
Natural Product-Based Glycolysis Inhibitors as a Therapeutic Strategy for Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer

 , , , , and
, , , , and 
Abstract
1. Introduction
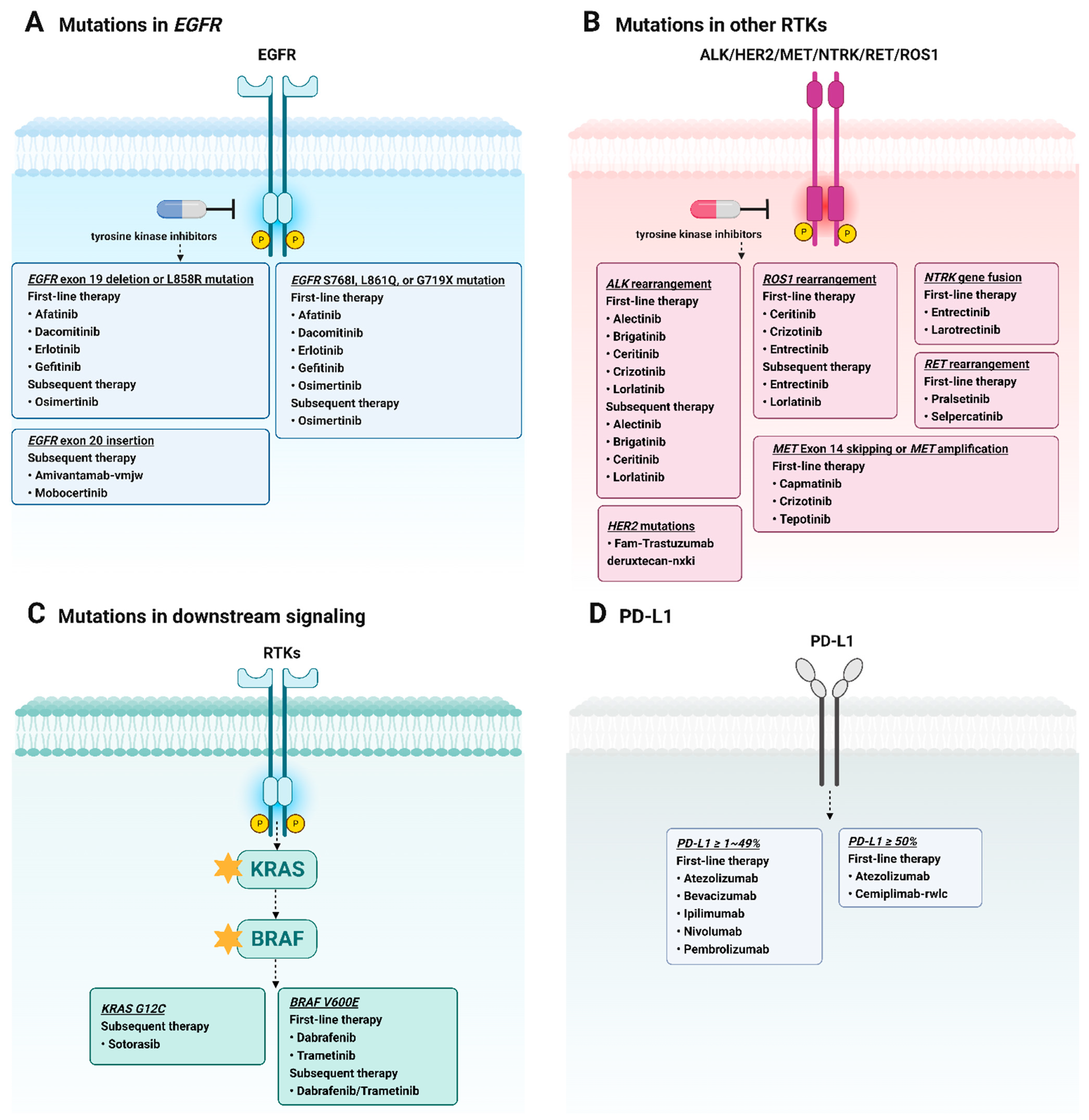
2. Targeted Therapy in NSCLC
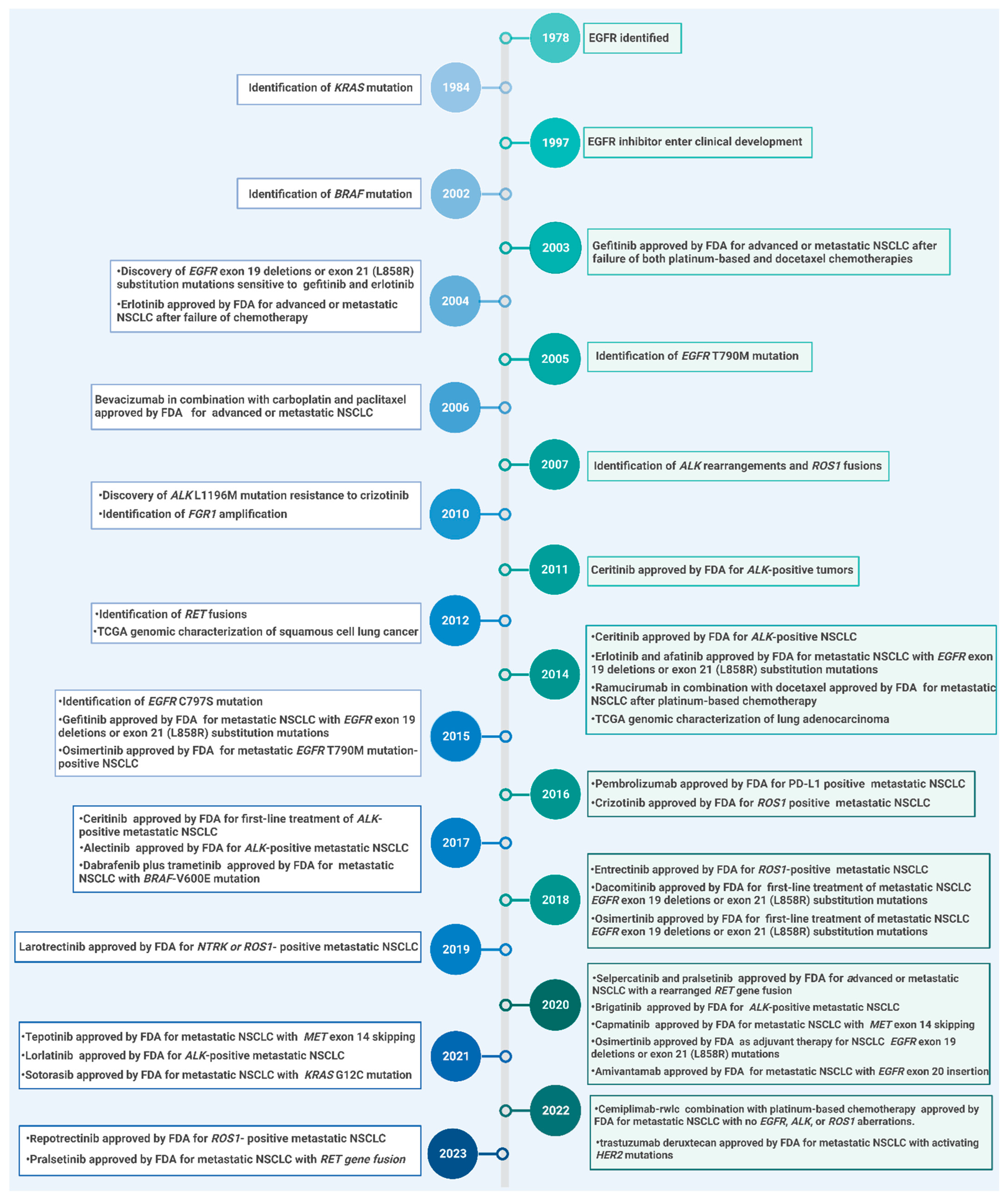
3. EGFR-TKIs in NSCLC Treatment
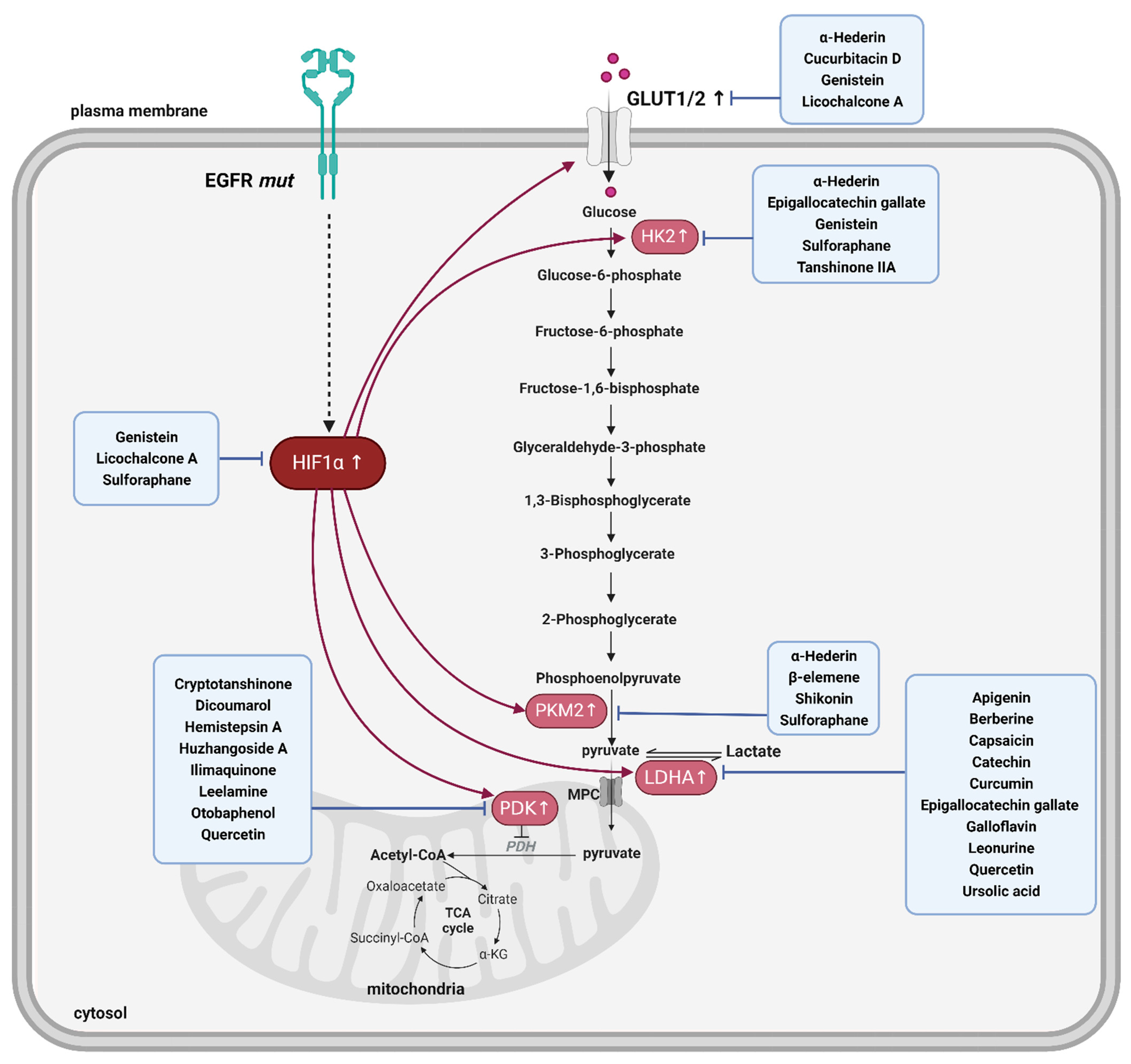
4. Enhanced Glycolysis in EGFR-TKI-Resistant NSCLC
5. Advantages of PDK Inhibition against EGFR-TKI Resistance and Inhibitors from Natural Products
6. Natural Product-Derived LDHA Inhibitors and Their Advantage against EGFR-TKI Resistance
7. Natural Products Suppressing Other Glycolytic Enzymes and Their Use for EGFR-TKI Resistance
8. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| 2-DG | 2-deoxy-d-glucose |
| ALK | Anaplastic lymphoma kinase |
| ATP | Adenosine triphosphate |
| BRAF | V-raf murine sarcoma viral oncogene homolog B1 |
| c-Myc | Cellular-myelocytomatosis oncogene |
| DCA | Dichloroacetate |
| DNA | Deoxyribonucleic acid |
| EGCG | Epigallocatechin gallate |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| FBP1 | Fructose-1,6-bisphosphatase |
| FDA | Food and Drug Administration |
| GLUT1 | Glucose transporter 1 |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF-1α | Hypoxia-inducible factor-1 alpha |
| HK2 | Hexokinase 2 |
| KRAS | Kirsten rat sarcoma virus |
| LDH | Lactate dehydrogenase |
| LDHA | Lactate dehydrogenase A |
| MET | Proto-oncogene, receptor tyrosine kinase |
| NAD | Nicotinamide adenine dinucleotide |
| NCCN | National Comprehensive Cancer Network |
| ND | Not determined |
| NSCLC | Non-small cell lung cancer |
| NTRK | Neurotrophic tyrosine receptor kinase |
| OXPHOS | Oxidative phosphorylation |
| OR | Overall survival |
| PDC | Pyruvate dehydrogenase complex |
| PDK | Pyruvate dehydrogenase kinase |
| PD-L1 | Programmed cell death ligand 1 |
| PEP | Phosphoenolpyruvate |
| PI3K | Phosphatidylinositol 3-kinase |
| PKM2 | Pyruvate kinase M2 |
| RET | RET proto-oncogene |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| ROS1 | ROS proto-oncogene 1, receptor tyrosine kinase |
| SCLC | Small cell lung cancer |
| SFN | Sulforaphane |
| TK | Tyrosine kinase |
| TKI | Tyrosine kinase inhibitor |
References
- Wang, M.; Herbst, R.S.; Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med. 2021, 27, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship; Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2008; pp. 584–594. [Google Scholar]
- Van Meerbeeck, J.P.; Fennell, D.A.; De Ruysscher, D.K. Small-cell lung cancer. Lancet 2011, 378, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Halliday, P.R.; Blakely, C.M.; Bivona, T.G. Emerging targeted therapies for the treatment of non-small cell lung cancer. Curr. Oncol. Rep. 2019, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yang, M.; Liang, N.; Li, S. Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR mutations in non-small cell lung cancer: Perplexity and solution. Oncol. Rep. 2017, 37, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Yuan, J.-Q.; Wang, K.-F.; Fu, X.-H.; Han, X.-R.; Threapleton, D.; Yang, Z.-Y.; Mao, C.; Tang, J.-L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Kobayashi, K.; Usui, K.; Maemondo, M.; Okinaga, S.; Mikami, I.; Ando, M.; Yamazaki, K.; Saijo, Y.; Gemma, A. First-line gefitinib for patients with advanced non-small-cell lung cancer harboring epidermal growth factor receptor mutations without indication for chemotherapy. J. Clin. Oncol. 2009, 27, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Varghese, E.; Samuel, S.M.; Líšková, A.; Samec, M.; Kubatka, P.; Büsselberg, D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers 2020, 12, 2252. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Jeoung, N.H.; Park, K.-G.; Lee, I.-K. Transcriptional regulation of pyruvate dehydrogenase kinase. Diabetes Metab. J. 2012, 36, 328–335. [Google Scholar] [CrossRef]
- Lu, C.-W.; Lin, S.-C.; Chen, K.-F.; Lai, Y.-Y.; Tsai, S.-J. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 2008, 283, 28106–28114. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Kim, M.; Choi, H.J.; Jin, J.S.; Lee, S.O.; Bae, S.J.; Ryu, D.; Ha, K.T. The Oral Administration of Sanguisorba officinalis Extract Improves Physical Performance through LDHA Modulation. Molecules 2021, 26, 1579. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Kim, M.; Kim, H.J.; Jang, S.B.; Bae, S.J.; Lee, I.K.; Ryu, D.; Ha, K.T. Targeting Lactate Dehydrogenase A with Catechin Resensitizes SNU620/5FU Gastric Cancer Cells to 5-Fluorouracil. Int. J. Mol. Sci. 2021, 22, 5406. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chard Dunmall, L.S.; Cheng, Z.; Wang, Y.; Si, L. Natural products targeting glycolysis in cancer. Front. Pharmacol. 2022, 13, 1036502. [Google Scholar] [CrossRef]
- Zhao, M.; Wei, F.; Sun, G.; Wen, Y.; Xiang, J.; Su, F.; Zhan, L.; Nian, Q.; Chen, Y.; Zeng, J. Natural compounds targeting glycolysis as promising therapeutics for gastric cancer: A review. Front. Pharmacol. 2022, 13, 1004383. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Han, J.H.; Bae, S.-J.; Lee, J.R.; Kim, Y.W.; Jang, S.B. Hemistepsin A suppresses colorectal cancer growth through inhibiting pyruvate dehydrogenase kinase activity. Sci. Rep. 2020, 10, 21940. [Google Scholar] [CrossRef]
- Kwak, C.-H.; Lee, J.-H.; Kim, E.-Y.; Han, C.W.; Kim, K.-J.; Lee, H.; Cho, M.; Jang, S.B.; Kim, C.-H.; Chung, T.-W. Huzhangoside A suppresses tumor growth through inhibition of pyruvate dehydrogenase kinase activity. Cancers 2019, 11, 712. [Google Scholar] [CrossRef]
- Kwak, C.-H.; Jin, L.; Han, J.H.; Han, C.W.; Kim, E.; Cho, M.; Chung, T.-W.; Bae, S.-J.; Jang, S.B.; Ha, K.-T. Ilimaquinone induces the apoptotic cell death of cancer cells by reducing pyruvate dehydrogenase kinase 1 activity. Int. J. Mol. Sci. 2020, 21, 6021. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef]
- Araghi, M.; Mannani, R.; Heidarnejad maleki, A.; Hamidi, A.; Rostami, S.; Safa, S.H.; Faramarzi, F.; Khorasani, S.; Alimohammadi, M.; Tahmasebi, S. Recent advances in non-small cell lung cancer targeted therapy; an update review. Cancer Cell Int. 2023, 23, 162. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A. Non–small cell lung cancer, version 3.2022, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 497–530. [Google Scholar] [CrossRef] [PubMed]
- Dungo, R.T.; Keating, G.M. Afatinib: First global approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Dacomitinib: First global approval. Drugs 2018, 78, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Johnson, J.R.; Chattopadhyay, S.; Tang, S.; Justice, R.; Sridhara, R.; Pazdur, R. Approval summary: Erlotinib maintenance therapy of advanced/metastatic non-small cell lung cancer (NSCLC). Oncologist 2010, 15, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Blumenthal, G.M.; Yuan, W.; He, K.; Keegan, P.; Pazdur, R. FDA approval of gefitinib for the treatment of patients with metastatic EGFR mutation–positive non–small cell lung cancer. Clin. Cancer Res. 2016, 22, 1307–1312. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Yang, J.; Lee, C.K.; Kurata, T.; Kim, D.-W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.Y.; Kim, S.W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef]
- Herden, M.; Waller, C.F. Alectinib. Recent Results Cancer Res. 2018, 211, 247–256. [Google Scholar]
- Markham, A. Brigatinib: First global approval. Drugs 2017, 77, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.F.; Van De Kerkhove, C.; Wauters, E.; Van Mol, P. Capmatinib for the treatment of non-small cell lung cancer. Expert Rev. Anticancer Ther. 2019, 19, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.C.; Neal, J.W. Crizotinib as first line therapy for advanced ALK-positive non-small cell lung cancers. Transl. Lung Cancer Res. 2015, 4, 639. [Google Scholar] [PubMed]
- Shaw, A.T.; Yasothan, U.; Kirkpatrick, P. Crizotinib. Nat. Rev. Drug Discov. 2011, 10, 897–898. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Pizzutilo, E.G.; Marrapese, G.; Tosi, F.; Cerea, G.; Siena, S. Entrectinib for the treatment of metastatic NSCLC: Safety and efficacy. Expert Rev. Anticancer Ther. 2020, 20, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Larotrectinib: First global approval. Drugs 2019, 79, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A. First-line lorlatinib or crizotinib in advanced ALK-positive lung cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef]
- Markham, A. Pralsetinib: First approval. Drugs 2020, 80, 1865–1870. [Google Scholar] [CrossRef]
- Markham, A. Selpercatinib: First approval. Drugs 2020, 80, 1119–1124. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J. Tepotinib in non–small-cell lung cancer with MET exon 14 skipping mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef]
- Odogwu, L.; Mathieu, L.; Blumenthal, G.; Larkins, E.; Goldberg, K.B.; Griffin, N.; Bijwaard, K.; Lee, E.Y.; Philip, R.; Jiang, X. FDA approval summary: Dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring BRAF V600E mutations. Oncologist 2018, 23, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Akinboro, O.; Larkins, E.; Pai-Scherf, L.H.; Mathieu, L.N.; Ren, Y.; Cheng, J.; Fiero, M.H.; Fu, W.; Bi, Y.; Kalavar, S. FDA Approval summary: Pembrolizumab, atezolizumab, and cemiplimab-rwlc as single agents for first-line treatment of Advanced/Metastatic PD-L1–high NSCLC. Clin. Cancer Res. 2022, 28, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Gootenberg, J.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin®) plus carboplatin and paclitaxel as first-line treatment of advanced/metastatic recurrent nonsquamous non-small cell lung cancer. Oncologist 2007, 12, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Vellanki, P.J.; Mulkey, F.; Jaigirdar, A.A.; Rodriguez, L.; Wang, Y.; Xu, Y.; Zhao, H.; Liu, J.; Howe, G.; Wang, J. FDA Approval Summary: Nivolumab with Ipilimumab and Chemotherapy for Metastatic Non–small Cell Lung Cancer, A Collaborative Project Orbis ReviewFDA Approval: Nivolumab with Ipilimumab and Chemotherapy. Clin. Cancer Res. 2021, 27, 3522–3527. [Google Scholar] [CrossRef]
- Kazandjian, D.; Suzman, D.L.; Blumenthal, G.; Mushti, S.; He, K.; Libeg, M.; Keegan, P.; Pazdur, R. FDA approval summary: Nivolumab for the treatment of metastatic non-small cell lung cancer with progression on or after platinum-based chemotherapy. Oncologist 2016, 21, 634–642. [Google Scholar] [CrossRef]
- Reck, M. Pembrolizumab as first-line therapy for metastatic non-small-cell lung cancer. Immunotherapy 2018, 10, 93–105. [Google Scholar] [CrossRef]
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Necitumumab: First Global Approval. Drugs 2016, 76, 283–289. [Google Scholar] [CrossRef]
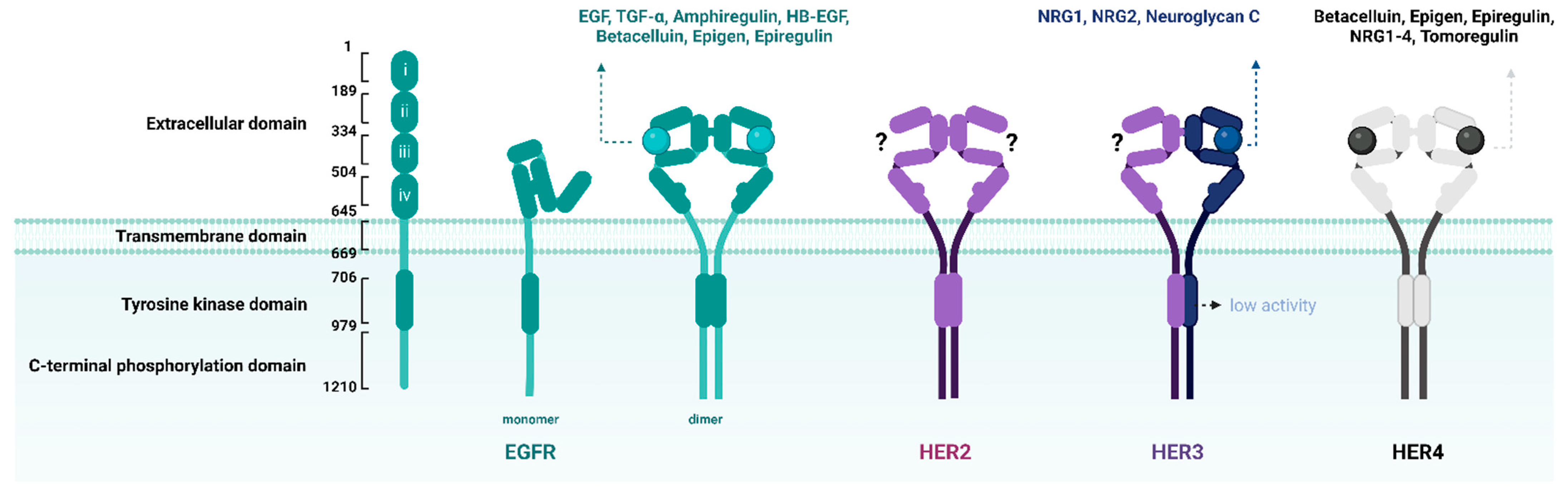
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, S21–S26. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y. The EGFR family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Lester, J.F. Tyrosine Kinase Inhibitors for the Treatment of EGFR Mutation-Positive Non-Small-Cell Lung Cancer: A Clash of the Generations. Clin. Lung Cancer 2020, 21, e216–e228. [Google Scholar] [CrossRef]
- Wieduwilt, M.; Moasser, M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.W.; Cho, H.-S.; Eigenbrot, C.; Ferguson, K.M.; Garrett, T.P.; Leahy, D.J.; Lemmon, M.A.; Sliwkowski, M.X.; Ward, C.W.; Yokoyama, S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 2003, 12, 541–552. [Google Scholar] [CrossRef]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Siegelin, M.D.; Borczuk, A.C. Epidermal growth factor receptor mutations in lung adenocarcinoma. Lab. Investig. 2014, 94, 129–137. [Google Scholar] [CrossRef]
- Huang, S.-F.; Cheng, S.-D.; Chien, H.-T.; Liao, C.-T.; Chen, I.-H.; Wang, H.-M.; Chuang, W.-Y.; Wang, C.-Y.; Hsieh, L.-L. Relationship between epidermal growth factor receptor gene copy number and protein expression in oral cavity squamous cell carcinoma. Oral Oncol. 2012, 48, 67–72. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, I.; Planchard, D. Next-Generation EGFR Tyrosine Kinase Inhibitors for Treating EGFR-Mutant Lung Cancer beyond First Line. Front. Med. 2016, 3, 76. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Fu, Y.N.; Lin, C.H.; Yang, S.T.; Hu, S.F.; Chen, Y.T.; Tsai, S.F.; Huang, S.F. Distinctive activation patterns in constitutively active and gefitinib-sensitive EGFR mutants. Oncogene 2006, 25, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Kancha, R.K.; von Bubnoff, N.; Peschel, C.; Duyster, J. Functional analysis of epidermal growth factor receptor (EGFR) mutations and potential implications for EGFR targeted therapy. Clin. Cancer Res. 2009, 15, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; Chang, C.F.; Huang, C.Y.; Yang, C.T.; Kuo, C.S.; Fang, Y.F.; Hsu, P.C.; Wu, C.E. The survival after discontinuation of EGFR-TKIs due to intolerable adverse events in patients with EGFR-mutated non-small cell lung cancer. Thorac. Cancer 2023, 14, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung CancersMechanisms of Acquired Resistance to EGFR-TKI Therapy. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Campo, M.; Gerber, D.; Gainor, J.F.; Heist, R.S.; Temel, J.S.; Shaw, A.T.; Fidias, P.; Muzikansky, A.; Engelman, J.A.; Sequist, L.V. Acquired resistance to first-line afatinib and the challenges of prearranged progression biopsies. J. Thorac. Oncol. 2016, 11, 2022–2026. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncoll 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Fujino, T.; Nishino, M.; Koga, T.; Chiba, M.; Sesumi, Y.; Ohara, S.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. EGFR T790M and C797S Mutations as Mechanisms of Acquired Resistance to Dacomitinib. J. Thorac. Oncol. 2018, 13, 727–731. [Google Scholar] [CrossRef]
- Asahina, H.; Tanaka, K.; Morita, S.; Maemondo, M.; Seike, M.; Okamoto, I.; Oizumi, S.; Kagamu, H.; Takahashi, K.; Kikuchi, T. A Phase II Study of Osimertinib Combined With Platinum Plus Pemetrexed in Patients With EGFR-Mutated Advanced Non–Small-cell Lung Cancer: The OPAL Study (NEJ032C/LOGIK1801). Clin. Lung Cancer 2021, 22, 147–151. [Google Scholar] [CrossRef]
- Shi, K.; Wang, G.; Pei, J.; Zhang, J.; Wang, J.; Ouyang, L.; Wang, Y.; Li, W. Emerging strategies to overcome resistance to third-generation EGFR inhibitors. J. Hematol. Oncol. 2022, 15, 94. [Google Scholar] [CrossRef] [PubMed]
- Khozin, S.; Blumenthal, G.M.; Jiang, X.; He, K.; Boyd, K.; Murgo, A.; Justice, R.; Keegan, P.; Pazdur, R. US Food and Drug Administration approval summary: Erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist 2014, 19, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Mobocertinib: First Approval. Drugs 2021, 81, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L. Osimertinib: First global approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. FDA Approves Osimertinib for First-Line Treatment of Metastatic NSCLC with Most Common EGFR Mutations; Food and Drug Administration: White Oak, MD, USA, 2018.
- Koch, A.L.; Vellanki, P.J.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Shen, Y.L.; Xia, H.; Li, Y.; Liu, J.; Zirkelbach, J.F. FDA Approval Summary: Osimertinib for Adjuvant Treatment of Surgically Resected Non–Small Cell Lung Cancer, a Collaborative Project Orbis ReviewFDA Approval: Adjuvant Osimertinib for EGFR-Mutated NSCLC. Clin. Cancer Res. 2021, 27, 6638–6643. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.; Xu, R.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shi, J.; Wang, X.; Zhou, L.; Wang, Q.; Xie, Y.; Peng, C.; Kuang, L.; Yang, D.; Yang, J.; et al. An antioxidant feedforward cycle coordinated by linker histone variant H1.2 and NRF2 that drives nonsmall cell lung cancer progression. Proc. Natl. Acad. Sci. USA 2023, 120, e2306288120. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Shcherba, M.; Pendurti, G.; Liang, Y.; Piperdi, B.; Perez-Soler, R. Targeting the PI3K/AKT/mTOR pathway: Potential for lung cancer treatment. Lung Cancer Manag. 2014, 3, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.T.; Rose, M.; Boucher, D.; Plowman, J.; Molloy, C.; Fisher, M.; O’Leary, C.; Richard, D.J.; O’Byrne, K.J.; Bolderson, E. The Therapeutic Potential of DNA Damage Repair Pathways and Genomic Stability in Lung Cancer. Front. Oncol. 2020, 10, 1256. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Fan, S.; Wu, M.; Zuo, Z.; Li, X.; Jiang, L.; Shen, Q.; Xu, P.; Zeng, L.; Zhou, Y.; et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat. Commun. 2019, 10, 4892. [Google Scholar] [CrossRef] [PubMed]
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernández-Pérez, D.; Isella, C.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M. Increased lactate secretion by cancer cells sustains non-cell-autonomous adaptive resistance to MET and EGFR targeted therapies. Cell Metab. 2018, 28, 848–865.e6. [Google Scholar] [CrossRef]
- Keam, B.; Lee, S.J.; Kim, T.M.; Paeng, J.C.; Lee, S.H.; Kim, D.W.; Jeon, Y.K.; Chung, D.H.; Kang, K.W.; Chung, J.K.; et al. Total Lesion Glycolysis in Positron Emission Tomography Can Predict Gefitinib Outcomes in Non-Small-Cell Lung Cancer with Activating EGFR Mutation. J. Thorac. Oncol. 2015, 10, 1189–1194. [Google Scholar] [CrossRef]
- Suzuki, S.; Okada, M.; Takeda, H.; Kuramoto, K.; Sanomachi, T.; Togashi, K.; Seino, S.; Yamamoto, M.; Yoshioka, T.; Kitanaka, C. Involvement of GLUT1-mediated glucose transport and metabolism in gefitinib resistance of non-small-cell lung cancer cells. Oncotarget 2018, 9, 32667–32679. [Google Scholar] [CrossRef]
- Wang, J.; Ye, C.; Chen, C.; Xiong, H.; Xie, B.; Zhou, J.; Chen, Y.; Zheng, S.; Wang, L. Glucose transporter GLUT1 expression and clinical outcome in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 16875–16886. [Google Scholar] [CrossRef]
- Kim, S.M.; Yun, M.R.; Hong, Y.K.; Solca, F.; Kim, J.H.; Kim, H.J.; Cho, B.C. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol. Cancer Ther. 2013, 12, 2145–2156. [Google Scholar] [CrossRef]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef] [PubMed]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, D.; Chen, X.; He, L.; Li, T.; Xu, X.; Li, M. Nuclear PKM2 contributes to gefitinib resistance via upregulation of STAT3 activation in colorectal cancer. Sci. Rep. 2015, 5, 16082. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Lee, E.J.; Park, W.; Ha, K.T.; Chung, H.S. Natural compounds as lactate dehydrogenase inhibitors: Potential therapeutics for lactate dehydrogenase inhibitors-related diseases. Front. Pharmacol. 2023, 14, 1275000. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Q.; Fu, Y.L.; Zhang, J.; Zhang, K.Y.; Ma, J.; Tang, J.Y.; Zhang, Z.W.; Zhou, Z.Y. Targeting glycolysis in non-small cell lung cancer: Promises and challenges. Front. Pharmacol. 2022, 13, 1037341. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chen, H.; Han, R.; Li, L.; Lu, C.; Hao, S.; Wang, Y.; He, Y. Hexokinases II-mediated glycolysis governs susceptibility to crizotinib in ALK-positive non-small cell lung cancer. Thorac. Cancer 2021, 12, 3184–3193. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, S.L.; Hu, X.; Tam, K.Y. Inhibition of pyruvate dehydrogenase kinase 1 enhances the anti-cancer effect of EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Eur. J. Pharmacol. 2018, 838, 41–52. [Google Scholar] [CrossRef]
- Ma, R.; Li, X.; Gong, S.; Ge, X.; Zhu, T.; Ge, X.; Weng, L.; Tao, Q.; Guo, J. Dual Roles of Lactate in EGFR-TKI-Resistant Lung Cancer by Targeting GPR81 and MCT1. J. Oncol. 2022, 2022, 3425841. [Google Scholar] [CrossRef]
- Jeoung, N.H. Pyruvate dehydrogenase kinases: Therapeutic targets for diabetes and cancers. Diabetes Metab. J. 2015, 39, 188. [Google Scholar] [CrossRef]
- Schell, J.C.; Olson, K.A.; Jiang, L.; Hawkins, A.J.; Van Vranken, J.G.; Xie, J.; Egnatchik, R.A.; Earl, E.G.; DeBerardinis, R.J.; Rutter, J. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol. Cell 2014, 56, 400–413. [Google Scholar] [CrossRef]
- Liu, T.; Yin, H. PDK1 promotes tumor cell proliferation and migration by enhancing the Warburg effect in non-small cell lung cancer. Oncol. Rep. 2017, 37, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and Mitochondrial Respiration: A Neglected ConnectionMitochondrial Role in Tumor Progression. Cancer Res. 2015, 75, 3687–3691. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Wei, S.; Kim, B.S.; Kim, B.; Bae, S.J.; Chae, Y.C.; Ryu, D.; Ha, K.T. Diversity and complexity of cell death: A historical review. Exp. Mol. Med. 2023, 55, 1573–1594. [Google Scholar] [CrossRef] [PubMed]
- Steussy, C.N.; Popov, K.M.; Bowker-Kinley, M.M.; Sloan, R.B.; Harris, R.A.; Hamilton, J.A. Structure of pyruvate dehydrogenase kinase: Novel folding pattern for a serine protein kinase. J. Biol. Chem. 2001, 276, 37443–37450. [Google Scholar] [CrossRef]
- Michelakis, E.; Webster, L.; Mackey, J. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef]
- Yang, Z.; Tam, K.Y. Anti-cancer synergy of dichloroacetate and EGFR tyrosine kinase inhibitors in NSCLC cell lines. Eur. J. Pharmacol. 2016, 789, 458–467. [Google Scholar] [CrossRef]
- Dyrstad, S.E.; Lotsberg, M.L.; Tan, T.Z.; Pettersen, I.K.; Hjellbrekke, S.; Tusubira, D.; Engelsen, A.S.; Daubon, T.; Mourier, A.; Thiery, J.P. Blocking aerobic glycolysis by targeting pyruvate dehydrogenase kinase in combination with EGFR TKI and ionizing radiation increases therapeutic effect in non-small cell lung cancer cells. Cancers 2021, 13, 941. [Google Scholar] [CrossRef]
- Xu, H.; He, Y.; Ma, J.; Zhao, Y.; Liu, Y.; Sun, L.; Su, J. Inhibition of pyruvate dehydrogenase kinase-1 by dicoumarol enhances the sensitivity of hepatocellular carcinoma cells to oxaliplatin via metabolic reprogramming. Int. J. Oncol. 2020, 57, 733–742. [Google Scholar] [CrossRef]
- Tambe, Y.; Terado, T.; Kim, C.J.; Mukaisho, K.I.; Yoshida, S.; Sugihara, H.; Tanaka, H.; Chida, J.; Kido, H.; Yamaji, K. Antitumor activity of potent pyruvate dehydrogenase kinase 4 inhibitors from plants in pancreatic cancer. Mol. Carcinog. 2019, 58, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-Y.; Li, Y.; Jiang, D.; Zhao, J.; Ge, J.-F. Anticancer effect and apoptosis induction by quercetin in the human lung cancer cell line A-549. Mol. Med. Rep. 2012, 5, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhuang, M.; Zhong, C.; Peng, J.; Wang, X.; Li, J.; Chen, Z.; Huang, Y. Baicalein reverses hypoxia-induced 5-FU resistance in gastric cancer AGS cells through suppression of glycolysis and the PTEN/Akt/HIF-1alpha signaling pathway. Oncol. Rep. 2015, 33, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Ding, X.; Wu, J.; Liu, S.; Sun, W.; Nie, M.; Pan, X.; Zou, X. beta-Asarone Increases Chemosensitivity by Inhibiting Tumor Glycolysis in Gastric Cancer. Evid. Based Complement Altern. Med. 2020, 2020, 6981520. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Jin, J.M.; Liang, X.H.; Yu, M.Z.; Yang, C.; Huang, F.; Wu, H.; Zhang, B.B.; Fei, X.Y.; Wang, Z.T.; et al. Helichrysetin inhibits gastric cancer growth by targeting c-Myc/PDHK1 axis-mediated energy metabolism reprogramming. Acta Pharmacol. Sin. 2022, 43, 1581–1593. [Google Scholar] [CrossRef]
- Jiao, L.; Wang, S.; Zheng, Y.; Wang, N.; Yang, B.; Wang, D.; Yang, D.; Mei, W.; Zhao, Z.; Wang, Z. Betulinic acid suppresses breast cancer aerobic glycolysis via caveolin-1/NF-kappaB/c-Myc pathway. Biochem. Pharmacol. 2019, 161, 149–162. [Google Scholar] [CrossRef]
- Jin, J.; Qiu, S.; Wang, P.; Liang, X.; Huang, F.; Wu, H.; Zhang, B.; Zhang, W.; Tian, X.; Xu, R.; et al. Cardamonin inhibits breast cancer growth by repressing HIF-1alpha-dependent metabolic reprogramming. J. Exp. Clin. Cancer Res. 2019, 38, 377. [Google Scholar] [CrossRef]
- Aicher, T.D.; Damon, R.E.; Koletar, J.; Vinluan, C.C.; Brand, L.J.; Gao, J.; Shetty, S.S.; Kaplan, E.L.; Mann, W.R. Triterpene and diterpene inhibitors of pyruvate dehydrogenase kinase (PDK). Bioorganic Med. Chem. Lett. 1999, 9, 2223–2228. [Google Scholar] [CrossRef]
- Ha, K.-T. Composition for Preventing or Treating Cancer, and Containing Otobaphenol as Active Component. WO2018026210A1, 8 February 2018. [Google Scholar]
- Dahiya, R.; Mohammad, T.; Roy, S.; Anwar, S.; Gupta, P.; Haque, A.; Khan, P.; Kazim, S.N.; Islam, A.; Ahmad, F. Investigation of inhibitory potential of quercetin to the pyruvate dehydrogenase kinase 3: Towards implications in anticancer therapy. Int. J. Biol. Macromol. 2019, 136, 1076–1085. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef]
- Livesey, A.; Garty, F.; Shipman, A.; Shipman, K. Lactate dehydrogenase in dermatology practice. Clin. Exp. Dermatol. 2020, 45, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Hols, P.; Ramos, A.; Hugenholtz, J.; Delcour, J.; de Vos, W.M.; Santos, H.; Kleerebezem, M. Acetate utilization in Lactococcus lactis deficient in lactate dehydrogenase: A rescue pathway for maintaining redox balance. J. Bacteriol. 1999, 181, 5521–5526. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.S.; Goldberg, E. Computational analyses of mammalian lactate dehydrogenases: Human, mouse, opossum and platypus LDHs. Comput. Biol. Chem. 2009, 33, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Urbańska, K.; Orzechowski, A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. Int. J. Mol. Sci. 2019, 20, 2085. [Google Scholar] [CrossRef] [PubMed]
- Kane, D.A. Lactate oxidation at the mitochondria: A lactate-malate-aspartate shuttle at work. Front. Neurosci. 2014, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Read, J.; Winter, V.; Eszes, C.; Sessions, R.; Brady, R. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins Struct. Funct. Bioinform. 2001, 43, 175–185. [Google Scholar] [CrossRef]
- Imagawa, T.; Yamamoto, E.; Sawada, M.; Okamoto, M.; Uehara, M. Expression of lactate dehydrogenase-A and-B messenger ribonucleic acids in chick glycogen body. Poult. Sci. 2006, 85, 1232–1238. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef]
- Klein, R.; Nagy, O.; Tóthová, C.; Chovanová, F. Clinical and Diagnostic Significance of Lactate Dehydrogenase and Its Isoenzymes in Animals. Vet. Med. Int. 2020, 2020, 5346483. [Google Scholar] [CrossRef]
- Yang, C.; Pan, R.Y.; Guan, F.; Yuan, Z. Lactate metabolism in neurodegenerative diseases. Neural Regen Res. 2024, 19, 69–74. [Google Scholar] [CrossRef]
- Zhu, W.; Ma, Y.; Guo, W.; Lu, J.; Li, X.; Wu, J.; Qin, P.; Zhu, C.; Zhang, Q. Serum Level of Lactate Dehydrogenase is Associated with Cardiovascular Disease Risk as Determined by the Framingham Risk Score and Arterial Stiffness in a Health-Examined Population in China. Int. J. Gen. Med. 2022, 15, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lu, W.; Garcia-Prieto, C.; Huang, P. The Warburg effect and its cancer therapeutic implications. J. Bioenerg. Biomembr. 2007, 39, 267–274. [Google Scholar] [CrossRef]
- Mishra, D.; Banerjee, D. Lactate Dehydrogenases as Metabolic Links between Tumor and Stroma in the Tumor Microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef] [PubMed]
- Inomata, M.; Hayashi, R.; Tanaka, H.; Shimokawa, K.; Tokui, K.; Taka, C.; Okazawa, S.; Kambara, K.; Ichikawa, T.; Yamada, T.; et al. Elevated levels of plasma lactate dehydrogenase is an unfavorable prognostic factor in patients with epidermal growth factor receptor mutation-positive non-small cell lung cancer, receiving treatment with gefitinib or erlotinib. Mol. Clin. Oncol. 2016, 4, 774–778. [Google Scholar] [CrossRef]
- Gong, T.; Liu, J.; Jiang, J.; Zhai, Y.F.; Wu, C.M.; Ma, C.; Wen, B.L.; Yan, X.Y.; Zhang, X.; Wang, D.M.; et al. The role of lactate deshydrogenase levels on non-small cell lung cancer prognosis: A meta-analysis. Cell. Mol. Biol. (Noisy-Le-Grand) 2019, 65, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Korga, A.; Ostrowska, M.; Jozefczyk, A.; Iwan, M.; Wojcik, R.; Zgorka, G.; Herbet, M.; Vilarrubla, G.G.; Dudka, J. Apigenin and hesperidin augment the toxic effect of doxorubicin against HepG2 cells. BMC Pharmacol. Toxicol. 2019, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Li, J.; Li, Y.; Zhang, Y.; Du, Q.; Hao, P.; Li, J.; Cao, X.; Li, L. Berberine protects against simulated ischemia/reperfusion injury-induced H9C2 cardiomyocytes apoptosis in vitro and myocardial ischemia/reperfusion-induced apoptosis in vivo by regulating the mitophagy-mediated HIF-1α/BNIP3 pathway. Front. Pharmacol. 2020, 11, 367. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.P.; Yun, H.J.; Choi, J.H.; Han, E.H.; Kim, H.G.; Song, G.Y.; Kwon, K.i.; Jeong, T.C.; Jeong, H.G. Suppression of EGF-induced tumor cell migration and matrix metalloproteinase-9 expression by capsaicin via the inhibition of EGFR-mediated FAK/Akt, PKC/Raf/ERK, p38 MAPK, and AP-1 signaling. Mol. Nutr. Food Res. 2011, 55, 594–605. [Google Scholar] [CrossRef]
- Chen, X.Q.; Hu, T.; Han, Y.; Huang, W.; Yuan, H.B.; Zhang, Y.T.; Du, Y.; Jiang, Y.W. Preventive Effects of Catechins on Cardiovascular Disease. Molecules 2016, 21, 1759. [Google Scholar] [CrossRef]
- Wang, K.; Fan, H.; Chen, Q.; Ma, G.; Zhu, M.; Zhang, X.; Zhang, Y.; Yu, J. Curcumin inhibits aerobic glycolysis and induces mitochondrial-mediated apoptosis through hexokinase II in human colorectal cancer cells in vitro. Anticancer Drugs 2015, 26, 15–24. [Google Scholar] [CrossRef]
- Chen, P.; Huang, H.P.; Wang, Y.; Jin, J.; Long, W.G.; Chen, K.; Zhao, X.H.; Chen, C.G.; Li, J. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J. Exp. Clin. Cancer Res. 2019, 38, 254. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, Z.; Zhu, F.; Fan, X.; Wu, X.; Zhao, H.; Jiang, L. Curcumin lowers erlotinib resistance in non-small cell lung carcinoma cells with mutated EGF receptor. Oncol. Res. 2013, 21, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.Y.; Zhang, L.; Yee, J.K.; Go, V.W.; Lee, W.N. Metabolic Consequences of LDHA inhibition by Epigallocatechin Gallate and Oxamate in MIA PaCa-2 Pancreatic Cancer Cells. Metabolomics 2015, 11, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.; Tighiouart, M.; Lee, J.E.; Shin, H.J.; Khuri, F.R.; Yang, C.S.; Chen, Z.; Shin, D.M. Synergistic inhibition of head and neck tumor growth by green tea (−)-epigallocatechin-3-gallate and EGFR tyrosine kinase inhibitor. Int. J. Cancer 2008, 123, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Rahman, M.A.; Chen, Z.G.; Saba, N.F.; Khuri, F.R.; Shin, D.M.; Ruhul Amin, A.R. Combination of erlotinib and EGCG induces apoptosis of head and neck cancers through posttranscriptional regulation of Bim and Bcl-2. Apoptosis 2015, 20, 986–995. [Google Scholar] [CrossRef]
- Grosse, F.; Nasheuer, H.P.; Scholtissek, S.; Schomburg, U. Lactate dehydrogenase and glyceraldehyde-phosphate dehydrogenase are single-stranded DNA-binding proteins that affect the DNA-polymerase-alpha-primase complex. Eur. J. Biochem. 1986, 160, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Fiume, L.; Vettraino, M.; Carnicelli, D.; Arfilli, V.; Di Stefano, G.; Brigotti, M. Galloflavin prevents the binding of lactate dehydrogenase A to single stranded DNA and inhibits RNA synthesis in cultured cells. Biochem. Biophys. Res. Commun. 2013, 430, 466–469. [Google Scholar] [CrossRef]
- Liu, X.; Pan, L.; Chen, P.; Zhu, Y. Leonurine improves ischemia-induced myocardial injury through antioxidative activity. Phytomedicine 2010, 17, 753–759. [Google Scholar] [CrossRef]
- Maurya, A.K.; Vinayak, M. Quercetin regresses Dalton’s lymphoma growth via suppression of PI3K/AKT signaling leading to upregulation of p53 and decrease in energy metabolism. Nutr. Cancer 2015, 67, 354–363. [Google Scholar] [CrossRef]
- Mlala, S.; Oyedeji, A.O.; Gondwe, M.; Oyedeji, O.O. Ursolic acid and its derivatives as bioactive agents. Molecules 2019, 24, 2751. [Google Scholar] [CrossRef]
- Wang, S.; Chang, X.; Zhang, J.; Li, J.; Wang, N.; Yang, B.; Pan, B.; Zheng, Y.; Wang, X.; Ou, H. Ursolic acid inhibits breast cancer metastasis by suppressing glycolytic metabolism via activating sp1/caveolin-1 signaling. Front. Oncol. 2021, 11, 745584. [Google Scholar] [CrossRef] [PubMed]
- Sikander, M.; Malik, S.; Chauhan, N.; Khan, P.; Kumari, S.; Kashyap, V.K.; Khan, S.; Ganju, A.; Halaweish, F.T.; Yallapu, M.M. Cucurbitacin D reprograms glucose metabolic network in prostate cancer. Cancers 2019, 11, 364. [Google Scholar] [CrossRef] [PubMed]
- Yar Saglam, A.; Alp, E.; Elmazoglu, Z.; Menevse, S. Treatment with cucurbitacin B alone and in combination with gefitinib induces cell cycle inhibition and apoptosis via EGFR and JAK/STAT pathway in human colorectal cancer cell lines. Hum. Exp. Toxicol. 2016, 35, 526–543. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Dai, W.; Zhang, Q.; Feng, J.; Wu, L.; Liu, T.; Yu, Q.; Xu, S.; Wang, W. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br. J. Cancer 2017, 117, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Keyhanmanesh, R.; Saadat, S.; Mohammadi, M.; Shahbazfar, A.A.; Fallahi, M. The protective effect of α-hederin, the active constituent of Nigella sativa, on lung inflammation and blood cytokines in ovalbumin sensitized Guinea pigs. Phytother. Res. 2015, 29, 1761–1767. [Google Scholar] [CrossRef]
- Fang, C.; Liu, Y.; Chen, L.; Luo, Y.; Cui, Y.; Zhang, N.; Liu, P.; Zhou, M.; Xie, Y. α-Hederin inhibits the growth of lung cancer A549 cells in vitro and in vivo by decreasing SIRT6 dependent glycolysis. Pharm. Biol. 2021, 59, 11–20. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, W.; Huang, S.; Ni, W.; Wei, Z.; Cao, Y.; Yu, S.; Jia, Q.; Wu, Y.; Chai, C. Beta-elemene inhibits breast cancer metastasis through blocking pyruvate kinase M2 dimerization and nuclear translocation. J. Cell. Mol. Med. 2019, 23, 6846–6858. [Google Scholar] [CrossRef]
- Li, J.; Dai, P.; Sun, J.; Yu, W.; Han, W.; Li, K. FBP1 induced by β-elemene enhances the sensitivity of gefitinib in lung cancer. Thorac. Cancer 2023, 14, 371–380. [Google Scholar] [CrossRef]
- Park, M.K.; Ji, J.; Haam, K.; Han, T.-H.; Lim, S.; Kang, M.-J.; Lim, S.S.; Ban, H.S. Licochalcone A inhibits hypoxia-inducible factor-1α accumulation by suppressing mitochondrial respiration in hypoxic cancer cells. Biomed. Pharmacother. 2021, 133, 111082. [Google Scholar] [CrossRef]
- Han, S.; Li, X.; Gan, Y.; Li, W. Licochalcone A promotes the ubiquitination of c-met to abrogate gefitinib resistance. BioMed Res. Int. 2022, 2022, 5687832. [Google Scholar] [CrossRef]
- Yoon, Y.; Kim, Y.-O.; Jeon, W.-K.; Park, H.-J.; Sung, H.J. Tanshinone IIA isolated from Salvia miltiorrhiza BUNGE induced apoptosis in HL60 human premyelocytic leukemia cell line. J. Ethnopharmacol. 1999, 68, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gao, F.; Zhao, Q.; Zuo, H.; Liu, W.; Li, W. Tanshinone IIA inhibits oral squamous cell carcinoma via reducing Akt-c-Myc signaling-mediated aerobic glycolysis. Cell Death Dis. 2020, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sung, B.; Kang, Y.J.; Hwang, S.Y.; Kim, M.J.; Yoon, J.H.; Im, E.; Kim, N.D. Sulforaphane inhibits hypoxia-induced HIF-1α and VEGF expression and migration of human colon cancer cells. Int. J. Oncol. 2015, 47, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; He, C.; Zheng, S.; Wu, C.; Ren, M.; Shan, Y. AKT1/HK2 Axis-mediated Glucose Metabolism: A Novel Therapeutic Target of Sulforaphane in Bladder Cancer. Mol. Nutr. Food Res. 2022, 66, e2100738. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Yu, Z.-Y.; Chuang, Y.-S.; Huang, R.-M.; Wang, T.-C.V. Sulforaphane attenuates EGFR signaling in NSCLC cells. J. Biomed. Sci. 2015, 22, 38. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Li, M.; Liu, W.-B.; Zhou, Z.-S.; Zhang, R.; Li, J.-L.; Zhou, K.-C. Epigallocatechin gallate inhibits human tongue carcinoma cells via HK2-mediated glycolysis. Oncol. Rep. 2015, 33, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Liang, Y.C.; Lin-shiau, S.Y.; Chen, C.F.; Lin, J.K. Suppression of extracellular signals and cell proliferation through EGF receptor binding by (−)-epigallocatechin gallate in human A431 epidermoid carcinoma cells. J. Cell Biochem. 1997, 67, 55–65. [Google Scholar] [CrossRef]
- Adachi, S.; Nagao, T.; To, S.; Joe, A.K.; Shimizu, M.; Matsushima-Nishiwaki, R.; Kozawa, O.; Moriwaki, H.; Maxfield, F.R.; Weinstein, I.B. (−)-Epigallocatechin gallate causes internalization of the epidermal growth factor receptor in human colon cancer cells. Carcinogenesis 2008, 29, 1986–1993. [Google Scholar] [CrossRef]
- Meng, J.; Chang, C.; Chen, Y.; Bi, F.; Ji, C.; Liu, W. EGCG overcomes gefitinib resistance by inhibiting autophagy and augmenting cell death through targeting ERK phosphorylation in NSCLC. OncoTargets Ther. 2019, 12, 6033. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Labrie, M.; Brugge, J.S.; Mills, G.B.; Zervantonakis, I.K. Therapy resistance: Opportunities created by adaptive responses to targeted therapies in cancer. Nat. Rev. Cancer 2022, 22, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Wu, J.; Liu, B. Therapeutic strategies of dual-target small molecules to overcome drug resistance in cancer therapy. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2023, 1878, 188866. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Cho, M.; Kim, B.-S.; Han, J.H.; Park, S.; Lee, I.-K.; Ryu, D.; Kim, J.H.; Bae, S.-J.; Ha, K.-T. Drug evaluation based on phosphomimetic PDHA1 reveals the complexity of activity-related cell death in A549 non-small cell lung cancer cells. BMB Rep. 2021, 54, 563. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Roy, S.; Kumaravel, S.; Sharma, A.; Duran, C.L.; Bayless, K.J.; Chakraborty, S. Hypoxic tumor microenvironment: Implications for cancer therapy. Exp. Biol. Med. 2020, 245, 1073–1086. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Demain, A.L.; Vaishnav, P. Natural products for cancer chemotherapy. Microb. Biotechnol. 2011, 4, 687–699. [Google Scholar] [CrossRef]
- Tan, G.; Gyllenhaal, C.; Soejarto, D.D. Biodiversity as a source of anticancer drugs. Curr. Drug Targets 2006, 7, 265–277. [Google Scholar] [CrossRef]
- Rahmani, A.H.; Babiker, A.Y.; Anwar, S. Hesperidin, a Bioflavonoid in Cancer Therapy: A Review for a Mechanism of Action through the Modulation of Cell Signaling Pathways. Molecules 2023, 28, 5152. [Google Scholar] [CrossRef] [PubMed]
- Yenigun, O.M.; Thanassi, M. Capsaicin: An Uncommon Exposure and Unusual Treatment. Clin. Pract. Cases Emerg. Med. 2019, 3, 219–221. [Google Scholar] [CrossRef]
- Mereles, D.; Hunstein, W. Epigallocatechin-3-gallate (EGCG) for clinical trials: More pitfalls than promises? Int. J. Mol. Sci. 2011, 12, 5592–5603. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E.; et al. The Therapeutic Potential of Apigenin. Int. J. Mol. Sci. 2019, 20, 1305. [Google Scholar] [CrossRef]
- Lozanovski, V.J.; Polychronidis, G.; Gross, W.; Gharabaghi, N.; Mehrabi, A.; Hackert, T.; Schemmer, P.; Herr, I. Broccoli sprout supplementation in patients with advanced pancreatic cancer is difficult despite positive effects-results from the POUDER pilot study. Investig. New Drugs 2020, 38, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Ouyang, Y.; Kong, Y.; Min, Y.; Xiao, J.; Li, S.; Zhou, M.; Feng, N.; Zhang, L. Catechin Inhibits the Release of Advanced Glycation End Products during Glycated Bovine Serum Albumin Digestion and Corresponding Mechanisms In Vitro. J. Agric. Food Chem. 2021, 69, 8807–8818. [Google Scholar] [CrossRef]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A Review of Its Effects on Human Health. Foods 2017, 6, 92. [Google Scholar] [CrossRef]
- Xing, L.; Tan, Z.R.; Cheng, J.L.; Huang, W.H.; Zhang, W.; Deng, W.; Yuan, C.S.; Zhou, H.H. Bioavailability and pharmacokinetic comparison of tanshinones between two formulations of Salvia miltiorrhiza in healthy volunteers. Sci. Rep. 2017, 7, 4709. [Google Scholar] [CrossRef]
- Solnier, J.; Zhang, Y.; Kuo, Y.C.; Du, M.; Roh, K.; Gahler, R.; Wood, S.; Chang, C. Characterization and Pharmacokinetic Assessment of a New Berberine Formulation with Enhanced Absorption In Vitro and in Human Volunteers. Pharmaceutics 2023, 15, 2567. [Google Scholar] [CrossRef]
- Song, M.; Lee, D.; Lee, T.; Lee, S. Determination of leelamine in mouse plasma by LC-MS/MS and its pharmacokinetics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 931, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-L.; Qin, Z.-M.; Cai, H.-D.; Tan, Y.-F.; Zhang, X.-P.; Luo, Y.-C.; Li, B.; Chen, F.; Zhang, J.-Q. Determination of α-hederin in rat plasma using liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) and its application to a pharmacokinetic study. Anal. Methods 2015, 7, 2155–2161. [Google Scholar] [CrossRef]
- Hao, H.; Wang, G.; Cui, N.; Li, J.; Xie, L.; Ding, Z. Pharmacokinetics, absorption and tissue distribution of tanshinone IIA solid dispersion. Planta Med. 2006, 72, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhao, W.; Wang, X.; Sun, Y.; Chen, X. A pharmacological review of dicoumarol: An old natural anticoagulant agent. Pharmacol. Res. 2020, 160, 105193. [Google Scholar] [CrossRef] [PubMed]
- Jinhua, W. Ursolic acid: Pharmacokinetics process in vitro and in vivo, a mini review. Arch. Pharm. 2019, 352, e1800222. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kulkarni, K.; Zhu, W.; Hu, M. Bioavailability and pharmacokinetics of genistein: Mechanistic studies on its ADME. Anticancer. Agents Med. Chem. 2012, 12, 1264–1280. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Gong, T.; Liu, M.; Ren, S.; Yang, H.; Zeng, S.; Zhao, H.; Chen, L.; Ming, T.; Meng, X.; et al. Shikonin, a naphthalene ingredient: Therapeutic actions, pharmacokinetics, toxicology, clinical trials and pharmaceutical researches. Phytomedicine 2022, 94, 153805. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Zeng, Y.; Zeng, Z.; Zhang, N.; Li, C.; Zeng, Y.; You, Y.; Wang, S.; Chen, X.; Sui, X.; et al. Drug delivery systems for elemene, its main active ingredient β-elemene, and its derivatives in cancer therapy. Int. J. Nanomed. 2018, 13, 6279–6296. [Google Scholar] [CrossRef]
- Li, T.; Ye, W.; Huang, B.; Lu, X.; Chen, X.; Lin, Y.; Wen, C.; Wang, X. Determination and pharmacokinetic study of echinatin by UPLC-MS/MS in rat plasma. J. Pharm. Biomed. Anal. 2019, 168, 133–137. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Z.; Yang, Y.; Adu-Frimpong, M.; Chen, L.; Ji, H.; Toreniyazov, E.; Wang, Q.; Yu, J.; Xu, X. Preparation, characterization, pharmacokinetics, and antirenal injury activity studies of Licochalcone A-loaded liposomes. J. Food Biochem. 2022, 46, e14007. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Structure | Drug Type | FDA Approval |
|---|---|---|---|
| Erlotinib (TarcevaTM) | 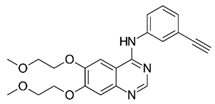 | 1st-generation EGFR-TKI | 1st-line, NSCLC with EGFR19D/EGFRL858R [74] |
| Gefitinib (IressaTM) | 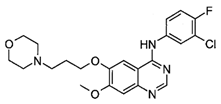 | 1st-generation EGFR-TKI | 1st-line, NSCLC with EGFR19D/EGFRL858R [28] |
| Afatinib (GilotrifTM) |  | 2nd-generation EGFR-TKI | 1st-line, NSCLC with EGFR19D/EGFRL858R [25] |
| Dacomitinib (VizimproTM) | 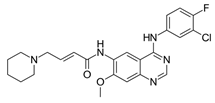 | 2nd-generation EGFR-TKI | 1st-line, NSCLC with EGFR19D/EGFRL858R [26] |
| Mobocertinib (ExkivityTM) | 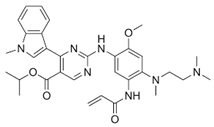 | 3rd-generation EGFR-TKI | NSCLC with EGFR exon20 insertion [75] |
| Osimertinib (TagrissoTM) | 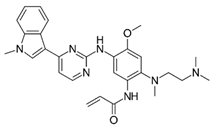 | 3rd-generation EGFR-TKI | 2nd-line, NSCLC with EGFRT790M [76] 1st-line, NSCLC with EGFR19D/EGFRL858R [77] adjuvant therapy for NSCLC [78] |
| PDK Inhibitor | Structure | Property | Origin | Clinical Trials for NSCLC | Reference |
|---|---|---|---|---|---|
| Cryptotanshinone | 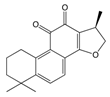 | IC50: PDK2 (11 µM), PDK4 (>30 µM) | Salvia miltiorrhiza | ND | [113] |
| Dicoumarol | 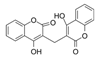 | IC50: PDK1 (19.42 μM) | Melilotus officinalis | ND | [112] |
| Hemistepsin A |  | ND | Hemistepta lyrate | ND | [18] |
| Huzhangoside A | 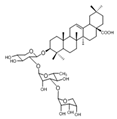 | ND | Anemone rivularis | ND | [19] |
| Ilimaquinone |  | ND | Smenospongia cerebriformis | ND | [20] |
| Leelamine |  | IC50: 9.5 µM | bark of pine trees | ND | [120] |
| Otobaphenol | 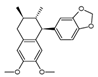 | ND | Myristica fragrans | ND | [121] |
| Quercetin | 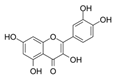 | IC50: PDK3 (~9.5 μM), | flavonoid glycosides from fruits and vegetables | ND | [122] |
| LDHA Inhibitor | Structure | Property | Origin | Clinical Trials for NSCLC | Reference |
|---|---|---|---|---|---|
| Apigenin | 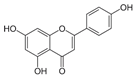 | IC50: LDHA (0.042 mM) | Flavonoid from fruits, vegetables, and herbs | ND | [139] |
| Berberine |  | ND | Goldenseal (Hydrastis canadensis) | NCT03486496 | [140] |
| Capsaicin |  | ND | Capsicum annuum | ND | [141] |
| Catechin | 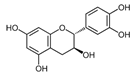 | IC50: LDHA (40.69 μM) | Camellia sinensis | NCT00573885 NCT00611650 | [14] |
| Curcumin | 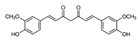 | ND | Curcuma longa | NCT02321293 NCT01048983 | [143] |
| Epigallocatechin gallate | 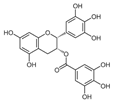 | ND | Camellia sinensis | ND | [146] |
| Galloflavin |  | IC50: LDHA (5.46 μM) | Flavonoid from food and vegetables | ND | [150] |
| Leonurine | 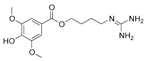 | ND | Leonurus cardiaca | ND | [151] |
| Quercetin |  | ND | Quercus, Flavonoid glycosides from fruits and vegetables | ND | [152] |
| Ursolic acid |  | ND | Triterpenoid from citrus fruits and vegetables | ND | [154] |
| Glycolysis Inhibitor | Structure | Effect | Origin | Clinical Trials for NSCLC | Reference |
|---|---|---|---|---|---|
| α-Hederin | 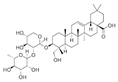 | GLUT1, PKM2, LDHA, and HK2↓ | Hedera helix | ND | [159] |
| β-elemene | 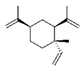 | PKM2↓ | Curcuma aromatica | ND | [160] |
| Cucurbitacin D | 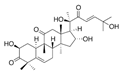 | Glut1↓ | Cucurbitaceae | ND | [155] |
| Epigallocatechin gallate | 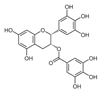 | HK2↓ | Green tea | ND | [169] |
| Genistein |  | HIF-1α, GLUT1, and HK2↓ | Lupin, fava beans, soybeans, kudzu, and psoralea | NCT01628471 NCT00769990 | [157] |
| Licochalcone A |  | HIF-1α, PDK1, and GLUT1↓ | Glycyrrhiza uralensis | ND | [162] |
| Shikonin | 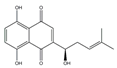 | PKM2↓ | lithospermum erythrorhizon | ND | [174] |
| Sulforaphane | 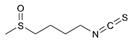 | HIF-1α, HK2, and PKM2↓ | Broccoli | ND | [166,167] |
| Tanshinone IIA | 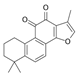 | HK2↓ | Salvia miltiorrhiza | ND | [165] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, W.; Han, J.H.; Wei, S.; Yang, E.-S.; Cheon, S.-Y.; Bae, S.-J.; Ryu, D.; Chung, H.-S.; Ha, K.-T. Natural Product-Based Glycolysis Inhibitors as a Therapeutic Strategy for Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2024, 25, 807. https://doi.org/10.3390/ijms25020807
Park W, Han JH, Wei S, Yang E-S, Cheon S-Y, Bae S-J, Ryu D, Chung H-S, Ha K-T. Natural Product-Based Glycolysis Inhibitors as a Therapeutic Strategy for Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2024; 25(2):807. https://doi.org/10.3390/ijms25020807
Chicago/Turabian StylePark, Wonyoung, Jung Ho Han, Shibo Wei, Eun-Sun Yang, Se-Yun Cheon, Sung-Jin Bae, Dongryeol Ryu, Hwan-Suck Chung, and Ki-Tae Ha. 2024. "Natural Product-Based Glycolysis Inhibitors as a Therapeutic Strategy for Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 25, no. 2: 807. https://doi.org/10.3390/ijms25020807
APA StylePark, W., Han, J. H., Wei, S., Yang, E.-S., Cheon, S.-Y., Bae, S.-J., Ryu, D., Chung, H.-S., & Ha, K.-T. (2024). Natural Product-Based Glycolysis Inhibitors as a Therapeutic Strategy for Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 25(2), 807. https://doi.org/10.3390/ijms25020807