris, A.L.; Pezzella, F. Non-angiogenic tumours and their influence on cancer biology. Nat. Rev. Cancer 2018, 18, 323–336. [Google Scholar] [CrossRef]
- Park-Windhol, C.; Ng, Y.S.; Yang, J.; Primo, V.; Saint-Geniez, M.; D’Amore, P.A. Endomucin inhibits VEGF-induced endothelial cell migration, growth, and morphogenesis by modulating VEGFR2 signaling. Sci. Rep. 2017, 7, 17138. [Google Scholar] [CrossRef] [PubMed]
- Baeriswyl, V.; Christofori, G. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef]
- Badodekar, N.; Sharma, A.; Patil, V.; Telang, G.; Sharma, R.; Patil, S.; Vyas, N.; Somasundaram, I. Angiogenesis induction in breast cancer: A paracrine paradigm. Cell Biochem. Funct. 2021, 39, 860–873. [Google Scholar] [CrossRef]
- Newport, E.L.; Pedrosa, A.R.; Njegic, A.; Hodivala-Dilke, K.M.; Munoz-Felix, J.M. Improved Immunotherapy Efficacy by Vascular Modulation. Cancers 2021, 13, 5207. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. alphaV integrins in angiogenesis and cancer. Cold Spring Harb. Perspect. Med. 2011, 1, a006478. [Google Scholar] [CrossRef] [PubMed]
- Ollauri-Ibanez, C.; Ayuso-Inigo, B.; Pericacho, M. Hot and Cold Tumors: Is Endoglin (CD105) a Potential Target for Vessel Normalization? Cancers 2021, 13, 1552. [Google Scholar] [CrossRef]
- D’Amico, G.; Munoz-Felix, J.M.; Pedrosa, A.R.; Hodivala-Dilke, K.M. “Splitting the matrix”: Intussusceptive angiogenesis meets MT1-MMP. EMBO Mol. Med. 2020, 12, e11663. [Google Scholar] [CrossRef]
- Gavard, J.; Patel, V.; Gutkind, J.S. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev. Cell 2008, 14, 25–36. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2017, 31, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Ayuso-Inigo, B.; Mendez-Garcia, L.; Pericacho, M.; Munoz-Felix, J.M. The Dual Effect of the BMP9-ALK1 Pathway in Blood Vessels: An Opportunity for Cancer Therapy Improvement? Cancers 2021, 13, 5412. [Google Scholar] [CrossRef] [PubMed]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Reynolds, A.R. Vessel co-option and resistance to anti-angiogenic therapy. Angiogenesis 2020, 23, 55–74. [Google Scholar] [CrossRef]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef]
- Bridgeman, V.L.; Wan, E.; Foo, S.; Nathan, M.R.; Welti, J.C.; Frentzas, S.; Vermeulen, P.B.; Preece, N.; Springer, C.J.; Powles, T.; et al. Preclinical Evidence That Trametinib Enhances the Response to Antiangiogenic Tyrosine Kinase Inhibitors in Renal Cell Carcinoma. Mol. Cancer Ther. 2016, 15, 172–183. [Google Scholar] [CrossRef]
- Jubb, A.M.; Cesario, A.; Ferguson, M.; Congedo, M.T.; Gatter, K.C.; Lococo, F.; Mule, A.; Pezzella, F. Vascular phenotypes in primary non-small cell lung carcinomas and matched brain metastases. Br. J. Cancer 2011, 104, 1877–1881. [Google Scholar] [CrossRef]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Kerbel, R.S. Implications of vessel co-option in sorafenib-resistant hepatocellular carcinoma. Chin. J. Cancer 2016, 35, 97. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kuczynski, E.A.; Yin, M.; Bar-Zion, A.; Lee, C.R.; Butz, H.; Man, S.; Daley, F.; Vermeulen, P.B.; Yousef, G.M.; Foster, F.S.; et al. Co-option of Liver Vessels and Not Sprouting Angiogenesis Drives Acquired Sorafenib Resistance in Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2016, 108, djw030. [Google Scholar] [CrossRef]
- Pezzella, F.; Pastorino, U.; Tagliabue, E.; Andreola, S.; Sozzi, G.; Gasparini, G.; Menard, S.; Gatter, K.C.; Harris, A.L.; Fox, S.; et al. Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am. J. Pathol. 1997, 151, 1417–1423. [Google Scholar] [PubMed]
- Cuypers, A.; Truong, A.K.; Becker, L.M.; Saavedra-Garcia, P.; Carmeliet, P. Tumor vessel co-option: The past & the future. Front. Oncol. 2022, 12, 965277. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010, 12, 233–242. [Google Scholar] [CrossRef]
- Jeong, H.S.; Jones, D.; Liao, S.; Wattson, D.A.; Cui, C.H.; Duda, D.G.; Willett, C.G.; Jain, R.K.; Padera, T.P. Investigation of the Lack of Angiogenesis in the Formation of Lymph Node Metastases. J. Natl. Cancer Inst. 2015, 107, djv155. [Google Scholar] [CrossRef]
- Leenders, W.P.; Kusters, B.; Verrijp, K.; Maass, C.; Wesseling, P.; Heerschap, A.; Ruiter, D.; Ryan, A.; de Waal, R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin. Cancer Res. 2004, 10, 6222–6230. [Google Scholar] [CrossRef]
- Rada, M.; Hassan, N.; Lazaris, A.; Metrakos, P. The molecular mechanisms underlying neutrophil infiltration in vessel co-opting colorectal cancer liver metastases. Front. Oncol. 2022, 12, 1004793. [Google Scholar] [CrossRef]
- Rada, M.; Lazaris, A.; Kapelanski-Lamoureux, A.; Mayer, T.Z.; Metrakos, P. Tumor microenvironment conditions that favor vessel co-option in colorectal cancer liver metastases: A theoretical model. Semin. Cancer Biol. 2021, 71, 52–64. [Google Scholar] [CrossRef]
- Latacz, E.; Caspani, E.; Barnhill, R.; Lugassy, C.; Verhoef, C.; Grunhagen, D.; Van Laere, S.; Fernandez Moro, C.; Gerling, M.; Dirix, M.; et al. Pathological features of vessel co-option versus sprouting angiogenesis. Angiogenesis 2020, 23, 43–54. [Google Scholar] [CrossRef]
- Sardari Nia, P.; Hendriks, J.; Friedel, G.; Van Schil, P.; Van Marck, E. Distinct angiogenic and non-angiogenic growth patterns of lung metastases from renal cell carcinoma. Histopathology 2007, 51, 354–361. [Google Scholar] [CrossRef]
- Kuhn, E.; Morbini, P.; Cancellieri, A.; Damiani, S.; Cavazza, A.; Comin, C.E. Adenocarcinoma classification: Patterns and prognosis. Pathologica 2018, 110, 5–11. [Google Scholar]
- Suzuki, S.; Aokage, K.; Hishida, T.; Yoshida, J.; Kuwata, T.; Yamauchi, C.; Tsuboi, M.; Ishii, G. Interstitial growth as an aggressive growth pattern in primary lung cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Passalidou, E.; Trivella, M.; Singh, N.; Ferguson, M.; Hu, J.; Cesario, A.; Granone, P.; Nicholson, A.G.; Goldstraw, P.; Ratcliffe, C.; et al. Vascular phenotype in angiogenic and non-angiogenic lung non-small cell carcinomas. Br. J. Cancer 2002, 86, 244–249. [Google Scholar] [CrossRef]
- Sardari Nia, P.; Colpaert, C.; Vermeulen, P.; Weyler, J.; Pezzella, F.; Van Schil, P.; Van Marck, E. Different growth patterns of non-small cell lung cancer represent distinct biologic subtypes. Ann. Thorac. Surg. 2008, 85, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Szabo, V.; Bugyik, E.; Dezso, K.; Ecker, N.; Nagy, P.; Timar, J.; Tovari, J.; Laszlo, V.; Bridgeman, V.L.; Wan, E.; et al. Mechanism of tumour vascularization in experimental lung metastases. J. Pathol. 2015, 235, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, E.E.; Andersen, S.; Rakaee, M.; Pedersen, M.I.; Lombardi, A.P.; Pohl, M.; Kilvaer, T.; Busund, L.T.; Pezzella, F.; Donnem, T. Impact of microvessel patterns and immune status in NSCLC: A non-angiogenic vasculature is an independent negative prognostic factor in lung adenocarcinoma. Front. Oncol. 2023, 13, 1157461. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.A.; De Rooij, L.; Cuypers, A.; Rohlenova, K.; Dumas, S.J.; Garcia-Caballero, M.; Meta, E.; Amersfoort, J.; Taverna, F.; Becker, L.M.; et al. Tumor vessel co-option probed by single-cell analysis. Cell Rep. 2021, 35, 109253. [Google Scholar] [CrossRef]
- Carbonell, W.S.; Ansorge, O.; Sibson, N.; Muschel, R. The vascular basement membrane as “soil” in brain metastasis. PLoS ONE 2009, 4, e5857. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Rajky, O.; Winkler, F.; Bartsch, R.; Furtner, J.; Hainfellner, J.A.; Goodman, S.L.; Weller, M.; Schittenhelm, J.; Preusser, M. Invasion patterns in brain metastases of solid cancers. Neuro Oncol. 2013, 15, 1664–1672. [Google Scholar] [CrossRef]
- Garcia-Gomez, P.; Valiente, M. Vascular co-option in brain metastasis. Angiogenesis 2020, 23, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Sontheimer, H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef]
- Yadav, V.N.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; DeCarvalho, A.C.; Mikkelsen, T.; Castro, M.G.; Lowenstein, P.R. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016, 7, 83701–83719. [Google Scholar] [CrossRef]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.C.; et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 2018, 33, 874–889.e7. [Google Scholar] [CrossRef]
- Lindberg, O.R.; McKinney, A.; Engler, J.R.; Koshkakaryan, G.; Gong, H.; Robinson, A.E.; Ewald, A.J.; Huillard, E.; David James, C.; Molinaro, A.M.; et al. GBM heterogeneity as a function of variable epidermal growth factor receptor variant III activity. Oncotarget 2016, 7, 79101–79116. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krusche, B.; Ottone, C.; Clements, M.P.; Johnstone, E.R.; Goetsch, K.; Lieven, H.; Mota, S.G.; Singh, P.; Khadayate, S.; Ashraf, A.; et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. Elife 2016, 5, e14845. [Google Scholar] [CrossRef]
- Bugyik, E.; Dezso, K.; Reiniger, L.; Laszlo, V.; Tovari, J.; Timar, J.; Nagy, P.; Klepetko, W.; Dome, B.; Paku, S. Lack of angiogenesis in experimental brain metastases. J. Neuropathol. Exp. Neurol. 2011, 70, 979–991. [Google Scholar] [CrossRef]
- Carbonell, W.S.; DeLay, M.; Jahangiri, A.; Park, C.C.; Aghi, M.K. beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013, 73, 3145–3154. [Google Scholar] [CrossRef]
- di Tomaso, E.; Snuderl, M.; Kamoun, W.S.; Duda, D.G.; Auluck, P.K.; Fazlollahi, L.; Andronesi, O.C.; Frosch, M.P.; Wen, P.Y.; Plotkin, S.R.; et al. Glioblastoma recurrence after cediranib therapy in patients: Lack of “rebound” revascularization as mode of escape. Cancer Res. 2011, 71, 19–28. [Google Scholar] [CrossRef]
- Voutouri, C.; Kirkpatrick, N.D.; Chung, E.; Mpekris, F.; Baish, J.W.; Munn, L.L.; Fukumura, D.; Stylianopoulos, T.; Jain, R.K. Experimental and computational analyses reveal dynamics of tumor vessel cooption and optimal treatment strategies. Proc. Natl. Acad. Sci. USA 2019, 116, 2662–2671. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Kim, J.; Ozawa, T.; Zhang, M.; Westphal, M.; Deen, D.F.; Shuman, M.A. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2000, 2, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef]
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.J.; Yadav, V.N.; Motsch, S.; Koschmann, C.; Calinescu, A.A.; Mineharu, Y.; Camelo-Piragua, S.I.; Orringer, D.; Bannykh, S.; Nichols, W.S.; et al. Mechanisms of glioma formation: Iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 2014, 16, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Young, G.S.; Setayesh, K.; Muzikansky, A.; Klufas, R.; Ross, G.L.; Ciampa, A.S.; Ebbeling, L.G.; Levy, B.; Drappatz, J.; et al. Bevacizumab for recurrent malignant gliomas: Efficacy, toxicity, and patterns of recurrence. Neurology 2008, 70, 779–787. [Google Scholar] [CrossRef]
- Fischer, I.; Cunliffe, C.H.; Bollo, R.J.; Raza, S.; Monoky, D.; Chiriboga, L.; Parker, E.C.; Golfinos, J.G.; Kelly, P.J.; Knopp, E.A.; et al. High-grade glioma before and after treatment with radiation and Avastin: Initial observations. Neuro Oncol. 2008, 10, 700–708. [Google Scholar] [CrossRef]
- Narayana, A.; Kelly, P.; Golfinos, J.; Parker, E.; Johnson, G.; Knopp, E.; Zagzag, D.; Fischer, I.; Raza, S.; Medabalmi, P.; et al. Antiangiogenic therapy using bevacizumab in recurrent high-grade glioma: Impact on local control and patient survival. J. Neurosurg. 2009, 110, 173–180. [Google Scholar] [CrossRef]
- Schulte, J.D.; Aghi, M.K.; Taylor, J.W. Anti-angiogenic therapies in the management of glioblastoma. Chin. Clin. Oncol. 2021, 10, 37. [Google Scholar] [CrossRef]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y.; et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Pedraza, M.; Fernandez, M. Interplay Between Macrophages and Angiogenesis: A Double-Edged Sword in Liver Disease. Front. Immunol. 2019, 10, 2882. [Google Scholar] [CrossRef]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A pathogenic crosstalk between tumor cells and pericytes. PLoS ONE 2014, 9, e101402. [Google Scholar] [CrossRef]
- Morath, I.; Hartmann, T.N.; Orian-Rousseau, V. CD44: More than a mere stem cell marker. Int. J. Biochem. Cell Biol. 2016, 81, 166–173. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Dudley, A.C. Models and molecular mechanisms of blood vessel co-option by cancer cells. Angiogenesis 2020, 23, 17–25. [Google Scholar] [CrossRef] [PubMed]
- van Dam, P.J.; van der Stok, E.P.; Teuwen, L.A.; Van den Eynden, G.G.; Illemann, M.; Frentzas, S.; Majeed, A.W.; Eefsen, R.L.; Coebergh van den Braak, R.R.J.; Lazaris, A.; et al. International consensus guidelines for scoring the histopathological growth patterns of liver metastasis. Br. J. Cancer 2017, 117, 1427–1441. [Google Scholar] [CrossRef]
- Meyer, Y.; Bohlok, A.; Hoppener, D.; Galjart, B.; Doukas, M.; Grunhagen, D.J.; Labar, A.; Lucidi, V.; Vermeulen, P.B.; Verhoef, C.; et al. Histopathological growth patterns of resected non-colorectal, non-neuroendocrine liver metastases: A retrospective multicenter study. Clin. Exp. Metastasis 2022, 39, 433–442. [Google Scholar] [CrossRef]
- Latacz, E.; Hoppener, D.; Bohlok, A.; Leduc, S.; Tabaries, S.; Fernandez Moro, C.; Lugassy, C.; Nystrom, H.; Bozoky, B.; Floris, G.; et al. Histopathological growth patterns of liver metastasis: Updated consensus guidelines for pattern scoring, perspectives and recent mechanistic insights. Br. J. Cancer 2022, 127, 988–1013. [Google Scholar] [CrossRef]
- Fleischer, J.R.; Schmitt, A.M.; Haas, G.; Xu, X.; Zeisberg, E.M.; Bohnenberger, H.; Kuffer, S.; Teuwen, L.A.; Karras, P.J.; Beissbarth, T.; et al. Molecular differences of angiogenic versus vessel co-opting colorectal cancer liver metastases at single-cell resolution. Mol. Cancer 2023, 22, 17. [Google Scholar] [CrossRef] [PubMed]
- Zaharia, C.; Veen, T.; Lea, D.; Kanani, A.; Alexeeva, M.; Soreide, K. Histopathological Growth Pattern in Colorectal Liver Metastasis and The Tumor Immune Microenvironment. Cancers 2022, 15, 181. [Google Scholar] [CrossRef]
- Haas, G.; Fan, S.; Ghadimi, M.; De Oliveira, T.; Conradi, L.C. Different Forms of Tumor Vascularization and Their Clinical Implications Focusing on Vessel Co-option in Colorectal Cancer Liver Metastases. Front. Cell Dev. Biol. 2021, 9, 612774. [Google Scholar] [CrossRef]
- Lazaris, A.; Amri, A.; Petrillo, S.K.; Zoroquiain, P.; Ibrahim, N.; Salman, A.; Gao, Z.H.; Vermeulen, P.B.; Metrakos, P. Vascularization of colorectal carcinoma liver metastasis: Insight into stratification of patients for anti-angiogenic therapies. J. Pathol. Clin. Res. 2018, 4, 184–192. [Google Scholar] [CrossRef]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Anti-angiogenesis: New concept for therapy of solid tumors. Ann. Surg. 1972, 175, 409–416. [Google Scholar] [CrossRef]
- Wong, P.P.; Bodrug, N.; Hodivala-Dilke, K.M. Exploring Novel Methods for Modulating Tumor Blood Vessels in Cancer Treatment. Curr. Biol. 2016, 26, R1161–R1166. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.B.; Bergers, G. CANCER. Tumor angiogenesis, from foe to friend. Science 2015, 349, 694–695. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Huang, M.; Lin, Y.; Wang, C.; Deng, L.; Chen, M.; Assaraf, Y.G.; Chen, Z.S.; Ye, W.; Zhang, D. New insights into antiangiogenic therapy resistance in cancer: Mechanisms and therapeutic aspects. Drug Resist. Updat. 2022, 64, 100849. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Wong, P.P.; Demircioglu, F.; Ghazaly, E.; Alrawashdeh, W.; Stratford, M.R.; Scudamore, C.L.; Cereser, B.; Crnogorac-Jurcevic, T.; McDonald, S.; Elia, G.; et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell 2015, 27, 123–137. [Google Scholar] [CrossRef]
- Tavora, B.; Reynolds, L.E.; Batista, S.; Demircioglu, F.; Fernandez, I.; Lechertier, T.; Lees, D.M.; Wong, P.P.; Alexopoulou, A.; Elia, G.; et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 2014, 514, 112–116. [Google Scholar] [CrossRef]
- Bridges, E.; Harris, A.L. Vascular-promoting therapy reduced tumor growth and progression by improving chemotherapy efficacy. Cancer Cell 2015, 27, 7–9. [Google Scholar] [CrossRef]
- Reynolds, L.E.; Wyder, L.; Lively, J.C.; Taverna, D.; Robinson, S.D.; Huang, X.; Sheppard, D.; Hynes, R.O.; Hodivala-Dilke, K.M. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat. Med. 2002, 8, 27–34. [Google Scholar] [CrossRef]
- Coelho, A.L.; Gomes, M.P.; Catarino, R.J.; Rolfo, C.; Lopes, A.M.; Medeiros, R.M.; Araujo, A.M. Angiogenesis in NSCLC: Is vessel co-option the trunk that sustains the branches? Oncotarget 2017, 8, 39795–39804. [Google Scholar] [CrossRef]
- Magnussen, A.L.; Mills, I.G. Vascular normalisation as the stepping stone into tumour microenvironment transformation. Br. J. Cancer 2021, 125, 324–336. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenic therapy for cancer: Current and emerging concepts. Oncology 2005, 19, 7–16. [Google Scholar]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Schmittnaegel, M.; De Palma, M. Reprogramming Tumor Blood Vessels for Enhancing Immunotherapy. Trends Cancer 2017, 3, 809–812. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, Q.; Zheng, Z.; Liu, S.; Meng, L.; Dong, L.; Jiang, X. Vascular normalization in immunotherapy: A promising mechanisms combined with radiotherapy. Biomed. Pharmacother. 2021, 139, 111607. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, J.; Melamed, A.; Worley, M.; Gockley, A.; Jones, D.; Nia, H.T.; Zhang, Y.; Stylianopoulos, T.; Kumar, A.S.; et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2019, 116, 2210–2219. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef]
- Takara, K.; Eino, D.; Ando, K.; Yasuda, D.; Naito, H.; Tsukada, Y.; Iba, T.; Wakabayashi, T.; Muramatsu, F.; Kidoya, H.; et al. Lysophosphatidic Acid Receptor 4 Activation Augments Drug Delivery in Tumors by Tightening Endothelial Cell-Cell Contact. Cell Rep. 2017, 20, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Okamoto, K.; Adachi, Y.; Semba, T.; Uesugi, M.; Ozawa, Y.; Tohyama, O.; Uehara, T.; Kimura, T.; Watanabe, H.; et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014, 105, 1334–1342. [Google Scholar] [CrossRef]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef]
- Yao, H.; Price, T.T.; Cantelli, G.; Ngo, B.; Warner, M.J.; Olivere, L.; Ridge, S.M.; Jablonski, E.M.; Therrien, J.; Tannheimer, S.; et al. Leukaemia hijacks a neural mechanism to invade the central nervous system. Nature 2018, 560, 55–60. [Google Scholar] [CrossRef]
- Jahangiri, A.; Aghi, M.K.; Carbonell, W.S. beta1 integrin: Critical path to antiangiogenic therapy resistance and beyond. Cancer Res. 2014, 74, 3–7. [Google Scholar] [CrossRef]
- Kiefel, H.; Bondong, S.; Hazin, J.; Ridinger, J.; Schirmer, U.; Riedle, S.; Altevogt, P. L1CAM: A major driver for tumor cell invasion and motility. Cell Adhes. Migr. 2012, 6, 374–384. [Google Scholar] [CrossRef]
- Zecchini, S.; Bianchi, M.; Colombo, N.; Fasani, R.; Goisis, G.; Casadio, C.; Viale, G.; Liu, J.; Herlyn, M.; Godwin, A.K.; et al. The differential role of L1 in ovarian carcinoma and normal ovarian surface epithelium. Cancer Res. 2008, 68, 1110–1118. [Google Scholar] [CrossRef]
- Toden, S.; Okugawa, Y.; Jascur, T.; Wodarz, D.; Komarova, N.L.; Buhrmann, C.; Shakibaei, M.; Boland, C.R.; Goel, A. Curcumin mediates chemosensitization to 5-fluorouracil through miRNA-induced suppression of epithelial-to-mesenchymal transition in chemoresistant colorectal cancer. Carcinogenesis 2015, 36, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Park, C.C.; Zhang, H.; Pallavicini, M.; Gray, J.W.; Baehner, F.; Park, C.J.; Bissell, M.J. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006, 66, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Park, C.C.; Zhang, H.J.; Yao, E.S.; Park, C.J.; Bissell, M.J. Beta1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer Res. 2008, 68, 4398–4405. [Google Scholar] [CrossRef]
- Nam, J.M.; Ahmed, K.M.; Costes, S.; Zhang, H.; Onodera, Y.; Olshen, A.B.; Hatanaka, K.C.; Kinoshita, R.; Ishikawa, M.; Sabe, H.; et al. beta1-Integrin via NF-kappaB signaling is essential for acquisition of invasiveness in a model of radiation treated in situ breast cancer. Breast Cancer Res. 2013, 15, R60. [Google Scholar] [CrossRef]
- Khalili, P.; Arakelian, A.; Chen, G.; Plunkett, M.L.; Beck, I.; Parry, G.C.; Donate, F.; Shaw, D.E.; Mazar, A.P.; Rabbani, S.A. A non-RGD-based integrin binding peptide (ATN-161) blocks breast cancer growth and metastasis in vivo. Mol. Cancer Ther. 2006, 5, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, F.; Lu, F.; Xu, S.; Hu, W.; Huang, J.; Gu, Q.; Sun, X. The antiangiogenic effects of integrin alpha5beta1 inhibitor (ATN-161) in vitro and in vivo. Invest. Ophthalmol. Vis. Sci. 2011, 52, 7213–7220. [Google Scholar] [CrossRef]
- Stoeltzing, O.; Liu, W.; Reinmuth, N.; Fan, F.; Parry, G.C.; Parikh, A.A.; McCarty, M.F.; Bucana, C.D.; Mazar, A.P.; Ellis, L.M. Inhibition of integrin alpha5beta1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int. J. Cancer 2003, 104, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, M.E.; Kimmel, K.A.; Gallo, J.; Cardoso, T.; Brown, M.M.; Hudes, G.; Lewis, N.; Weiner, L.; Lam, G.N.; Brown, S.C.; et al. Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), a beta integrin antagonist, in patients with solid tumours. Br. J. Cancer 2006, 94, 1621–1626. [Google Scholar] [CrossRef]
- Giordano, M.; Cavallaro, U. Different Shades of L1CAM in the Pathophysiology of Cancer Stem Cells. J. Clin. Med. 2020, 9, 1502. [Google Scholar] [CrossRef]
- Cho, S.; Lee, T.S.; Song, I.H.; Kim, A.R.; Lee, Y.J.; Kim, H.; Hwang, H.; Jeong, M.S.; Kang, S.G.; Hong, H.J. Combination of anti-L1 cell adhesion molecule antibody and gemcitabine or cisplatin improves the therapeutic response of intrahepatic cholangiocarcinoma. PLoS ONE 2017, 12, e0170078. [Google Scholar] [CrossRef]
- Nam, J.K.; Kim, A.R.; Choi, S.H.; Kim, J.H.; Choi, K.J.; Cho, S.; Lee, J.W.; Cho, H.J.; Kwon, Y.W.; Cho, J.; et al. An antibody against L1 cell adhesion molecule inhibits cardiotoxicity by regulating persistent DNA damage. Nat. Commun. 2021, 12, 3279. [Google Scholar] [CrossRef]
- Pham, K.; Luo, D.; Siemann, D.W.; Law, B.K.; Reynolds, B.A.; Hothi, P.; Foltz, G.; Harrison, J.K. VEGFR inhibitors upregulate CXCR4 in VEGF receptor-expressing glioblastoma in a TGFbetaR signaling-dependent manner. Cancer Lett. 2015, 360, 60–67. [Google Scholar] [CrossRef]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069. [Google Scholar] [CrossRef]
- Infanger, D.W.; Cho, Y.; Lopez, B.S.; Mohanan, S.; Liu, S.C.; Gursel, D.; Boockvar, J.A.; Fischbach, C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013, 73, 7079–7089. [Google Scholar] [CrossRef]
- Otsubo, T.; Iwaya, K.; Mukai, Y.; Mizokami, Y.; Serizawa, H.; Matsuoka, T.; Mukai, K. Involvement of Arp2/3 complex in the process of colorectal carcinogenesis. Mod. Pathol. 2004, 17, 461–467. [Google Scholar] [CrossRef]
- Rada, M.; Kapelanski-Lamoureux, A.; Tsamchoe, M.; Petrillo, S.; Lazaris, A.; Metrakos, P. Angiopoietin-1 Upregulates Cancer Cell Motility in Colorectal Cancer Liver Metastases through Actin-Related Protein 2/3. Cancers 2022, 14, 2540. [Google Scholar] [CrossRef]
- Ibrahim, N.S.; Lazaris, A.; Rada, M.; Petrillo, S.K.; Huck, L.; Hussain, S.; Ouladan, S.; Gao, Z.H.; Gregorieff, A.; Essalmani, R.; et al. Angiopoietin1 Deficiency in Hepatocytes Affects the Growth of Colorectal Cancer Liver Metastases (CRCLM). Cancers 2019, 12, 35. [Google Scholar] [CrossRef]
- Duran, C.L.; Borriello, L.; Karagiannis, G.S.; Entenberg, D.; Oktay, M.H.; Condeelis, J.S. Targeting Tie2 in the Tumor Microenvironment: From Angiogenesis to Dissemination. Cancers 2021, 13, 5730. [Google Scholar] [CrossRef]
- Chanez-Paredes, S.; Montoya-Garcia, A.; Schnoor, M. Cellular and pathophysiological consequences of Arp2/3 complex inhibition: Role of inhibitory proteins and pharmacological compounds. Cell Mol. Life Sci. 2019, 76, 3349–3361. [Google Scholar] [CrossRef]
- Fokin, A.I.; Chuprov-Netochin, R.N.; Malyshev, A.S.; Romero, S.; Semenova, M.N.; Konyushkin, L.D.; Leonov, S.V.; Semenov, V.V.; Gautreau, A.M. Synthesis, Screening and Characterization of Novel Potent Arp2/3 Inhibitory Compounds Analogous to CK-666. Front. Pharmacol. 2022, 13, 896994. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Trompetto, B.; Tan, X.H.M.; Scott, M.B.; Hu, K.H.; Deeds, E.; Butte, M.J.; Chiou, P.Y.; Rowat, A.C. Differential Contributions of Actin and Myosin to the Physical Phenotypes and Invasion of Pancreatic Cancer Cells. Cell Mol. Bioeng. 2020, 13, 27–44. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, X.; Chen, C.; Liu, B.; Ren, B.; Wang, L.; Zhao, K.; Yu, S.; Ming, H. Expression of the Arp2/3 complex in human gliomas and its role in the migration and invasion of glioma cells. Oncol. Rep. 2013, 30, 2127–2136. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Han, Y.M.; Choi, J.; Lee, Y.J.; Yun, J.; Lee, S.K.; Lee, C.W.; Kang, J.S.; Chi, S.W.; Moon, J.H.; et al. Benproperine, an ARPC2 inhibitor, suppresses cancer cell migration and tumor metastasis. Biochem. Pharmacol. 2019, 163, 46–59. [Google Scholar] [CrossRef]
- Jang, H.J.; Yoon, Y.J.; Choi, J.; Lee, Y.J.; Lee, S.; Cho, W.; Byun, W.G.; Park, S.B.; Han, D.C.; Kwon, B.M. S-Benproperine, an Active Stereoisomer of Benproperine, Suppresses Cancer Migration and Tumor Metastasis by Targeting ARPC2. Pharmaceuticals 2022, 15, 1462. [Google Scholar] [CrossRef]
- Choi, J.; Lee, Y.J.; Yoon, Y.J.; Kim, C.H.; Park, S.J.; Kim, S.Y.; Doo Kim, N.; Cho Han, D.; Kwon, B.M. Pimozide suppresses cancer cell migration and tumor metastasis through binding to ARPC2, a subunit of the Arp2/3 complex. Cancer Sci. 2019, 110, 3788–3801. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Tungsukruthai, S.; Petpiroon, N.; Chanvorachote, P. Molecular Mechanisms of Breast Cancer Metastasis and Potential Anti-metastatic Compounds. Anticancer. Res. 2018, 38, 2607–2618. [Google Scholar] [CrossRef]
- Ganesh, K.; Massague, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef]
- Rada, M.; Kapelanski-Lamoureux, A.; Petrillo, S.; Tabaries, S.; Siegel, P.; Reynolds, A.R.; Lazaris, A.; Metrakos, P. Runt related transcription factor-1 plays a central role in vessel co-option of colorectal cancer liver metastases. Commun. Biol. 2021, 4, 950. [Google Scholar] [CrossRef]
- Rada, M.; Tsamchoe, M.; Kapelanski-Lamoureux, A.; Hassan, N.; Bloom, J.; Petrillo, S.; Kim, D.H.; Lazaris, A.; Metrakos, P. Cancer Cells Promote Phenotypic Alterations in Hepatocytes at the Edge of Cancer Cell Nests to Facilitate Vessel Co-Option Establishment in Colorectal Cancer Liver Metastases. Cancers 2022, 14, 1318. [Google Scholar] [CrossRef]
- Maione, F.; Capano, S.; Regano, D.; Zentilin, L.; Giacca, M.; Casanovas, O.; Bussolino, F.; Serini, G.; Giraudo, E. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J. Clin. Invest. 2012, 122, 1832–1848. [Google Scholar] [CrossRef]
- Hammers, H.J.; Verheul, H.M.; Salumbides, B.; Sharma, R.; Rudek, M.; Jaspers, J.; Shah, P.; Ellis, L.; Shen, L.; Paesante, S.; et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: Evidence from a xenograft study. Mol. Cancer Ther. 2010, 9, 1525–1535. [Google Scholar] [CrossRef]
- van Malenstein, H.; Dekervel, J.; Verslype, C.; Van Cutsem, E.; Windmolders, P.; Nevens, F.; van Pelt, J. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013, 329, 74–83. [Google Scholar] [CrossRef]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.T.; Mock, K.; Ruh, M.; Schuler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef]
- Namba, T.; Kodama, R.; Moritomo, S.; Hoshino, T.; Mizushima, T. Zidovudine, an anti-viral drug, resensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine by inhibition of the Akt-GSK3beta-Snail pathway. Cell Death Dis. 2015, 6, e1795. [Google Scholar] [CrossRef]
- Wen, Z.; Feng, S.; Wei, L.; Wang, Z.; Hong, D.; Wang, Q. Evodiamine, a novel inhibitor of the Wnt pathway, inhibits the self-renewal of gastric cancer stem cells. Int. J. Mol. Med. 2015, 36, 1657–1663. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, L.; Hu, C.; Liang, S.; Fei, X.; Yan, N.; Zhang, Y.; Zhang, F. WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int. J. Oncol. 2016, 48, 1175–1186. [Google Scholar] [CrossRef]
- Qin, G.; Xu, F.; Qin, T.; Zheng, Q.; Shi, D.; Xia, W.; Tian, Y.; Tang, Y.; Wang, J.; Xiao, X.; et al. Palbociclib inhibits epithelial-mesenchymal transition and metastasis in breast cancer via c-Jun/COX-2 signaling pathway. Oncotarget 2015, 6, 41794–41808. [Google Scholar] [CrossRef]
- Xu, B.; Jiang, C.; Han, H.; Liu, H.; Tang, M.; Liu, L.; Ji, W.; Lu, X.; Yang, X.; Zhang, Y.; et al. Icaritin inhibits the invasion and epithelial-to-mesenchymal transition of glioblastoma cells by targeting EMMPRIN via PTEN/AKt/HIF-1alpha signalling. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wu, G.; Chang, C.; Zhu, F.; Xiao, Y.; Li, Q.; Zhang, T.; Zhang, L. Disulfiram inhibits TGF-beta-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-kappaB/Snail pathway. Oncotarget 2015, 6, 40907–40919. [Google Scholar] [CrossRef] [PubMed]
- Busaranon, K.; Plaimee, P.; Sritularak, B.; Chanvorachote, P. Moscatilin inhibits epithelial-to-mesenchymal transition and sensitizes anoikis in human lung cancer H460 cells. J. Nat. Med. 2016, 70, 18–27. [Google Scholar] [CrossRef]
- Hseu, Y.C.; Chang, C.T.; Gowrisankar, Y.V.; Chen, X.Z.; Lin, H.C.; Yen, H.R.; Yang, H.L. Zerumbone Exhibits Antiphotoaging and Dermatoprotective Properties in Ultraviolet A-Irradiated Human Skin Fibroblast Cells via the Activation of Nrf2/ARE Defensive Pathway. Oxid. Med. Cell Longev. 2019, 2019, 4098674. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, S.; Che, X.; Hou, K.; Ma, Y.; Li, C.; Wen, T.; Fan, Y.; Hu, X.; Liu, Y.; et al. Bufalin inhibits TGF-beta-induced epithelial-to-mesenchymal transition and migration in human lung cancer A549 cells by downregulating TGF-beta receptors. Int. J. Mol. Med. 2015, 36, 645–652. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, P.; Wang, H.; Hou, D.; Li, W.; Xiao, G.; Li, C. Inhibitory effects of metformin at low concentration on epithelial-mesenchymal transition of CD44(+)CD117(+) ovarian cancer stem cells. Stem Cell Res. Ther. 2015, 6, 262. [Google Scholar] [CrossRef]
- Popova, N.V.; Jucker, M. The Functional Role of Extracellular Matrix Proteins in Cancer. Cancers 2022, 14, 238. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Cai, B.; Zeng, M.; Hao, Y.; Giancotti, F.G.; Fu, B.M. Integrin beta4 signaling promotes mammary tumor cell adhesion to brain microvascular endothelium by inducing ErbB2-mediated secretion of VEGF. Ann. Biomed. Eng. 2011, 39, 2223–2241. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef]
- Wong, C.C.; Zhang, H.; Gilkes, D.M.; Chen, J.; Wei, H.; Chaturvedi, P.; Hubbi, M.E.; Semenza, G.L. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J. Mol. Med. 2012, 90, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Belhabib, I.; Zaghdoudi, S.; Lac, C.; Bousquet, C.; Jean, C. Extracellular Matrices and Cancer-Associated Fibroblasts: Targets for Cancer Diagnosis and Therapy? Cancers 2021, 13, 3466. [Google Scholar] [CrossRef]
- Kalli, M.; Stylianopoulos, T. Defining the Role of Solid Stress and Matrix Stiffness in Cancer Cell Proliferation and Metastasis. Front. Oncol. 2018, 8, 55. [Google Scholar] [CrossRef]
- Kalli, M.; Papageorgis, P.; Gkretsi, V.; Stylianopoulos, T. Solid Stress Facilitates Fibroblasts Activation to Promote Pancreatic Cancer Cell Migration. Ann. Biomed. Eng. 2018, 46, 657–669. [Google Scholar] [CrossRef]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef]
- Bondareva, A.; Downey, C.M.; Ayres, F.; Liu, W.; Boyd, S.K.; Hallgrimsson, B.; Jirik, F.R. The lysyl oxidase inhibitor, beta-aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PLoS ONE 2009, 4, e5620. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, S.; Li, W.; Chen, J.; Xiao, X.; Wang, Y.; Yan, G.; Chen, L. Inactivation of lysyl oxidase by beta-aminopropionitrile inhibits hypoxia-induced invasion and migration of cervical cancer cells. Oncol. Rep. 2013, 29, 541–548. [Google Scholar] [CrossRef]
- Ninomiya, G.; Yamada, S.; Hayashi, M.; Takeda, S.; Suenaga, M.; Takami, H.; Kanda, M.; Iwata, N.; Niwa, Y.; Tanaka, C.; et al. Significance of Lysyl oxidase-like 2 gene expression on the epithelial-mesenchymal status of hepatocellular carcinoma. Oncol. Rep. 2018, 39, 2664–2672. [Google Scholar] [CrossRef]
- Tanaka, N.; Yamada, S.; Sonohara, F.; Suenaga, M.; Hayashi, M.; Takami, H.; Niwa, Y.; Hattori, N.; Iwata, N.; Kanda, M.; et al. Clinical Implications of Lysyl Oxidase-Like Protein 2 Expression in Pancreatic Cancer. Sci. Rep. 2018, 8, 9846. [Google Scholar] [CrossRef]
- Rodriguez, H.M.; Vaysberg, M.; Mikels, A.; McCauley, S.; Velayo, A.C.; Garcia, C.; Smith, V. Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J. Biol. Chem. 2010, 285, 20964–20974. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B., 3rd; Wainberg, Z.A.; Hecht, J.R.; Vyushkov, D.; Dong, H.; Bendell, J.; Kudrik, F. A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist 2017, 22, 241-e5. [Google Scholar] [CrossRef]
- Hecht, J.R.; Benson, A.B., 3rd; Vyushkov, D.; Yang, Y.; Bendell, J.; Verma, U. A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab in Combination with FOLFIRI for the Second-Line Treatment of Metastatic KRAS Mutant Colorectal Adenocarcinoma. Oncologist 2017, 22, 243-e23. [Google Scholar] [CrossRef] [PubMed]
- Haj-Shomaly, J.; Vorontsova, A.; Barenholz-Cohen, T.; Levi-Galibov, O.; Devarasetty, M.; Timaner, M.; Raviv, Z.; Cooper, T.J.; Soker, S.; Hasson, P.; et al. T Cells Promote Metastasis by Regulating Extracellular Matrix Remodeling following Chemotherapy. Cancer Res. 2022, 82, 278–291. [Google Scholar] [CrossRef]
- Li, M.; Xing, S.; Zhang, H.; Shang, S.; Li, X.; Ren, B.; Li, G.; Chang, X.; Li, Y.; Li, W. A matrix metalloproteinase inhibitor enhances anti-cytotoxic T lymphocyte antigen-4 antibody immunotherapy in breast cancer by reprogramming the tumor microenvironment. Oncol. Rep. 2016, 35, 1329–1339. [Google Scholar] [CrossRef]
- Haas, N.B.; Quirt, I.; Hotte, S.; McWhirter, E.; Polintan, R.; Litwin, S.; Adams, P.D.; McBryan, T.; Wang, L.; Martin, L.P.; et al. Phase II trial of vorinostat in advanced melanoma. Invest. New Drugs 2014, 32, 526–534. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Mina, R.; Jakubowiak, A.J.; Zimmerman, T.L.; Wolf, J.J.; Lewis, C.; Gleason, C.; Sharp, C.; Martin, T.; Heffner, L.T.; et al. Combining carfilzomib and panobinostat to treat relapsed/refractory multiple myeloma: Results of a Multiple Myeloma Research Consortium Phase I Study. Blood Cancer J. 2019, 9, 3. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Effect | |
|---|---|---|
| Anti-angiogenic agents | Aflibercept | Anti-VEGF glicoprotein |
| Bevacizumab | Anti-VEGF monoclonal antibody | |
| Cabozantinib | Inhibitor of tyrosine kinase domain VEGFR | |
| Cediranib | Inhibitor of tyrosine kinase domain of VEGFR1, VEGFR2 and VEGFR3 | |
| Lenvatinib | Inhibitor of tyrosine kinase domain of VEGFR1, VEGFR2 and VEGFR3 | |
| Ramucirumab | Anti-VEGFR2 monoclonal antibody | |
| Regorafenib | Inhibitor of tyrosine kinase domain of VEGFR1, VEGFR2, VEGFR3 and TIE2 | |
| Sorafenib | Inhibitor of tyrosine kinase domain of VEGFR2 | |
| Sunitinib | Inhibitor of tyrosine kinase domain of VEGFR2 | |
| Pro-angiogenic agents | Vandetanib | Inhibitor of tyrosine kinase domain of VEGFR2 |
| Eribulin | Microtubule dynamics inhibitor | |
| ldCil | Increase levels of VEGFR2 | |
| Lysophosphatidic acid (LPA) | Tightens endothelial cell contacts | |
| Inhibitors of cancer cell–blood vessel adhesion | Ab417 | L1CAM antibody |
| AIIB2 | β1 integrin inhibitor | |
| ATN-161 | β1 integrin inhibitor | |
| OS2966 | β1 integrin inhibitor | |
| Inhibitors of cancer cell migration | Benproperine | Inhibitor of Arp2/3 complex |
| CK-666 | Inhibitor of Arp2/3 complex | |
| LY294002 | Inhibitor of PI3K/AKT | |
| Pimozide | Inhibitor of Arp2/3 complex | |
| Bufalin | Inhibits TGF-β-induced EMT | |
| Inhibitors of EMT | Curcumin | Inhibitor of EMT in 5-FU resistant cells |
| Disulfiram | Inhibits TGF-β pathway | |
| Evodiamine | Decrease the expression of Slug, Twist, Zeb1 and vimentin. | |
| Icaritin | Inhibits PTEN/Akt/HIF-1α pathway | |
| Metformin | Inhibits EMT | |
| Mocetinostat | Interfere with ZEB1 function | |
| Moscatilin | Inhibits Erk and Akt pathways | |
| Palbociclib | Regulates Jun/COX-2 pathway | |
| Pyrvinum paomate | Inhibits the Wnt pathway | |
| Zerumbone | Inhibits TGF-β-induced EMT | |
| Zidovudine | Inhibits Akt-GSK3b-Snail1 pathway | |
| ECM regulators | AB0023 | LOXL2 inhibitor |
| βAPN | LOXL2 inhibitor | |
| MMPs | Collagen degradation | |
| Simtuzumab | Anti-LOXL2 monoclonal antibody | |
| Hypoxia inhibitors | Panobinostat | Inhibitor of HIF-1α |
| Vorinostat | Inhibitor of HIF-1α |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrera-Aguado, I.; Marcos-Zazo, L.; Carrancio-Salán, P.; Guerra-Paes, E.; Sánchez-Juanes, F.; Muñoz-Félix, J.M. The Inhibition of Vessel Co-Option as an Emerging Strategy for Cancer Therapy. Int. J. Mol. Sci. 2024, 25, 921. https://doi.org/10.3390/ijms25020921
Carrera-Aguado I, Marcos-Zazo L, Carrancio-Salán P, Guerra-Paes E, Sánchez-Juanes F, Muñoz-Félix JM. The Inhibition of Vessel Co-Option as an Emerging Strategy for Cancer Therapy. International Journal of Molecular Sciences. 2024; 25(2):921. https://doi.org/10.3390/ijms25020921
Chicago/Turabian StyleCarrera-Aguado, Iván, Laura Marcos-Zazo, Patricia Carrancio-Salán, Elena Guerra-Paes, Fernando Sánchez-Juanes, and José M. Muñoz-Félix. 2024. "The Inhibition of Vessel Co-Option as an Emerging Strategy for Cancer Therapy" International Journal of Molecular Sciences 25, no. 2: 921. https://doi.org/10.3390/ijms25020921
APA StyleCarrera-Aguado, I., Marcos-Zazo, L., Carrancio-Salán, P., Guerra-Paes, E., Sánchez-Juanes, F., & Muñoz-Félix, J. M. (2024). The Inhibition of Vessel Co-Option as an Emerging Strategy for Cancer Therapy. International Journal of Molecular Sciences, 25(2), 921. https://doi.org/10.3390/ijms25020921