
Development of Good Manufacturing Practice-Compatible Isolation and Culture Methods for Human Olfactory Mucosa-Derived Mesenchymal Stromal Cells

 , and
, and 
Abstract
1. Introduction
2. Results
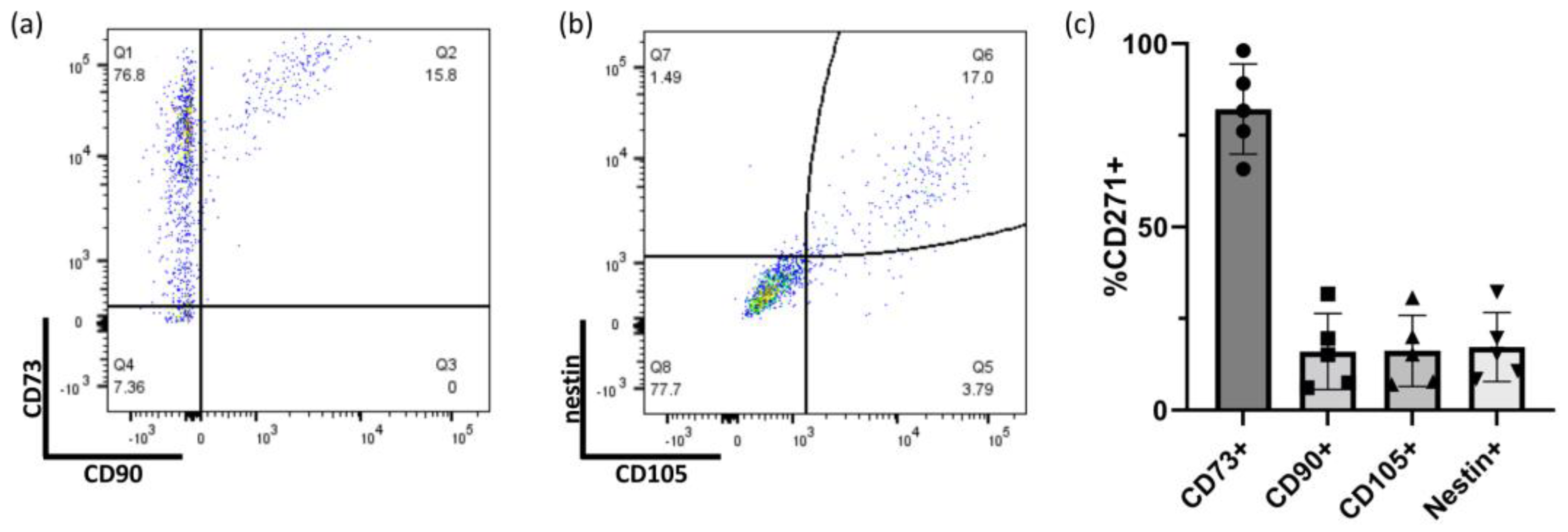
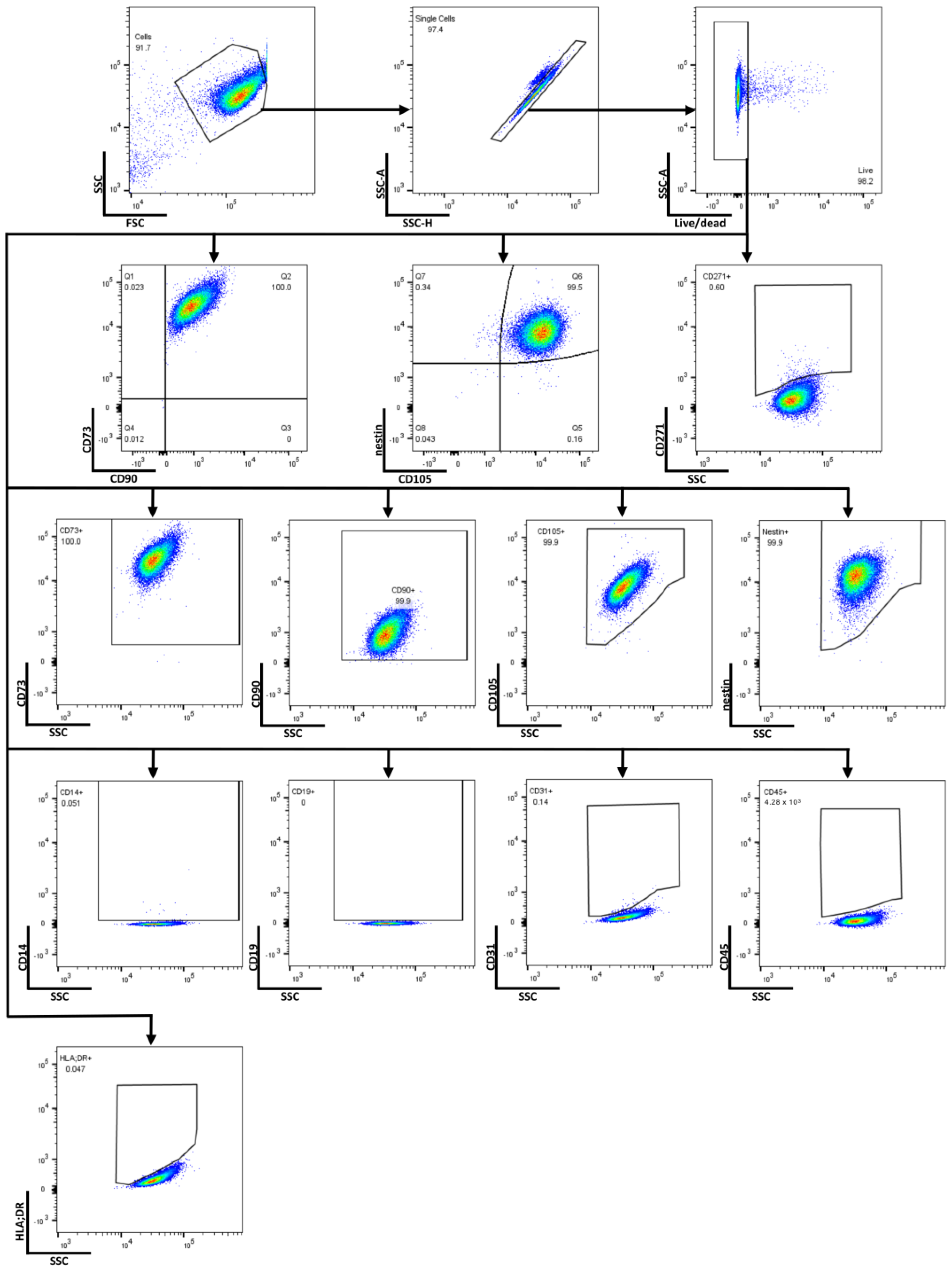
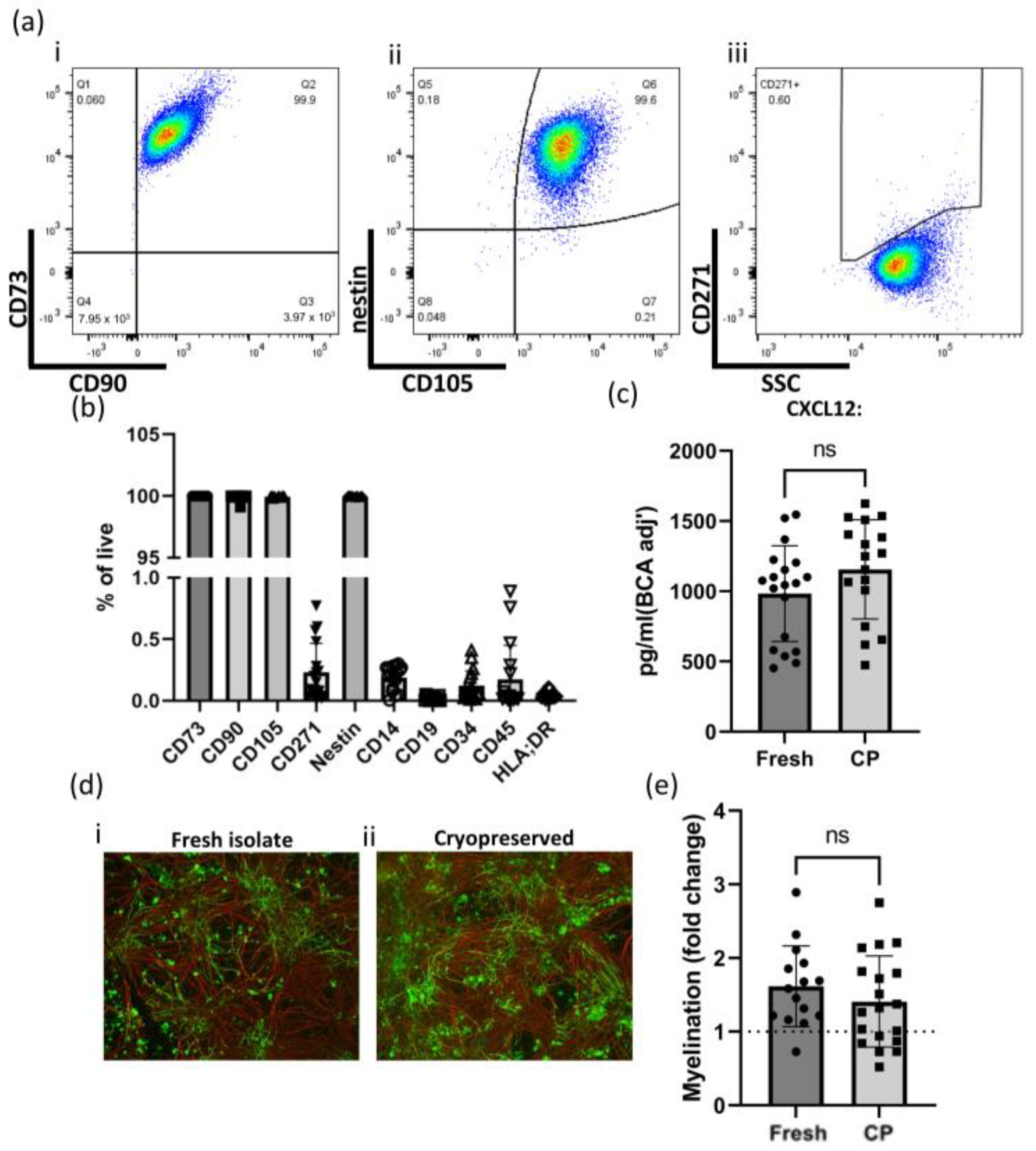
2.1. Determining MSC Identity of GMP-Compatible hOM-MSCs by Surface and Intracellular Marker Expression
2.2. Differentiation Capacity of GMP-Compatible hOM-MSCs
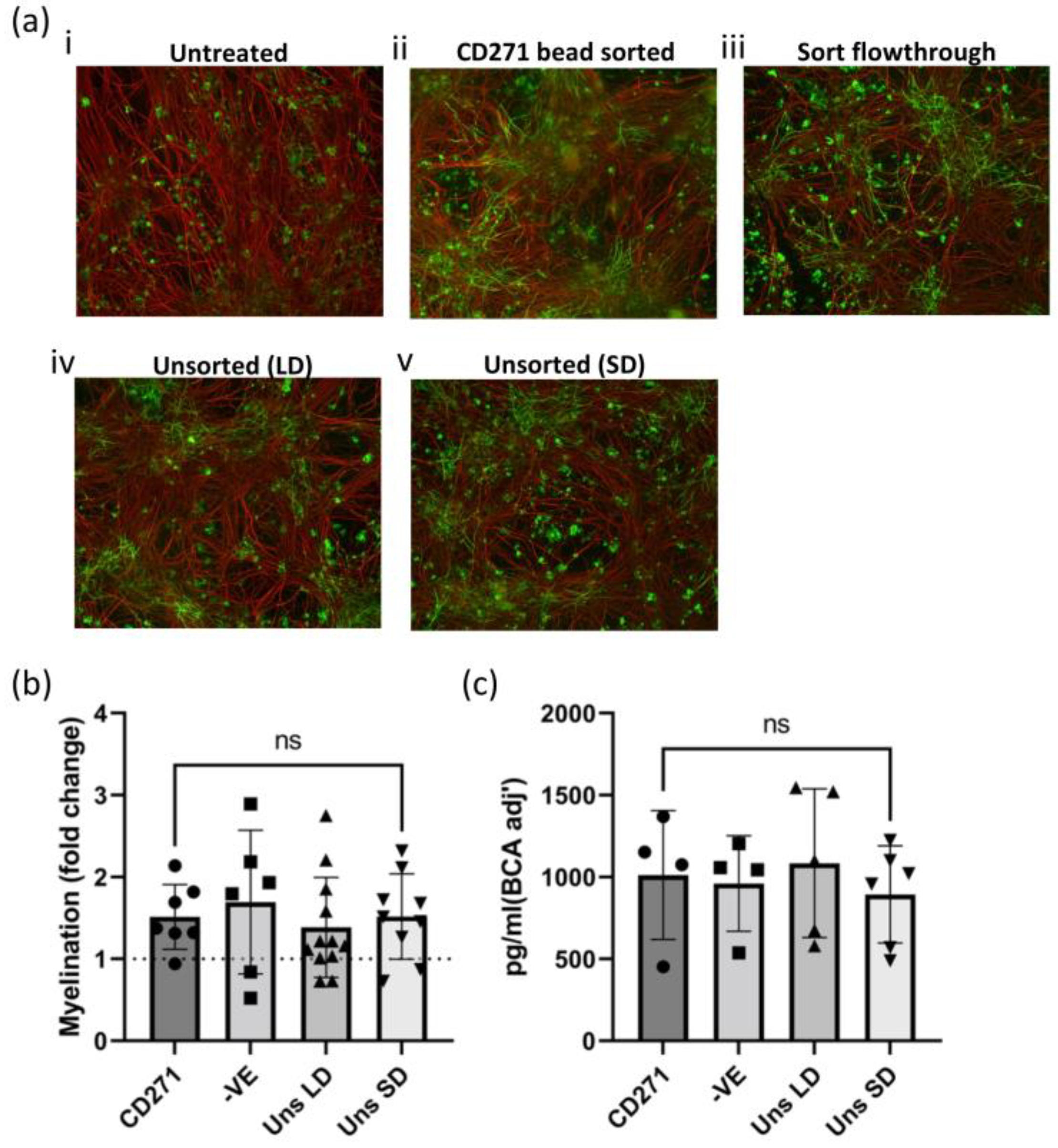
2.3. Characterization of the Pro-Myelinating Phenotype of GMP-Compatible hOM-MSCs In Vitro
2.4. Phenotypic Stability of GMP-Compatible hOM-MSCs Following Cryopreservation
2.5. Comparing hOM-MSCs Expanded with GMP-Compliant and Xeno-Derived Media Supplements
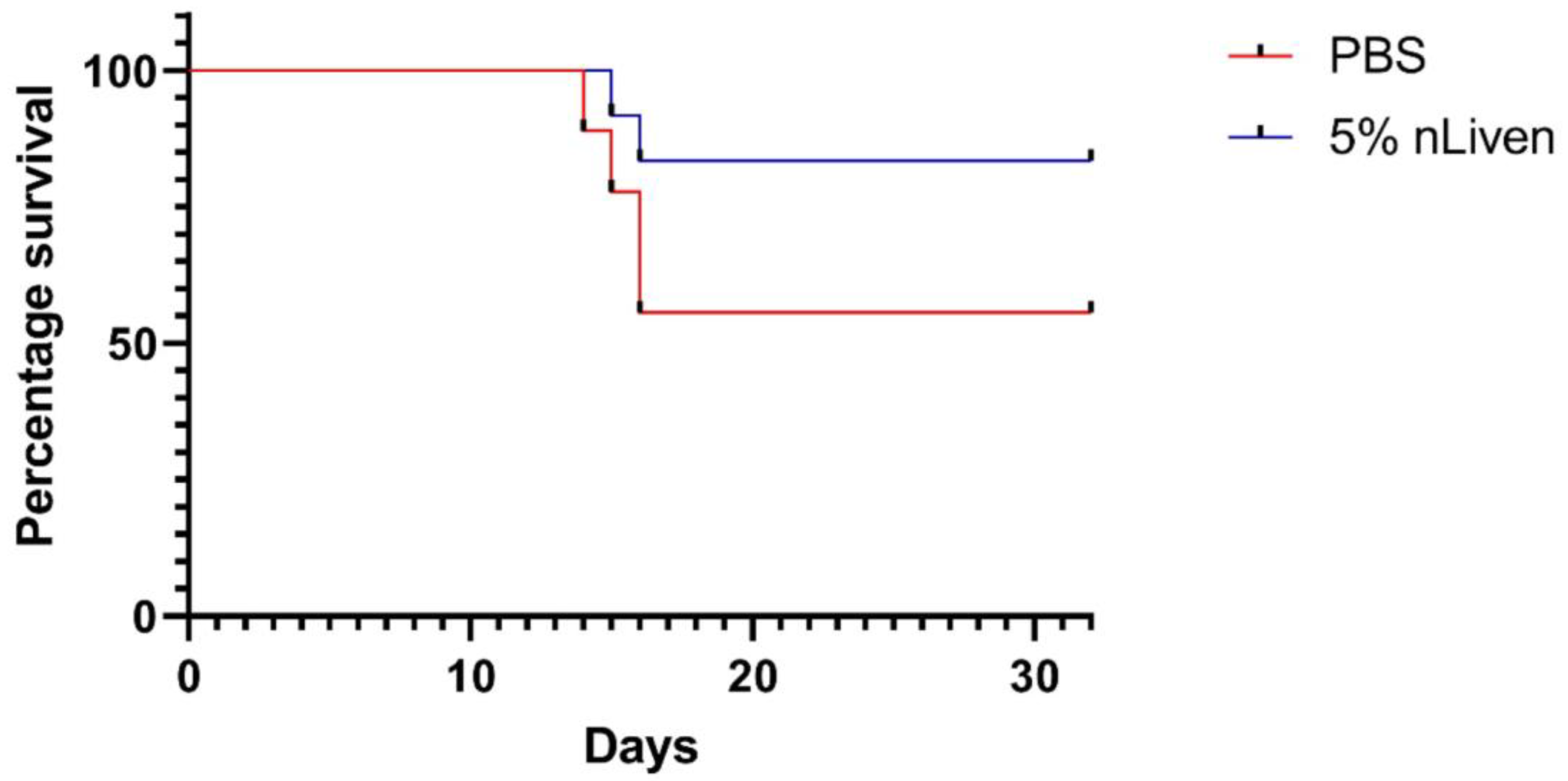
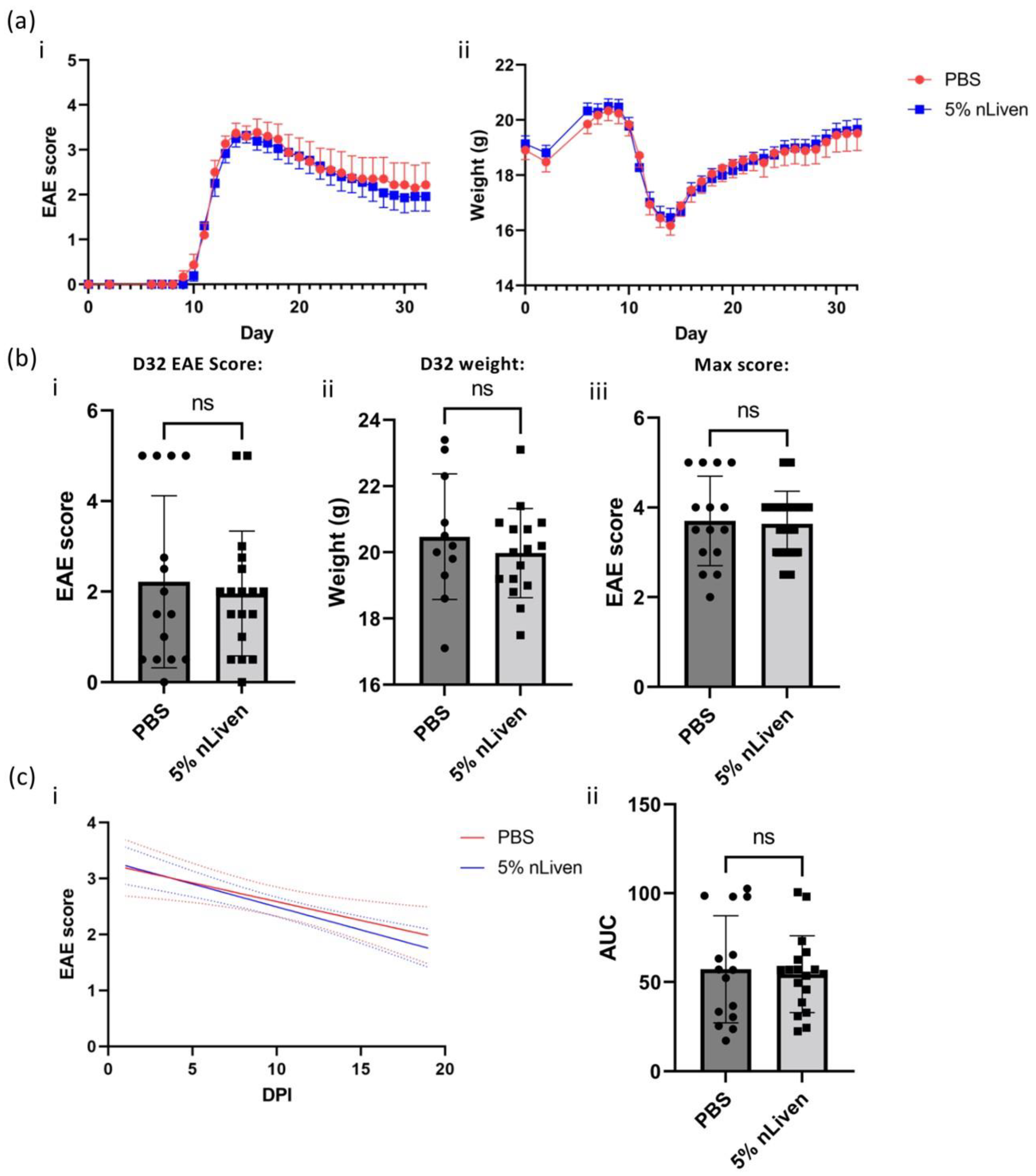
2.6. Evaluating In Vivo Efficacy of GMP-Compatible hOM-MSCs in the EAE Model
3. Discussion
4. Materials and Methods
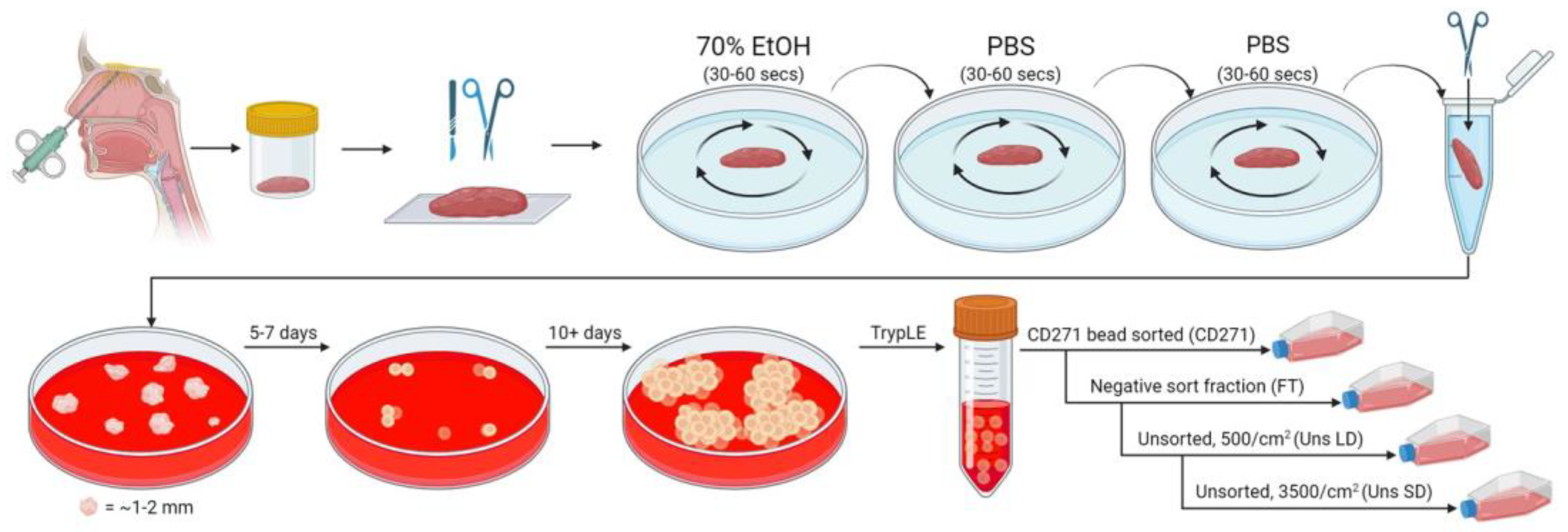
4.1. Nasal Biopsy Processing and hOM-MSC Cell Line Generation
4.2. Cell Culture
4.3. Flow Cytometry and Antibody Panel
4.4. Adipogenic/Osteogenic Differentiation
4.5. Oil Red O/Alizarin Red Staining
4.6. RNA Extractions, cDNA Conversions and qPCR Analysis
4.7. Conditioned Media and Myelinating Cultures
4.8. Immunocytochemistry
4.9. Microscopy and Image Analysis
4.10. Protein Extractions and ELISA Analysis
4.11. Cryopreservation and Recovery
4.12. EAE Induction
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Anjum, A.; Yazid, M.D.; Fauzi Daud, M.; Idris, J.; Ng, A.M.H.; Selvi Naicker, A.; Ismail, O.H.R.; Athi Kumar, R.K.; Lokanathan, Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. Int. J. Mol. Sci. 2020, 21, 7533. [Google Scholar] [CrossRef] [PubMed]
- Bhagavati, S. Autoimmune Disorders of the Nervous System: Pathophysiology, Clinical Features, and Therapy. Front. Neurol. 2021, 12, 664664. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, A.; Hadden, R.D.M.; Sarrigiannis, P.G.; Hadjivassiliou, M.; Zis, P. Pain in Chronic Inflammatory Demyelinating Polyradiculoneuropathy: A Systematic Review and Meta-Analysis. Pain Ther. 2019, 8, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Drulovic, J.; Basic-Kes, V.; Grgic, S.; Vojinovic, S.; Dincic, E.; Toncev, G.; Kezic, M.G.; Kisic-Tepavcevic, D.; Dujmovic, I.; Mesaros, S.; et al. The Prevalence of Pain in Adults with Multiple Sclerosis: A Multicenter Cross-Sectional Survey. Pain Med. 2015, 16, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Abakay, H.; Dogan, H.; Yetkin, M.F.; Guc, A.; Talay Calis, H.; Demir Caltekin, M. The effects of incontinence on functionality, disability, and quality of life in male and female patients with multiple sclerosis. Mult. Scler. Relat. Disord. 2022, 66, 104010. [Google Scholar] [CrossRef] [PubMed]
- Denys, P.; Castaño Botero, J.C.; Vita Nunes, R.L.; Wachs, B.; Mendes Gomes, C.; Krivoborodov, G.; Tu, L.M.; Del-Popolo, G.; Thompson, C.; Vilain, C.; et al. AbobotulinumtoxinA is effective in patients with urinary incontinence due to neurogenic detrusor overactivity regardless of spinal cord injury or multiple sclerosis etiology: Pooled analysis of two phase III randomized studies (CONTENT1 and CONTENT2). Neurourol. Urodyn. 2023, 42, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Bouhrara, M.; Reiter, D.A.; Bergeron, C.M.; Zukley, L.M.; Ferrucci, L.; Resnick, S.M.; Spencer, R.G. Evidence of demyelination in mild cognitive impairment and dementia using a direct and specific magnetic resonance imaging measure of myelin content. Alzheimer’s Dement. 2018, 14, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Schulz, D.; Kopp, B.; Kunkel, A.; Faiss, J.H. Cognition in the early stage of multiple sclerosis. J. Neurol. 2006, 253, 1002–1010. [Google Scholar] [CrossRef]
- Popović, D.B.; Sinkjær, T. Diseases and Injuries of the Central Nervous System Leading to Sensory-Motor Impairment. In Introduction to Neural Engineering for Motor Rehabilitation; Wiley: Hoboken, NJ, USA, 2013; pp. 3–20. [Google Scholar] [CrossRef]
- Shaw, M.A.; Gao, Z.; McElhinney, K.E.; Thornton, S.; Flick, M.J.; Lane, A.; Degen, J.L.; Ryu, J.K.; Akassoglou, K.; Mullins, E.S. Plasminogen Deficiency Delays the Onset and Protects from Demyelination and Paralysis in Autoimmune Neuroinflammatory Disease. J. Neurosci. 2017, 37, 3776–3788. [Google Scholar] [CrossRef]
- Zipser, C.M.; Cragg, J.J.; Guest, J.D.; Fehlings, M.G.; Jutzeler, C.R.; Anderson, A.J.; Curt, A. Cell-based and stem-cell-based treatments for spinal cord injury: Evidence from clinical trials. Lancet Neurol. 2022, 21, 659–670. [Google Scholar] [CrossRef]
- Badner, A.; Siddiqui, A.M.; Fehlings, M.G. Spinal cord injuries: How could cell therapy help? Expert Opin. Biol. Ther. 2017, 17, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.K.; Choi, I.; Jeon, S.R. Current Status and Future Strategies to Treat Spinal Cord Injury with Adult Stem Cells. J. Korean Neurosurg. Soc. 2020, 63, 153–162. [Google Scholar] [CrossRef]
- Laroni, A.; de Rosbo, N.K.; Uccelli, A. Mesenchymal stem cells for the treatment of neurological diseases: Immunoregulation beyond neuroprotection. Immunol. Lett. 2015, 168, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guan, H.; Zhang, Y.; Li, S.; Li, K.; Hu, S.; Zuo, E.; Zhang, C.; Zhang, X.; Gong, G.; et al. Bone marrow mesenchymal stem cells promote remyelination in spinal cord by driving oligodendrocyte progenitor cell differentiation via TNFα/RelB-Hes1 pathway: A rat model study of 2,5-hexanedione-induced neurotoxicity. Stem Cell Res. Ther. 2021, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Dörnen, J.; Dittmar, T. The Role of MSCs and Cell Fusion in Tissue Regeneration. Int. J. Mol. Sci. 2021, 22, 10980. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Tunger, A.; Wobus, M.; von Bonin, M.; Towers, R.; Bornhäuser, M.; Dazzi, F.; Wehner, R.; Schmitz, M. Immunomodulatory Properties of Mesenchymal Stromal Cells: An Update. Front. Cell Dev. Biol. 2021, 9, 637725. [Google Scholar] [CrossRef] [PubMed]
- Czapla, J.; Matuszczak, S.; Kulik, K.; Wiśniewska, E.; Pilny, E.; Jarosz-Biej, M.; Smolarczyk, R.; Sirek, T.; Zembala, M.O.; Zembala, M.; et al. The effect of culture media on large-scale expansion and characteristic of adipose tissue-derived mesenchymal stromal cells. Stem Cell Res. Ther. 2019, 10, 235. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kang, M.H.; Jang, J.E.; Lee, J.E.; Yang, Y.; Choi, J.Y.; Kang, H.S.; Lee, U.; Choung, J.W.; Jung, H.; et al. Comparative analysis of mesenchymal stem cells cultivated in serum free media. Sci. Rep. 2022, 12, 8620. [Google Scholar] [CrossRef]
- Kirkham, A.M.; Bailey, A.J.M.; Shorr, R.; Lalu, M.M.; Fergusson, D.A.; Allan, D.S. Systematic review and meta-analysis of randomized controlled trials of mesenchymal stromal cells to treat coronavirus disease 2019: Is it too late? Cytotherapy 2023, 25, 341–352. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, Y.; Wu, X.; Xu, X.; Niu, J. The assessment of mesenchymal stem cells therapy in acute on chronic liver failure and chronic liver disease: A systematic review and meta-analysis of randomized controlled clinical trials. Stem Cell Res. Ther. 2022, 13, 204. [Google Scholar] [CrossRef]
- Mohamed, S.A.; Howard, L.; McInerney, V.; Hayat, A.; Krawczyk, J.; Naughton, S.; Finnerty, A.; Holohan, M.; Duffy, A.; Moloney, T.; et al. Autologous bone marrow mesenchymal stromal cell therapy for “no-option” critical limb ischemia is limited by karyotype abnormalities. Cytotherapy 2020, 22, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Premer, C.; Blum, A.; Bellio, M.A.; Schulman, I.H.; Hurwitz, B.E.; Parker, M.; Dermarkarian, C.R.; DiFede, D.L.; Balkan, W.; Khan, A.; et al. Allogeneic Mesenchymal Stem Cells Restore Endothelial Function in Heart Failure by Stimulating Endothelial Progenitor Cells. EBioMedicine 2015, 2, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sun, Q.L.; Duan, L.J.; Yang, Y.D.; Gao, Y.S.; Zhao, D.Y.; Xiong, Y.; Wang, H.J.; Song, J.W.; Yang, K.T.; et al. Is cell transplantation a reliable therapeutic strategy for spinal cord injury in clinical practice? A systematic review and meta-analysis from 22 clinical controlled trials. Eur. Spine J. 2019, 28, 1092–1112, Correction in Eur. Spine J. 2020, 29, 2105. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.; Viswanathan, C. Establishment of a mesenchymal stem cell bank. Stem Cells Int. 2011, 2011, 905621. [Google Scholar] [CrossRef] [PubMed]
- Massey, C.J.; Diaz Del Valle, F.; Abuzeid, W.M.; Levy, J.M.; Mueller, S.; Levine, C.G.; Smith, S.S.; Bleier, B.S.; Ramakrishnan, V.R. Sample collection for laboratory-based study of the nasal airway and sinuses: A research compendium. Int. Forum Allergy Rhinol. 2020, 10, 303–313. [Google Scholar] [CrossRef]
- Lledo, P.M.; Valley, M. Adult Olfactory Bulb Neurogenesis. Cold Spring Harb. Perspect. Biol. 2016, 8, a018945. [Google Scholar] [CrossRef] [PubMed]
- Mackay-Sim, A.; Feron, F.; Cochrane, J.; Bassingthwaighte, L.; Bayliss, C.; Davies, W.; Fronek, P.; Gray, C.; Kerr, G.; Licina, P.; et al. Autologous olfactory ensheathing cell transplantation in human paraplegia: A 3-year clinical trial. Brain 2008, 131, 2376–2386. [Google Scholar] [CrossRef]
- Lindsay, S.L.; Johnstone, S.A.; Mountford, J.C.; Sheikh, S.; Allan, D.B.; Clark, L.; Barnett, S.C. Human mesenchymal stem cells isolated from olfactory biopsies but not bone enhance CNS myelination in vitro. Glia 2013, 61, 368–382. [Google Scholar] [CrossRef]
- Lindsay, S.L.; Johnstone, S.A.; McGrath, M.A.; Mallinson, D.; Barnett, S.C. Comparative miRNA-Based Fingerprinting Reveals Biological Differences in Human Olfactory Mucosa- and Bone-Marrow-Derived Mesenchymal Stromal Cells. Stem Cell Rep. 2016, 6, 729–742. [Google Scholar] [CrossRef]
- Nicolas, F.E.; Pais, H.; Schwach, F.; Lindow, M.; Kauppinen, S.; Moulton, V.; Dalmay, T. Experimental identification of microRNA-140 targets by silencing and overexpressing miR-140. RNA 2008, 14, 2513–2520. [Google Scholar] [CrossRef]
- Lindsay, S.L.; Toft, A.; Griffin, J.; Emraja, A.M.M.; Barnett, S.C.; Riddell, J.S. Human olfactory mesenchymal stromal cell transplants promote remyelination and earlier improvement in gait co-ordination after spinal cord injury. Glia 2017, 65, 639–656. [Google Scholar] [CrossRef]
- Lindsay, S.L.; Molęda, A.M.; MacLellan, L.M.; Keh, S.M.; McElroy, D.E.; Linington, C.; Goodyear, C.S.; Barnett, S.C. Human olfactory mesenchymal stromal cell transplantation ameliorates experimental autoimmune encephalomyelitis revealing an inhibitory role for IL16 on myelination. Acta Neuropathol. Commun. 2022, 10, 12. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Mushahary, D.; Spittler, A.; Kasper, C.; Weber, V.; Charwat, V. Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytom. Part A 2018, 93, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Ferrin, I.; Beloqui, I.; Zabaleta, L.; Salcedo, J.M.; Trigueros, C.; Martin, A.G. Isolation, Culture, and Expansion of Mesenchymal Stem Cells. In Stem Cell Banking: Concepts and Protocols; Crook, J.M., Ludwig, T.E., Eds.; Springer: New York, NY, USA, 2017; pp. 177–190. [Google Scholar] [CrossRef]
- Jaloux, C.; Bonnet, M.; Vogtensperger, M.; Witters, M.; Veran, J.; Giraudo, L.; Sabatier, F.; Michel, J.; Legré, R.; Guiraudie-Capraz, G.; et al. Human nasal olfactory stem cells, purified as advanced therapy medicinal products, improve neuronal differentiation. Front. Neurosci. 2022, 16, 1042276. [Google Scholar] [CrossRef]
- Skiles, M.L.; Marzan, A.J.; Brown, K.S.; Shamonki, J.M. Comparison of umbilical cord tissue-derived mesenchymal stromal cells isolated from cryopreserved material and extracted by explantation and digestion methods utilizing a split manufacturing model. Cytotherapy 2020, 22, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, R.R.; Cetrulo, K.J.; Cetrulo, C.L. Collagenase Impacts the Quantity and Quality of Native Mesenchymal Stem/Stromal Cells Derived during Processing of Umbilical Cord Tissue. Cell Transpl. 2018, 27, 181–193. [Google Scholar] [CrossRef]
- Yoon, J.H.; Roh, E.Y.; Shin, S.; Jung, N.H.; Song, E.Y.; Chang, J.Y.; Kim, B.J.; Jeon, H.W. Comparison of explant-derived and enzymatic digestion-derived MSCs and the growth factors from Wharton’s jelly. Biomed. Res. Int. 2013, 2013, 428726. [Google Scholar] [CrossRef]
- Meneghel, J.; Kilbride, P.; Morris, G.J. Cryopreservation as a Key Element in the Successful Delivery of Cell-Based Therapies-A Review. Front. Med. (Lausanne) 2020, 7, 592242. [Google Scholar] [CrossRef]
- Weng, L. Cell Therapy Drug Product Development: Technical Considerations and Challenges. J. Pharm. Sci. 2023, 112, 2615–2620. [Google Scholar] [CrossRef]
- Fehlings, M.G.; Vaccaro, A.; Wilson, J.R.; Singh, A.; Cadotte, D.W.; Harrop, J.S.; Aarabi, B.; Shaffrey, C.; Dvorak, M.; Fisher, C.; et al. Early versus delayed decompression for traumatic cervical spinal cord injury: Results of the Surgical Timing in Acute Spinal Cord Injury Study (STASCIS). PLoS ONE 2012, 7, e32037. [Google Scholar] [CrossRef] [PubMed]
- Bahsoun, S.; Coopman, K.; Akam, E.C. The impact of cryopreservation on bone marrow-derived mesenchymal stem cells: A systematic review. J. Transl. Med. 2019, 17, 397. [Google Scholar] [CrossRef] [PubMed]
- Winkel, A.; Jaimes, Y.; Melzer, C.; Dillschneider, P.; Hartwig, H.; Stiesch, M.; von der Ohe, J.; Strauss, S.; Vogt, P.M.; Hamm, A.; et al. Cell culture media notably influence properties of human mesenchymal stroma/stem-like cells from different tissues. Cytotherapy 2020, 22, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, S.; Moradi, B.; Frank, S.; Dreher, T.; Kämmerer, P.W.; Richter, W.; Gotterbarm, T. Different culture media affect growth characteristics, surface marker distribution and chondrogenic differentiation of human bone marrow-derived mesenchymal stromal cells. BMC Musculoskelet. Disord. 2013, 14, 223. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, R.; Medina-Ruiz, L.; Hayes, A.J.; Kelly, C.J.; Halawa, H.A.; Graham, G.J. Inflammatory Chemokine Receptors Support Inflammatory Macrophage and Dendritic Cell Maturation. Immunohorizons 2022, 6, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-H.K.; Ogando, C.R.; Wang See, C.; Chang, T.-Y.; Barabino, G.A. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res. Ther. 2018, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Pingen, M.; Bryden, S.R.; Pondeville, E.; Schnettler, E.; Kohl, A.; Merits, A.; Fazakerley, J.K.; Graham, G.J.; McKimmie, C.S. Host Inflammatory Response to Mosquito Bites Enhances the Severity of Arbovirus Infection. Immunity 2016, 44, 1455–1469. [Google Scholar] [CrossRef]
- Sorensen, A.; Moffat, K.; Thomson, C.; Barnett, S.C. Astrocytes, but not olfactory ensheathing cells or Schwann cells, promote myelination of CNS axons in vitro. Glia 2008, 56, 750–763. [Google Scholar] [CrossRef]
- Yamamura, T.; Konola, J.T.; Wekerle, H.; Lees, M.B. Monoclonal antibodies against myelin proteolipid protein: Identification and characterization of two major determinants. J. Neurochem. 1991, 57, 1671–1680. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker: | Extra/Intracellular: | Clone: | Fluorophore: | Manufacturer: | mL/Sample: |
|---|---|---|---|---|---|
| CD73 | Extracellular | AD2 | PE | Biolegend | 0.25 |
| CD90 | Extracellular | 5E10 | AF700 | Biolegend | 1 |
| CD105 | Extracellular | 266 | BUV395 | BD Biosciences | 1 |
| CD271 | Extracellular | ME20.4 | PerCP/Cy5.5 | Biolegend | 1 |
| Nestin | Intracellular | 10C2 | AF488 | Biolegend | 1 |
| CD14 | Extracellular | HCD14 | PE/Cy7 | Biolegend | 0.5 |
| CD19 | Extracellular | HIB19 | BV785 | Biolegend | 0.5 |
| CD34 | Extracellular | 561 | BV421 | Biolegend | 0.5 |
| CD45 | Extracellular | HI30 | APC | Biolegend | 0.5 |
| HLA:DR | Extracellular | L243 | BV510 | Biolegend | 0.5 |
| GOI: | For/Rev: | Sequence: |
|---|---|---|
| GAPDH | F | GGTCACCAGGGCTGCTTTTA |
| GAPDH | R | TTCCCGTTCTCAGCCTTGAC |
| LPL | F | TCACAGCAGCAAAACCTTCATG |
| LPL | R | CAGCCAGTCCACCACAATGA |
| PPARg | F | AAGCCCTTCACTACTGTTGACTT |
| PPARg | R | GCAGGCTCCACTTTGATTGC |
| BGLAP | F | CCTCACACTCCTCGCCCTAT |
| BGLAP | R | CGCCTGGGTCTCTTCACTAC |
| RUNX2 | F | AACAGCCTCTTCAGCACAGT |
| RUNX2 | R | GCTCACGTCGCTCATTTTGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelly, C.J.; Lindsay, S.L.; Smith, R.S.; Keh, S.; Cunningham, K.T.; Thümmler, K.; Maizels, R.M.; Campbell, J.D.M.; Barnett, S.C. Development of Good Manufacturing Practice-Compatible Isolation and Culture Methods for Human Olfactory Mucosa-Derived Mesenchymal Stromal Cells. Int. J. Mol. Sci. 2024, 25, 743. https://doi.org/10.3390/ijms25020743
Kelly CJ, Lindsay SL, Smith RS, Keh S, Cunningham KT, Thümmler K, Maizels RM, Campbell JDM, Barnett SC. Development of Good Manufacturing Practice-Compatible Isolation and Culture Methods for Human Olfactory Mucosa-Derived Mesenchymal Stromal Cells. International Journal of Molecular Sciences. 2024; 25(2):743. https://doi.org/10.3390/ijms25020743
Chicago/Turabian StyleKelly, Christopher J., Susan L. Lindsay, Rebecca Sherrard Smith, Siew Keh, Kyle T. Cunningham, Katja Thümmler, Rick M. Maizels, John D. M. Campbell, and Susan C. Barnett. 2024. "Development of Good Manufacturing Practice-Compatible Isolation and Culture Methods for Human Olfactory Mucosa-Derived Mesenchymal Stromal Cells" International Journal of Molecular Sciences 25, no. 2: 743. https://doi.org/10.3390/ijms25020743
APA StyleKelly, C. J., Lindsay, S. L., Smith, R. S., Keh, S., Cunningham, K. T., Thümmler, K., Maizels, R. M., Campbell, J. D. M., & Barnett, S. C. (2024). Development of Good Manufacturing Practice-Compatible Isolation and Culture Methods for Human Olfactory Mucosa-Derived Mesenchymal Stromal Cells. International Journal of Molecular Sciences, 25(2), 743. https://doi.org/10.3390/ijms25020743